Background
Pediatric cancers typically present similar
pathohistological features to adult cancers, but at the same time,
they can have a strikingly different molecular signature.
Therefore, successful treatment of adult and pediatric cancers can
greatly differ (1,2). One of the most representative
molecular characteristics of pediatric cancers is a low mutational
burden, where either a single gene can be highly mutated or a gene
fusion can be formed as a byproduct of genomic rearrangements
(3,4). Gene fusions are the most frequent
oncogenic driver (and often unique driver) of many subtypes of
pediatric cancer and are thus typically used as a biomarker for
unequivocal diagnosis (1,5). In pediatric lymphomas, leukemias, and
soft tissue sarcomas, gene fusions are present in 90, 50, and 30%
of all cases, respectively (6).
Inactivation or knockout of the gene fusion can directly inhibit
tumor growth, implying that drugs selectively targeting the
chimeric oncoprotein should be developed (7).
Gene fusions can contribute to oncogenicity by
generating new chimeric proteins that can result either in the loss
of function of the original gene or the gain of function of the new
chimeric protein. The chimeric protein expression can diverge into
rearrangements of critical molecular pathways and thus disturb
normal cell function. Furthermore, the expression of the fusions
can also affect the expression profile of oncogenes and/or tumor
suppressor genes (3,4). The combinations and distributions of
preserved domains in gene fusions seem to be non-random (8). In general, a DNA-binding domain is at
the 3′-end of a fusion oncogene, and a potent proto-oncogene
(tyrosine kinase, transcription factor, or a histone modifier) is
at the 5′-end (9).
Nuclear receptor coactivators (NCoA) function as a
critical link between activated nuclear receptors (NR) and the
transcription machinery. They are responsible for transducing the
NR signals in the presence of the ligand, resulting in the
induction of the transcription of NR target genes (10,11). A
subset of NCoAs belong to the p160 coactivator family and are
essential coregulators in several physiological processes, such as
inflammatory and metabolic pathways, where they transform the
environmental signals into epigenome alterations and
transcriptional responses (12).
When a p160 family member becomes a partner gene in a new gene
fusion, the new chimeric protein becomes a strong oncogenic driver
through the regulation of transcription. Oncogenic fusions with
p160 family members at their C-terminal are very frequent in a
variety of pediatric malignancies (13–15).
These domains contain large intrinsically disordered regions that
lack hydrophobic pockets where small molecules could bind, making
chimeric proteins impossible to directly target with small molecule
inhibitors (16).
Here we provide an overview of the known literature
on NCoA1/2/3 structure, regulation, and function. Next, we explore
and comment on the role of p160 family members as a fusion partner
gene and a contributing factor in tumorigenesis. We are focused on
cancers that have at least one reported gene alteration involving a
p160 family member fused to another gene, and try to understand the
common approaches that could target these types of cancers. We then
further summarize recent research on these tumors, and explore
current and future treatment possibilities. Lastly, we provide
insights into technologies that could be utilized to directly
target these undruggable oncogenic fusions.
The structure and function of the p160
coactivator family
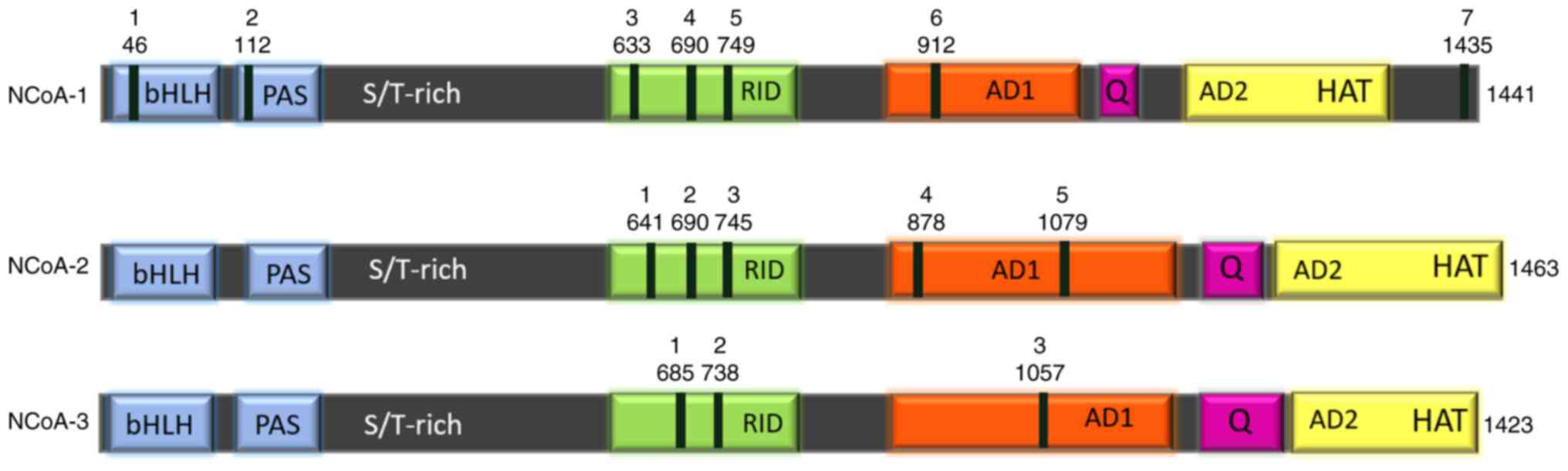
The p160 coactivator family consists of three
members: NCoA1 (SRC1), NCoA2 (SRC2/TIF2/GRIP1), and NCoA3
(SRC3/p-CIP/RAC3/ACTR/AIB1/TRAM-1) (17,18).
In humans, these genes present 54–58% of sequence identity, and
they are believed to have originated from gene duplication events
(19) (Fig. 1). The most conserved regions among
all three members are the basic helix-loop-helix (bHLH) and
Per/Arnt/Sim (PAS) domains, commonly annotated as bHLH/PAS at the
N-terminal end (20).
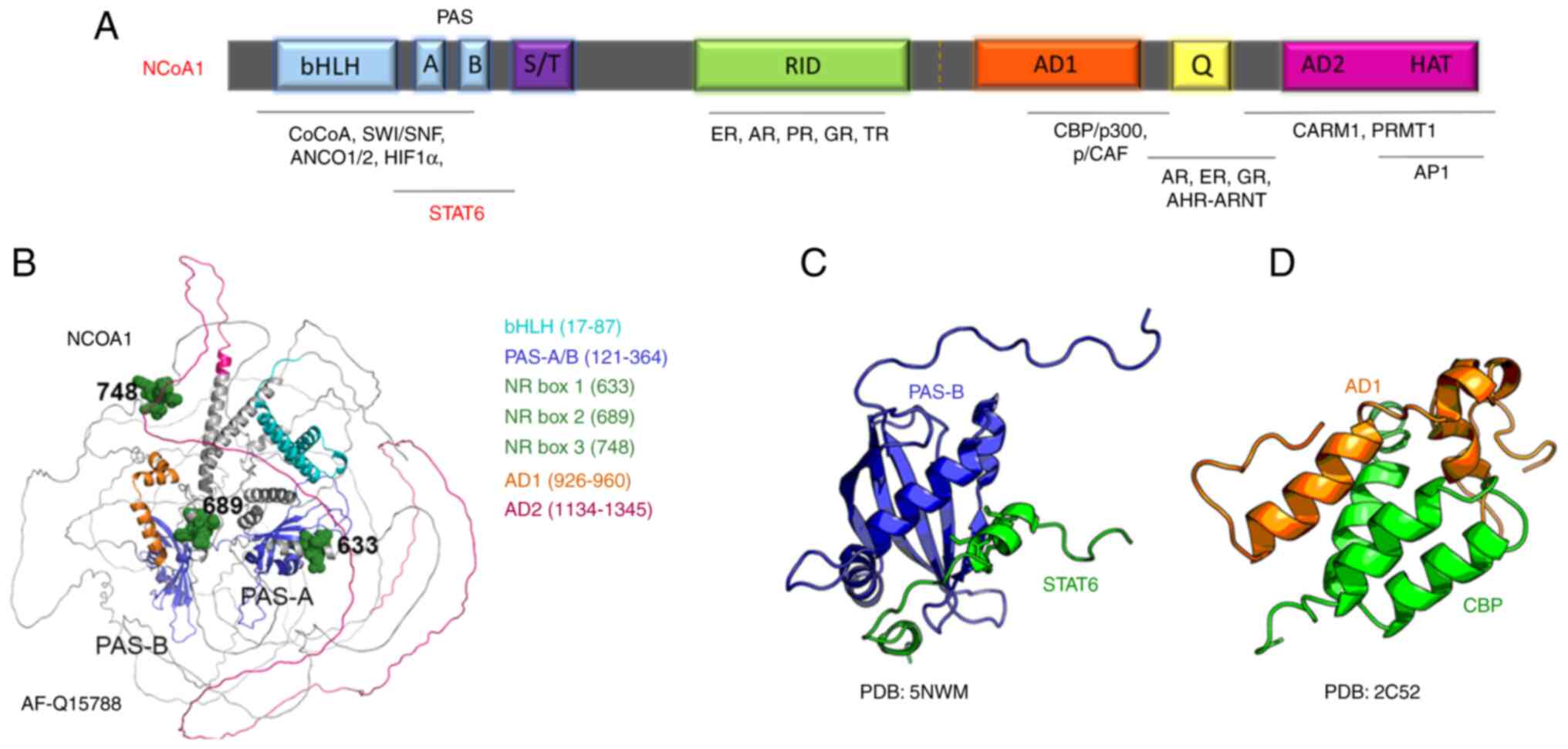
bHLH domains are known to mediate dimerization with
other transcription factors as well as DNA binding, signal sensing,
and signal transduction (20,21).
Nonetheless, DNA binding activity still hasn't been described for
p160 family members. The bHLH-PAS domain of p160 family members is
well characterized as a protein-protein interaction region, capable
of binding to secondary coregulators (including CoCoA, GAC 63, and
Flii), and transcription factors such as p53, MEF2C, TEAD2, and
STAT (20–22) (Fig.
2A). The PAS-B domain has been shown to interact with LXXLL
motifs, where L represents leucine and X stands for any amino acid.
These LXXLL motifs are located on the C-terminal domains of all
three NCoA homologs, where they contribute to their homo- and
hetero-dimerization, as well as dimerization with other proteins
containing LXXLL (23,24).
The serine and threonine-rich region (S/T-rich) that
follows the bHLH-PAS domain is a hotspot for posttranslational
modifications, important for p160 protein regulation (17). Immediately after the S/T-rich region
lies the receptor interaction domain (RID) that contains three
LXXLL motifs, called nuclear receptor boxes (NR boxes) (18). The three NR boxes are necessary for
binding to a hydrophobic pocket in the Nuclear Receptor
ligand-binding domain (LBD). This interaction represents the first
physical contact between NR and the coactivator before the signal
is transmitted to secondary coregulators (22).
The C-terminal region of p160 family members
consists of two activation domains, called AD1 and AD2, which act
as potent mediators of epigenetic enzymatic activities required to
modulate gene transcription (17,25).
The AD1 domain [also called CBP-interaction domain (CID) or
p300-interaction domain (PID)], recruits secondary coregulators
which are responsible for chromatin remodeling. The AD1 domain also
contains LXXLL motifs important for interaction with CBP/p300, AP1,
members of the bHLH-PAS protein family such as AHR, ARNT/HIF-1β,
and transcription factor NF-κB amongst others (26,27).
One of the roles of the AD1 is to recruit components of the RNA Pol
II transcription preinitiation complex and RNA helicase A, which
initiate the transcription (22,28).
The AD1 can also recruit histone methyltransferases such as
coactivator-associated arginine methyltransferase 1 (CARM1) and
protein arginine N-methyltransferase 1 (PRMT1), leading to
chromatin remodeling and decondensation (29).
Immediately after the AD2 domain, there is a weak
intrinsic histone acetyltransferase (HAT) activity contributing to
the acetylation of the downstream transcriptional machinery
components (30). Due to the HAT
activity, the NCoA homologs have HAT names, such as KAT13A for
NCoA1, KAT13B for NCoA2, and KAT13C for NCoA3 (31). Certain splicing isoforms (for
example, NcoA1a) contain an additional LXXLL motif in their extreme
C-terminal end, contributing to NR binding (32,33).
Finally, a Q-rich region with abundant glutamine repetitions lies
between AD1 and AD2 and is important for the mediation of
ligand-independent NR signal transduction activity (34,35).
The structural prediction by Alpha Fold for NCoA1
shows a structured bHLH/PAS domain, while the rest of the protein
presents a high component of unstructured regions (36) (Fig.
2B). Structural predictions of the other p160 family members
show a similar pattern. The crystal and NMR structures of the NCoA1
PAS-B domain in a complex with a STAT6-derived peptide were solved
(37,38) (Fig.
2C). Another NMR structure of the AD1 domain of NCoA1 showed
details of the interaction with a peptide derived from the CREB
binding protein (CBP) (Fig. 2D).
Additional structures of small peptides of NCoA1/2/3 with protein
interactors are available and demonstrate the ability of these
proteins to function as binding platforms for multiple proteins to
promote epigenetic modifications and transcription.
The stability and activity of p160 proteins can be
modulated by post-translational modifications (PMTs), such as
phosphorylation, sumoylation, ubiquitination, acetylation, and
methylation (39,40). Several phosphorylation sites have
been identified in Ser/Thr-Pro motifs, which are targets of
proline-directed kinases, including CDKs, MAPK, cAMP-PKA, and NF-κB
kinase-mediated signaling pathways (39,41,42).
The majority of these Ser/Thr phosphorylation sites are located
either in the S/T-rich region, while some sites reside in the
Q-rich domain at the C-t. Changes in phosphorylation state have
been shown to influence the NCoA preference for different NRs
(42–44). In addition, phosphorylation can
modulate the interaction with CBP/p300, and in some cases also
induce the degradation of some p160 members (41,45).
Tyrosine phosphorylation has also been implicated in
the regulation of NCoAs. For example, phosphorylation at Tyr-1357
by c-Abl kinase was reported to increase the binding of NCoA3 to
p300 and ERα, while decreasing its association with the repressor
CARM1. This phosphorylation site is conserved in NCoA2 while
missing in NCoA1 (46).
Ubiquitination plays an important role in the
stability of p160 family members. The addition of a long
polyubiquitin chain to the C-terminal region of p160 proteins
mediates their proteasomal degradation via the 26S
ubiquitin-proteasome pathway. The AD2 domain in NCoA2 was shown to
be essential for 26S proteasome degradation (47). Sumoylation of p160 family members
directs the subcellular localization and can affect protein-protein
interactions (48–50), while acetylation can have an impact
on the regulation of hormonal signaling (51). The methylation of p160 family
members occurs by CARM1 recruitment, leading to disruption of
CBP/p300/p160 interactions and transcriptional repression (52).
In short, the binding of the NR to a specific ligand
induces conformational changes in its ligand binding domain (LBD),
enabling the dissociation of corepressors, and binding of NCoAs
through its LXXLL motifs. This interaction is essential to mediate
the NR responses (53). Once the
NR-bounded NCoA is activated, it recruits CBP, p300, p/CAF, and
other transcriptional factors, leading to acetylation modulation of
core histones, and chromatin decondensation (54). Since histone acetylation is not
sufficient to activate the transcription of target genes, NCoA also
serves as an important scaffold for the assembly of the
transcription machinery and recruitment of transcription factors
(TFIIB, TBP, TAFs, TFIIH) at the promoter and/or enhancer regions
of NR targeted genes (55)
(Fig. 3).
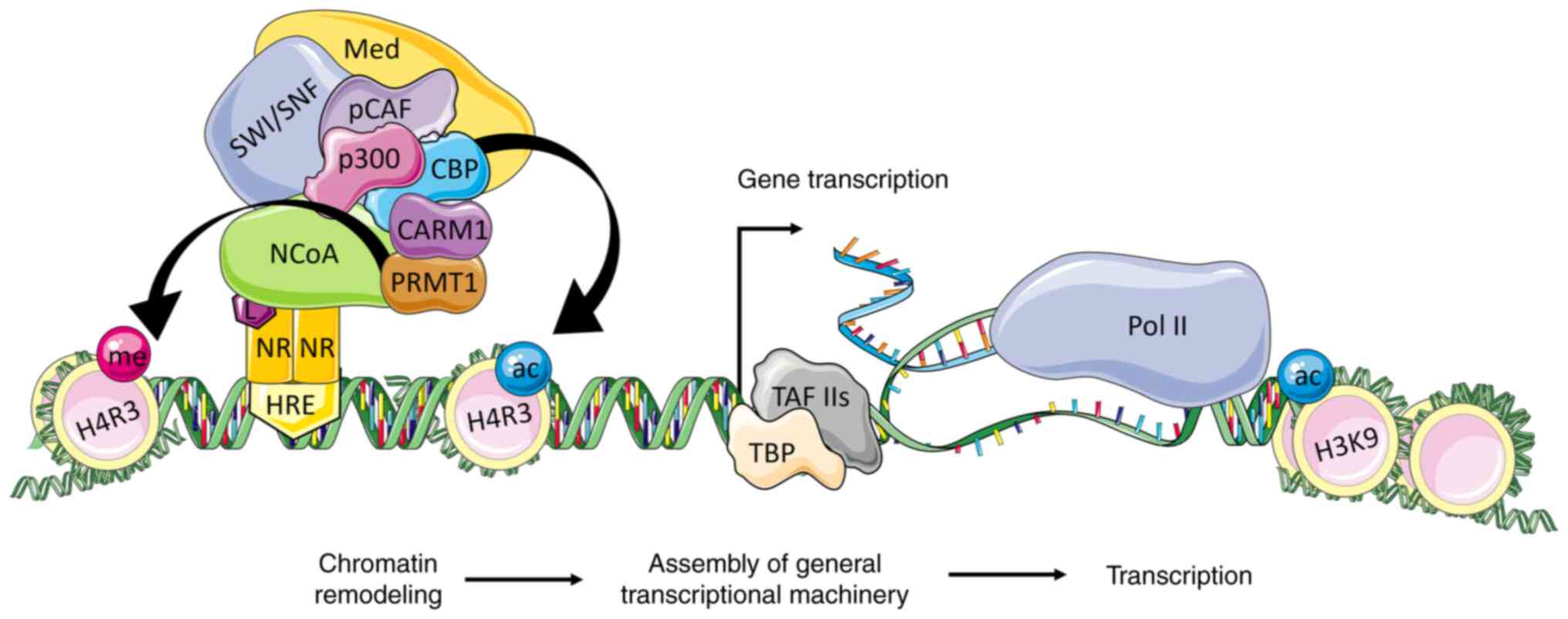 | Figure 3.The NCoA coactivation of
transcription in the ligand-dependent pathway. The p160 family
members interact via the RID domain with the ligand-activated
nuclear receptor that is bound to its HRE. The activated NCoA binds
CBP/p300 to its AD1 domain and CARM1/PRMT1 to its AD2 domain. The
CBP and p300 acetylate histones and facilitate the recruitment of
SWI/SNF complex for further chromatin remodeling. This leads to
changes in the DNA topology, exposing the regulatory DNA sequences
to the basal transcription machinery. The Med is activated by p300
and NCoA and facilitates the recruitment of TBP and TAFs to form
the link with RNA polymerase II and initiate the transcription of
target genes. NR, nuclear receptor; HRE, hormone-responsive
elements; L, ligand; NCoA, nuclear receptor coactivator; CBP, p300
and pCAF, histone acetyltransferases; CARM1 and PRMT1, histone
methyltransferases; SWI/SNF, ATP-dependent chromatin remodeling
complex; Med, mediator complex; TBP, TATA-box-binding protein;
TAFs, TBP associated factors; ac, acetylation; me, methylation;
H3K9, histone H3 Lys9; H4R3, H4 Arg3. |
The role of p160 protein domains in
fusion-driven cancers
All members of the p160 coactivator family have been
identified as partner genes in many aggressive gene-fusion-driven
cancers. Usually the truncated p160 members are positioned at the
C-terminal of the chimeric protein, where they retain their
C-terminal domains (AD1, Q-rich region, and AD2). The N-terminal
region of the chimeric protein is mostly a DNA-binding gene
partner. This makes the N-terminal domains of the newly generated
gene fusion a facilitator of the DNA binding to target locations,
while the C-terminal domains can recruit CBP/p300 and other
transcription factors, resulting in the reprogramming of the
cellular transcriptional profile (Table
I).
 | Table I.Fusion oncogenes with p160 family
members as a partner gene and their corresponding tumor type. |
Table I.
Fusion oncogenes with p160 family
members as a partner gene and their corresponding tumor type.
| First author,
year | Gene fusion | Translocation | Type | (Refs.) |
|---|
| Wachtel, 2004 | PAX3-NCoA1 |
t(2;2)(p23;q35) | Alveolar
habdomyosarcoma | (64) |
| Yoshida, 2004 | PAX3-NCoA2 |
t(2;8)(q36;q13)/ |
Alveolar/embryonal | (13) |
|
|
|
t(2;8)(q35;q13) |
rhabdomyosarcoma |
|
| Wachtel, 2004 | NCoA1-PAX3 |
t(2;2)(q35;p23) |
Rhabdomyosarcoma | (64) |
| Alaggio, 2006 | SRF-NCoA2 |
t(6;8)(p12;q11) | Spindle cell
rhabdomyosarcoma | (92) |
| Tan, 2020 | TEAD1-NCoA2 |
t(8;11)(q13;p15) | Spindle cell
rhabdomyosarcoma | (116) |
| Alaggio, 2004 | VGLL2-NCoA2 |
t(6;8)(q22;q13) | Spindle cell
rhabdomyosarcoma | (92) |
| Avenarius,
2020 | WHSC1L1-NCoA2 |
| Spindle cell
rhabdomyosarcoma | (117) |
| Argani, 2018 | MEIS1-NCoA2 |
t(2;8)(p14;q13.3) | Spindle cell
rhabdomyosarcoma | (59) |
| Bennett, 2020 | ESR1-NCoA2 |
t(8;20)(p13.3;q13.3) | Spindle cell
rhabdomyosarcoma | (118) |
| Piscuoglio,
2016 | ESR1-NCoA3 |
t(6;20)(q25.1;q13.3) | Müllerian
adenosarcomas | (60) |
| Bekers, 2017 | GTF2I-NCoA2 |
t(7;8)(q11;q13) | Soft tissue
angiofibroma | (58) |
| Panagopoulos,
2016 | NCoA2-ETV4 |
t(8;17)(q13;q21) | Soft tissue
angiofibroma | (119) |
| Bekers, 2017 | AHRR-NCoA2 |
t(5;8)(p15;q13) | Soft tissue
angiofibroma | (58) |
| Teramura, 2020 | AHRR-NCOA3 | t (5;8)
(p15;q13) | Spindle cell
sarcoma | (120) |
| Zhou, 2020 | ETV6-NCoA2 |
t(8;12)(q13;p13) | Acute myeloid
leukemia | (121) |
| Zhuravleva,
2008 | MOZ-NCoA2 | t(8;16)
(p11;p13) | Acute myeloid
leukemia | (122) |
| Esteyries,
2008 | MOZ-NCoA3 |
t(8;20)(p11;q13) | Acute myeloid
leukemia | (123) |
| Wang, 2012 | HEY1-NCoA2 |
t(8;8)(q13;q21) | Mesenchymal
chondrosarcoma | (56) |
| Chang, 2020 | GREB1-NCoA2 |
t(2;8)(p25;q13) | Uterine
sarcoma | (124) |
| Lacambra, 2019 | PRRX1-NCoA1 |
t(1;2)(q24.2;p23.3) | Fibroblastic
neoplasms | (63) |
| Lacambra, 2019 | PRRX1-NCoA2 |
t(1;8)(q24.2;q13.3) | Fibroblastic
neoplasms | (63) |
| Yu, 2016 | LACTB2-NCoA2 |
t(8;8)(q13;q13) | Colon-Rectum
adenocarcinoma | (125) |
| Cao, 2019 | NCoA1-ALK |
| Lung
adenocarcinoma | (126) |
| Yoshihara,
2015 | NCoA2-LEPROTL1 |
t(8;8)(p12;q13) | Lung
adenocarcinoma | (127) |
| Yoshihara,
2015 | NCoA2-XKR9 |
t(8;8)(q13;q13) | Lung
adenocarcinoma | (127) |
| Yoshihara,
2015 | NCoA2-NCALD |
t(8;8)(q13;q22) | Breast
adenocarcinoma | (127) |
| Yoshihara,
2015 | NCoA2-ARFGEF1 |
t(8;8)(q13;q13) | Breast
adenocarcinoma | (127) |
| Robinson, 2011 | NCoA2-ZNF704 |
t(8;8)(q13;q21) | Breast
adenocarcinoma | (128) |
| Yoshihara,
2015 | RAB10-NCoA1 |
t(2;2)(p23;p23) | Breast:
Adenocarcinoma | (127) |
| Yoshihara,
2015 | SH2D6-NCoA2 |
t(2;8)(p11;q13) | Bladder
transitional cell carcinoma | (127) |
| Yoshihara,
2015 | NCoA2-ST18 |
t(8;8)(q11;q13) | Melanoma | (127) |
The p160 RID domain is usually missing in the
fusions, making the chimeric proteins less likely to interact with
the ligand-dependent NR signaling pathways. In contrast,
ligand-independent pathways that rely on the Q-rich region and/or
LXXLL motifs could still be active (10,11).
For instance, in the case of several NRs, it has been reported that
the C-terminal LXXLL motifs in p160 members can contribute to
nearly wild-type binding efficiency to the LBD domain in the NR
such as estrogen receptors (ER), glucocorticoid receptors, retinoic
acid receptors, and retinoic X receptors (54). Furthermore, the splicing isoform of
NCoA1 (NCoA1a) is capable of binding glucocorticoid and androgen
receptors (AR) solely through its additional extreme C-terminal
LXXLL motif (32). These examples
suggest that the C-terminal domain of p160 members could mediate
some of the NR-dependent functions even in the absence of their RID
domain, which could be preserved in the p160-fusion-driven
malignancies.
Oncogenic gene fusions with NCoA as a gene
partner
Among the three p160 family members, NCoA2 is the
gene most frequently involved in the formation of oncogenic
fusions, predominantly in pediatric cancers (Table I). These fusions have been detected
in mesenchymal chondrosarcoma (56), variants of rhabdomyosarcoma
(57), soft tissue angiofibroma
(58), kidney spindle cell sarcoma
(59), uterine adenosarcoma
(60), ovarian sex cord tumor
(61), biphenotypic sinonasal
sarcoma (62), and myelogenous
leukemia/fibroblastic neoplasms (63). The other two family members have
likewise been implicated in mesenchymal lesions, but interestingly
they are mostly present in adult tumors. Translocation in NCoA1 and
NCoA3 has been observed in rhabdomyosarcoma (NCoA1) (64), uterine adenosarcoma (NCoA3)
(65), ovarian sex cord tumor
(NCoA3) (61), biphenotypic
sinonasal sarcoma (NCoA1) (62),
and myelogenous leukemia/fibroblastic neoplasms (NCoA1) (63), among others.
Mesenchymal chondrosarcoma
Mesenchymal chondrosarcoma (MCS) is a rare neoplasm
that is characterized by the presence of primitive mesenchymal
cells mixed with sections of cartilage differentiation. MCS
typically arises from bone, and current treatment includes surgical
resection coupled with cytotoxic chemotherapy (66). MCS is one of the most aggressive
subtypes of chondrosarcoma, evidenced by low survival rates and
limited treatment options (67).
MCS presents similar histological features to many other soft
tissue sarcomas, making its correct diagnosis significantly
challenging (2). The discovery of a
recurrent oncogenic fusion between HEY1 and NCoA2, occurring in
over 80% of MCS samples, made it possible to distinguish MCS and
has been used as a diagnostic biomarker (56,68).
In HEY1-NCoA2 fusion, the bHLH domain of HEY1, which strongly binds
DNA is preserved at the N-terminal end, while at the C-terminal
AD1, Q-rich, and AD2 domains of NCoA2 are preserved (69).
Pioneering IHC staining-based studies of MCS tumor
samples showed reactivity for PDGFR-α, PDGRF-β, c-KIT, Bcl-2,
cPKC-α, TGF- β1, SOX9, c-Jun, p-JNK, p-p38MAPK, IL-6, MMP2, TIMP2,
and collagen types II and X (68,70),
suggesting that these pathways could be targeted in new potential
therapeutic approaches. A recent study used iPSC-MSCs to
characterize the fusion binding sites in the genome via ChIP-seq,
and the transcriptional modifications induced. The authors found
that the DNA binding profile of the HEY1-NCoA2 fusion is very
similar to the binding profile of HEY1, confirming the hypothesis
that HEY1 directs DNA binding. However, HEY1 is typically a
transcriptional repressor, while the NCoA2 activation domains
preserved in the fusion result in transcriptional activation of
these HEY1-targeted genes. The HEY1-NCoA2 fusion binds to promoter
regions of the genes HES1, PDGFB, PDGFR-α, BCL-2, and SOX4. These
results are consistent with previous findings of MCS biology and
could help to develop new effective targeted therapies for this
disease (71).
A recent study showed that the expression of
HEY1-NCoA2 gene fusion in human primary chondrocytes promoted their
proliferation, enhanced the expression of PDGFR-α, PDGRF-β, SOX9,
LAMTOR1, MTOR, RHEB, PKC-α at the transcriptional level and the
expression of FGFR1, ABL1, AXL, COL2A1, PDGFR-α, and PDGRF-β at the
protein level (72). When cells
expressing the fusion were treated with the multi-kinase inhibitor
imatinib mesylate (targeting ABL, PDGRF-β, and c-KIT), a targeted
reduction in the cell population expressing the fusion was
observed. Interestingly, patient derived xenograft mouse models
(PDX) of MCS also responded to imatinib mesylate treatment,
suggesting that HEY1-NCoA2 fusion-expressing cells rely on
signaling pathways that are inhibited by this multikinase inhibitor
(72). An additional study
performed in a HEY1-NCoA2 expressing MCS cell line demonstrated
that BCL-2 inhibitors can sensitize MCS cells to chemotherapy,
which could have clinical importance, as MCS tumors have shown a
high reoccurrence rate after chemotherapy treatment (73).
A mouse model derived from mice embryonic
osteochondrogenic cells transduced with the HEY1-NCoA2 fusion
presented high rates of tumor development when implanted
subcutaneously in nude mice. The tumors recapitulated morphological
and molecular features of human mesenchymal chondrosarcoma,
including nuclear expression of SOX9, a master regulator of
chondrogenic differentiation. The cells expressing the fusion
presented upregulation of Notch signaling, HEY1, and HES1. Single
cell analysis of mouse mesenchymal chondrosarcoma suggested that
the fusion expression results in incomplete chondrogenic
differentiation, while ChIP seq analysis evidenced an association
of the HEY1-NCoA2 fusion with active enhancers and open chromatin
marks. A protein interaction with the Runx2 transcription factor
was identified, and co-regulation of transcripts by these two
proteins seems to be important for the altered transcriptional
profile observed in MCS. Finally, the authors explored the efficacy
of HDAC inhibitors to target in vivo and in vitro MCS
models and found that treatment of tumor cells with panobinostat
effectively reduced their growth and increased the apoptosis
(74).
Angiofibroma of soft tissue
Soft tissue angiofibroma is typically a benign
fibrovascular tumor that arises in the deep soft tissue of the
lower extremities and is characterized by the proliferation of
spindle cells with abundant collagenous stroma and prominent
branching thin-walled vessels. These tumors stain positive for
epithelial membrane antigen (EMA), desmin, CD34, CD68, CD163,
smooth muscle actin (SMA), and ER. Surgical resection is usually
sufficient for the successful management (75). In this tumor a gene fusion was
identified between the Aryl Hydrocarbon Receptor Repressor (AHRR)
and NCoA2, forming AHRR-NCoA2 (76). The AD1, Q-rich, and AD2 domains of
NCoA2 are preserved and fused to the N-terminal region of AHRR,
which includes a bHLH/PAS domain, important for dimerization and
DNA targeting (77).
The fusion of a repressor with the transactivation
domains of a p160 family member is expected to result in the
activation of genes that would normally be repressed by the AHRR.
Consistent with this hypothesis, a study using soft tissue
angiofibroma samples expressing the fusion gene found
overexpression of AHR target genes or genes associated with AHR
signaling, including CYP1A1, CYP1B1, and genes encoding toll-like
receptors (77). Particularly the
overexpression of CYP1A1 in many angiofibroma samples has recently
led to a proposal to utilize CYP1A1 as a diagnostic marker for
these tumors (78).
In other rare cases of soft tissue angiofibroma,
other gene fusions were also detected including GTF2I-NCoA2
(58), GAB1-ABL1 (58), and more recently, AHRR-NCoA3
(79).
Acute myeloid leukemia
Acute myeloid leukemia comprises a group of
heterogeneous cancers that typically harbor acquired somatic
mutations or genomic rearrangements. Translocations involving
chromosome 8 comprise approximately 2% of AML cases and include
several gene fusions with transcriptional coactivators, such as
MOZ-p300 and MOZ-NCoA2 (14,80,81).
Monocytic leukemia zinc finger (MOZ) belongs to the MYST family of
histone acetyltransferases, where in MOZ-NCoA2 fusion the two
N-terminal domains of MOZ, C4HC3 zinc finger domain, and the HAT
domain are preserved, while NCoA2 keeps its C-terminal activation
domains (80). The expression of
MOZ-NCoA2 was shown to be sufficient to immortalize myeloid
progenitors in vitro and to induce AML in vivo,
driven by the critical interaction between MOZ-NCoA2 and p300/CBP
(80,82). In addition, the MOZ-NCoA2 fusion was
shown to repress cell senescence in mice (83). This oncogenic fusion results in the
loss of NCoA2′s ability to respond to NR activation, while
constitutively enhancing transcription of MOZ target genes
(80).
Several studies focused on dissecting the mechanisms
by which this gene fusion promotes tumorigenesis. The bromodomain
containing protein Brpf1 was identified to direct the MOZ-NCoA2
fusion to the target loci, while M-CSFR and STAT5 signaling have
been shown to contribute to clonal expansion and stem cell
maintenance in these tumors (84–86). A
recent study provided evidence of a connection between
transcription factor MLL and the MOZ-NCoA2 fusion, resulting in the
constitutive activation of CpG-rich promoters, including higher
histone acetylation (HK329ac) at the Hox and Myc loci. The histone
methyltransferase DOT1L was also identified as an important
component of this system, as it helps to maintain the
transcriptionally active state of chromatin. Inhibition of DOT1L
and MLL induced differentiation of MOZ-NCoA2 transformed cells,
whereas inhibition of p300/CBP activity induced cytotoxicity
(87).
Another study using an AML mouse model expressing
the MOZ-NCoA2 fusion suggested that the components of the Polycomb
repressive complex 1 (PRC1) and E3 ubiquitin-protein ligase
(Ring1A, and Ring1B) maintain the stemness of cells in AML
(88). It has also been suggested
that the recruitment of lysine demethylase KDM4C by MOZ-NCoA2,
results in the removal of repressive methylation marks, promoting
the opening of chromatin. In parallel, recruitment of PRMT1 leads
to a high level of H4R3me2, also promoting the opening of chromatin
at the MOZ-NCoA2 binding loci, causing leukemia progression
(89).
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is a high-grade malignant
neoplasm of skeletal myoblast-like cells and is the most common
form of soft tissue sarcoma in children (90). RMS is divided into four subgroups:
embryonal rhabdomyosarcoma (ERMS), alveolar rhabdomyosarcoma
(ARMS), spindle cell/sclerosing rhabdomyosarcoma, and pleomorphic
rhabdomyosarcoma (91). About 70%
of ARMS are driven by gene fusions involving PAX3/7 and FOXO1.
However, PAX1-NCoA1/2 fusions have also been detected in some cases
of ARMS and ERMS. The in vitro studies on murine cell lines
grown in soft agar colony assays showed a transforming activity of
fusions that contain NCoA1/2 fusion partner, where the presence of
the NCoA's transactivation domain was crucial for the
transformation of cells (57,64).
In another study where the mouse myoblast cell line C2C12 was
transduced with PAX3-NCoA2, the fusion protein acted as a
transcriptional activator of PAX3-regulated genes. Differentiation
into myotubules was restrained, while cells exhibited higher
proliferation rates, motility, and induction of cell cycle
progression. Mice with injected transduced C2C12 cells were able to
form tumors that shared pathological features with ERMS samples. In
comparison with a similar model harboring the PAX3-FOXO1A fusion
gene, the PAX3-NCoA2 fusion presented a less aggressive phenotype
(13).
Besides PAX-NCoA fusions, other transcription
factors involved in skeletal muscle differentiation were also
reported in cases of spindle cell rhabdomyosarcoma, such as
SRF-NCoA2, TEAD1-NCoA2, and VGLL2-NCoA2 (91–93).
In all fusions mentioned above, the NCoA1/2 portion retains the
C-terminal AD1, Q-rich, and AD2 domains (57). The presence of these gene fusions in
spindle cell RMS cases seems to be correlated with a more favorable
prognosis when compared with cases that harbor MYOD1 mutations,
another common marker of spindle cell RMS (94).
Uterine tumors resembling ovarian
sex-cord tumors
Uterine tumors resembling ovarian sex-cord tumors
(UTROSCT) are rare mesenchymal neoplasms of unclear histogenesis.
Morphologically, UTROSCT presents features of sex cord elements,
and tumor cells can be arranged in cords, trabeculae, tubules,
clusters, or sheets that can present a reticular appearance
(95). Similar to MCS and RMS, it
has been suggested that malignant UTROSCT cells derive from
pluripotent mesenchymal cell precursors (96–99).
UTROSCT can harbor gene fusions with NCoA as a C-terminal or
N-terminal gene partner (15). In
comparison to previously described tumors which predominantly occur
in a pediatric population, these tumors mainly affect middle-aged
women (15,96).
Molecular analysis of 26 UTROSCT samples using FISH
and a targeted RNA sequencing method detected NCoA1/3
rearrangements with either ESR1 (estrogen receptor 1) or GREB1
(growth regulating estrogen receptor biding 1) in 81.8% of tumor
samples, with the most common gene fusion being ESR1-NCoA3
(15). GREB1 and ESR1 are key
factors in the sex hormone pathway and are highly expressed in
uterine tissue. Cells of UTROSCT tumors harboring fusion with
GREB1-NCoA2 have larger morphology, are more mitotically active and
exhibit more aggressive behavior (100). Because of the high occurrence of
NCoA1/3 gene fusions in those tumors, it has been suggested that
they could be used for the diagnosis of endometrial stromal
neoplasia with sex cord-like differentiation (15). More recently, an additional fusion
between GTF2A1 (general transcription-initiation factor IIA,
subunit 1) and NCoA2 was detected in UTROSCT, which further expands
the molecular rearrangements observed in these tumors (101). The ESR1-NCoA2/3 gene fusion can
also be present in rare Müllerian adenosarcomas in both benign
epithelial and malignant mesenchymal components (60,65,99).
A recent study of a 23-patient cohort showed
inconsistent expression of sex cord markers, epithelial markers,
smooth muscle markers, and hormone receptors in the different tumor
samples analyzed by immunohistochemistry. Expression of CD56, WT1,
SF-1, and CD99 was detected in a high percentage of analyzed
samples, and diffused expression of ER and PR was detected in all
cases. Although there was a high molecular variability amongst
samples, 5 different types of gene fusions were detected, all
containing NCoA fusion partners with GREB1-NCoA2 fusions being the
most common (102,103). A recent study found that malignant
UTROSCT is more likely to have higher mitotic activity, high
expression of stromal PD-L1, and a gene alteration involving NCoA2
(104).
Perspectives on new and existing
therapies
It has been shown that cancer cells can be addicted
to the fusion oncogenes. Especially in pediatric tumors, fusion
depletion can lead to cancer cell death, indicating that loss of a
fusion reverses the malignant progression (105). This feature makes NCoA-oncogenic
fusions attractive therapeutic targets. However, most NCoA-fused
oncogenes retain intrinsically disordered domains of C-terminal
NCoA partners, or even both fusion partners, which makes them
difficult to target with small molecules (16). In addition, there is a tight
regulation of p160 family members' physiological activities, as
they play an important role in sustaining normal cell homeostasis.
Therefore, targeting p160 members in gene fusions should be
specific to the fusion protein only.
The management of pediatric tumors, driven by
NCoA-fusion genes usually comprises surgical resection and
chemotherapy. Unfortunately, these treatments have often shown to
be ineffective, due to recurrence and the development of
resistance. Nonetheless, in recent years new creative therapeutic
approaches have emerged, that have the potential to bring new
therapeutic opportunities, as we describe in the next sections.
Inhibiting the fusion activity
The activity of NCoA proteins can be rapidly
modulated by post-translational modifications, namely
serine/threonine and tyrosine phosphorylation, sumoylation,
ubiquitylation, and methylation (Fig.
4). These modifications can be leveraged to manipulate the
activity of the fusion genes by inhibiting or promoting the
activities of the enzymes that perform the modifications. One
example is the conserved phosphorylation at Tyr1357 in the AD2
domain of NCoA3 and the equivalent position in NCoA2. This specific
tyrosine residue is phosphorylated by c-Abl kinase and results in
an altered interaction with CARM1, p300, and activated receptors
upon IGF1, EGF, and estrogen treatment (46). Tyr-1357 phosphorylation results in
decreased binding of AD2 to CARM1 and an increased affinity to p300
and steroid receptor interaction, enhancing NCoA transcription
activity (46). Inhibition of this
phosphorylation event using c-Abl inhibitors presents a potential
therapeutic opportunity that could be combined with other
approaches to target cancers dependent on the fusion
transcriptional activities (46,106).
Further characterization of posttranslational modifications and
their effect could help develop new therapeutic opportunities to
target modifications that control the fusion oncogenic
properties.
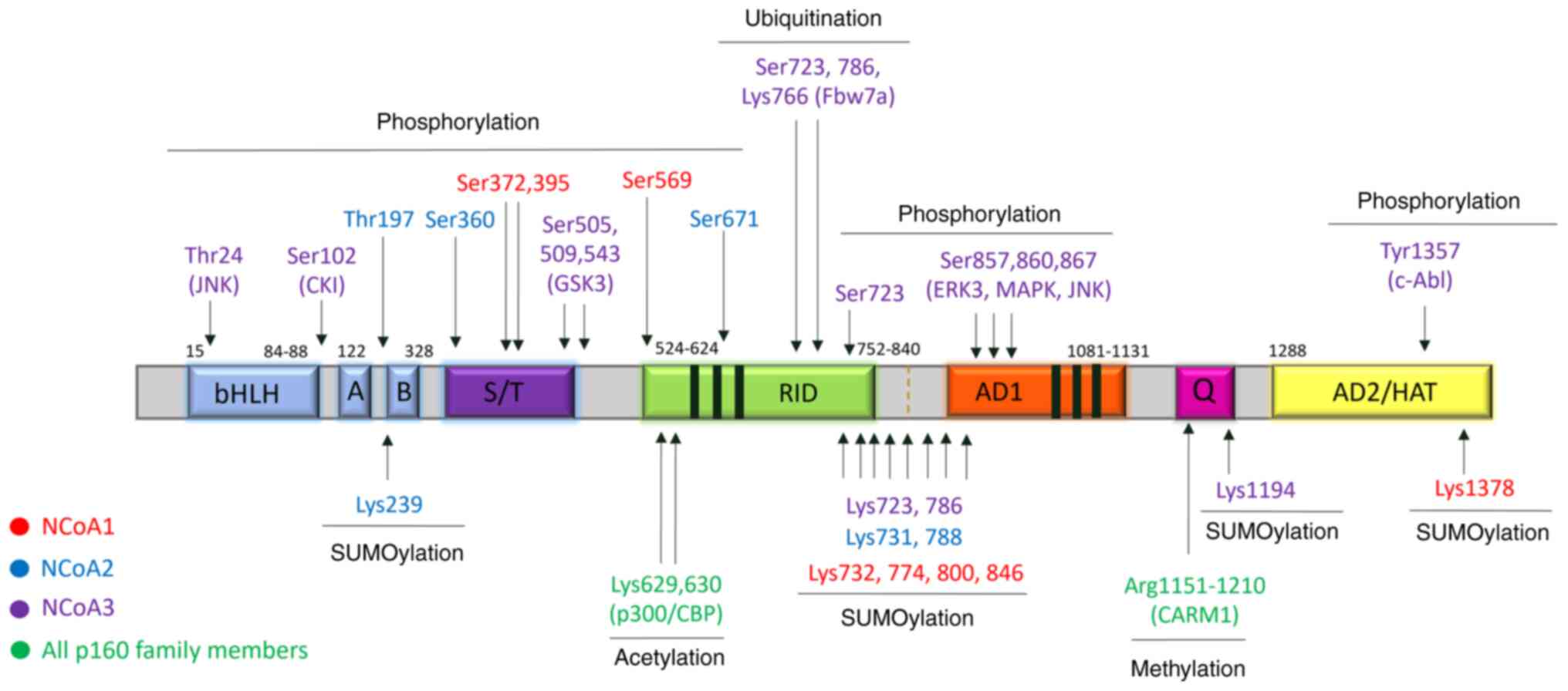 | Figure 4.Post-translational modifications of
NCoAs. The amino acids correspond to the phosphorylation,
ubiquitination, sumoylation, acetylation, and methylation sites and
are indicated above and under the diagram respectively, color-coded
corresponding to the p160 family member. The known enzymes
responsible for post-translational modification are marked in the
brackets under the amino acid residue sites. bHLH, basic
helix-loop-helix; PAS, Per-Arnt-Sim; S/T, serine and threonine
repetition region; RID, receptor-interacting domain; AD, activation
domain; HAT, histone acetyltransferase. |
Another approach to tackle the effects of the fusion
protein in tumorigenesis is to identify and target the functions of
transcriptionally activated genes that can significantly contribute
to tumor development. For example, multiple lines of evidence have
indicated the importance of wild-type kinases in contributing to
the maintenance of fusion-driven tumors (68,70–72).
Efforts towards understanding the molecular changes upon expression
of these fusions could reveal relevant druggable targets that are
crucial for the maintenance or development of tumorigenic and/or
metastatic properties. This would allow the repurposing of drugs
developed for other cancers or conditions in rare pediatric tumors
for which the de novo drug development may not be feasible.
Finally, drugs that alter the state of chromatin, like HDAC
inhibitors could help counteract the constitutive upregulation of
genes by the fusion transactivation domains (74).
Silencing the fusion
New direct therapeutic approaches, which decrease
the undruggable target's expression rather than its activity or
effectors are currently being investigated.
Antisense technologies are one of the most promising
approaches, based on the specific targeting of the RNA that is
causing a disease. In the case of fusion-driven pediatric cancers,
the targeted RNA can be the fusion's pre-mRNA or mRNA that drives
the tumor. Antisense technologies include single-stranded antisense
oligonucleotides (ASOs) or double-stranded antisense drugs
(siRNAs). Antisense refers to the mode of action of all these
drugs, that relies on the Watson and Crick base pairing of an oligo
nucleotide-like molecule with the target RNA (107). The binding of the drug to the
targeted RNA can typically result on the degradation of the RNA,
the inhibition of translation or the modulation of the pre mRNA
splicing. Some ASO therapies have already been approved and used in
clinics (such as ASO therapy for spinal muscular atrophy, Duchenne
muscular dystrophy, and hereditary transthyretin-mediated
amyloidosis, among others), and more are currently in development
stages for treating cancer (108).
CRISPR/Cas9 technologies can be used to produce
random genomic rearrangements that generate inactive forms of
targeted genes, including oncogenic fusions (109,110). A recent study showed the
feasibility of using gene editing to target gene fusions in cancer
of three independent PDX models of Ewing Sarcoma. Two intronic
sequences of the EWSR-FLI1 fusion were simultaneously targeted, one
on each partner gene. This led to either the elimination of crucial
fusion protein domains or changes in the gene-fusion reading frame,
without affecting the unfused gene's exonic sequences or protein
expression (111). This strategy
has the advantage of using the NHEJ pathway, which is active in all
cells, making it easy to use. The targeting of intronic regions
flanking the breaking point of the fusion makes the approach
suitable for patients with different breaking points. Finally, the
fact that exonic regions are not targeted makes the approach safer,
since exonic regions of the normal unfused alleles should remain
unmodified. The advantage of this method over other strategies
based on targeting the fusion region is that it should not affect
the natural unfused forms of the partner genes (112).
Targeted protein degradation also holds promise as a
new type of therapy for undruggable targets. In
proteolysis-targeting chimeras (PROTACs), the ubiquitin/proteasome
system is directed specifically toward a given protein to induce
its selective degradation (113,114). The PROTAC system utilizes
heterobifunctional molecules consisting of a binding ligand for the
protein of interest (such as chimeric oncoprotein) followed by a
small linker and a binding ligand for E3 ligase. Simultaneous
binding of the target protein and the E3 ligase to the PROTAC
results in ubiquitination and proteasomal degradation of the target
protein and release of the PROTAC, which can participate in another
targeting cycle (113). This
feature allows the PROTAC to be utilized in multiple targeting
cycles, reducing the concentration needed to achieve therapeutic
effects (114).
Several PROTACs are currently under different stages
of clinical trials, targeting the AR and ER among other proteins
(114). A PROTAC approach
targeting NCoA1 has recently been described, using a small peptide
(Y2L) that mimics the LXXLL helical fragment of STAT6, which has a
high affinity and specificity for the NCoA1 PAS-B domain. In the
PROTAC, Y2L is linked to a tetrapeptide (RLAA), an N-degron
fragment that binds the UBR box (a class of E3 ligases). The study
showed that the PROTAC was effective in inducing the specific
degradation of NCoA1, resulting in the impairment of NCoA1
transcriptional activity and suppression of cell invasion and
migration in vitro and in vivo (115).
Conclusion
Many pediatric cancers express oncogenic fusions as
the only driver of tumorigenesis. The p160 protein family members
have a prominent representation among these gene fusions. In some
cases, it has been demonstrated that only the expression of the
oncogenic fusion was sufficient to induce tumors. Conversely, the
inhibition or deletion of the oncogenic fusion in cancer cells led
to cancer cell death or cell differentiation, imposing the
importance of direct elimination of the fusion from these
tumors.
Classical therapeutic approaches with small
inhibitors rely on the manipulation of the activity of the
oncogenic fusion or the inhibition of its transcriptional targets.
The possible disadvantages of these approaches are the requirement
of a deep understanding of the regulatory mechanisms and molecular
pathways affected by the expression of the oncogenic fusion and the
selection for resistance to small inhibitors. In the near future,
more promising strategies could rely on targeting the expression of
gene fusion itself, using technologies based on CRISPR/Cas9,
antisense oligonucleotides, and proteolysis-targeting chimeras.
These technologies have a big potential not only to directly target
chimeric proteins that were traditionally considered ‘undruggable
targets’, but also to overcome drug resistance. Since other
pathologies are already benefiting from the progress of these new
approaches, it would be highly beneficial to profit from these
experiences in pediatric fusion-driven tumors as well.
Clinically, targeting the gene fusion expression
holds great promise for future therapies where its effects can be
addressed directly. In addition, detailed knowledge of the
molecular pathways affected (for example, recent progress on MCS)
suggests potential combinatorial therapies for efficient targeting
of the tumors. One clear example is the treatment of MCS cells with
BCL-2 inhibitors that sensitize MCS cells to chemotherapy, or the
proposed use of imatinib or panobinostat specifically in MCS. These
findings can rapidly evolve into clinical studies and provide
treatment alternatives while approaches that directly target the
expression of the fusion gene are being developed.
Acknowledgements
The authors would like to thank Mr. Niko Šafarič and
Dr Bojana Žvan (Society for the Prevention of Cerebral and Vascular
Diseases of Slovenia, Ljubljana, Slovenia) for their insights and
support.
Funding
The present study was funded by Northwell Health, Advancing
Women in Science and Medicine (grant no. 580694) awarded to Dr
Polona Safaric Tepes.
Availability of data and materials
Not applicable.
Authors' contributions
PST wrote, edited and reviewed the manuscript and
prepared the figures and tables. DS wrote, edited and reviewed the
manuscript. Data authentication is not applicable. Both authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Abl
|
Abelson murine leukemia virus
|
|
AHRR
|
aryl-hydrocarbon receptor
repressor
|
|
AML
|
acute myeloid leukemia
|
|
AR
|
androgen receptor
|
|
ARMS
|
alveolar rhabdomyosarcoma
|
|
ASO
|
antisense oligonucleotide
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
bHLH
|
basic helix-loop-helix domain
|
|
C2C12
|
mouse skeletal muscle cell line
|
|
CARM
|
coactivator associated arginine
methyltransferase
|
|
CBP
|
CREB binding protein
|
|
CYP1
|
cytochrome P450 family 1
|
|
ERMS
|
embryonal rhabdomyosarcoma
|
|
ESR1
|
estrogen receptor 1
|
|
GREB1
|
growth regulating estrogen receptor
binding 1
|
|
GTF2I
|
general transcription factor III
|
|
HEY1-NCoA2
|
gene fusion in mesenchymal
chondrosarcoma
|
|
IL
|
interleukin
|
|
KAT
|
acetylase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MCS
|
mesenchymal chondrosarcoma
|
|
NCoA
|
nuclear receptor coactivator
|
|
NR
|
nuclear receptor
|
|
PAS
|
Per-Arnt-Sim domain
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
|
PDX
|
patient derived xenograft
|
|
PRMT
|
protein arginine
methyltransferase
|
|
PROTAC
|
proteolysis-targeting chimera
|
|
RID
|
receptor interaction domain
|
|
TBP
|
TATA-box binding protein
|
|
TF
|
transcription factors
|
|
TGF
|
transforming growth factor
|
|
UTROSCT
|
uterine tumor resembling ovarian sex
cord tumor
|
References
|
1
|
Mertens F, Johansson B, Fioretos T and
Mitelman F: The emerging complexity of gene fusions in cancer. Nat
Rev Cancer. 15:371–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Folpe AL, Graham RP, Martinez A,
Schembri-Wismayer D, Boland J and Fritchie KJ: Mesenchymal
chondrosarcomas showing immunohistochemical evidence of
rhabdomyoblastic differentiation: A potential diagnostic pitfall.
Hum Pathol. 77:28–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Latysheva NS and Babu MM: Discovering and
understanding oncogenic gene fusions through data intensive
computational approaches. Nucleic Acids Res. 44:4487–4503. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mitelman F, Johansson B and Mertens F: The
impact of translocations and gene fusions on cancer causation. Nat
Rev Cancer. 7:233–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pugh TJ, Morozova O, Attiyeh EF,
Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M,
Kiezun A, et al: The genetic landscape of high-risk neuroblastoma.
Nat Genet. 45:279–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lobato MN, Metzler M, Drynan L, Forster A,
Pannell R and Rabbitts TH: Modeling chromosomal translocations
using conditional alleles to recapitulate initiating events in
human leukemias. J Natl Cancer Inst Monogr. 39:58–63. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cocco E, Scaltriti M and Drilon A: NTRK
fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin
Oncol. 15:731–747. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frenkel-Morgenstern M and Valencia A:
Novel domain combinations in proteins encoded by chimeric
transcripts. Bioinformatics. 28:i67–i74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Padmavathi G, Roy NK, Bordoloi D, Monisha
J and Kunnumakkara AB: ‘Basic concepts of fusion genes and their
classification’ in fusion genes and cancer. (World scientific,
2016), doi:10.1142/9789813200944_000210.1142/9789813200944_0002.
17–58
|
|
10
|
Webb P, Nguyen P, Shinsako J, Anderson C,
Feng W, Nguyen MP, Chen D, Huang SM, Subramanian S, McKinerney E,
et al: Estrogen receptor activation function 1 works by binding
p160 coactivator proteins. Mol Endocrinol. 12:1605–1618. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kushner PJ, Agard D, Feng WJ, Lopez G,
Schiau A, Uht R, Webb P and Greene G: Oestrogen receptor function
at classical and alternative response elements. Novartis Found
Symp. 230:20–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rollins DA, Coppo M and Rogatsky I:
Minireview: Nuclear receptor coregulators of the p160 family:
Insights into inflammation and metabolism. Mol Endocrinol.
29:502–517. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida H, Miyachi M, Sakamoto K, Ouchi K,
Yagyu S, Kikuchi K, Kuwahara Y, Tsuchiya K, Imamura T, Iehara T, et
al: PAX3-NCOA2 fusion gene has a dual role in promoting the
proliferation and inhibiting the myogenic differentiation of
rhabdomyosarcoma cells. Oncogene. 33:5601–5608. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin H, Glass J and Blanchard KJ: MOZ-TIF2
repression of nuclear receptor-mediated transcription requires
multiple domains in MOZ and in the CID domain of TIF2. Mol Cancer.
6:512007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goebel EA, Bonilla SH, Dong F, Dickson BC,
Hoang LN, Hardisson D, Lacambra MD, Lu FI, Fletcher CDM, Crum CP,
et al: Uterine tumor resembling ovarian sex cord tumor (UTROSCT): A
morphologic and molecular study of 26 cases confirms recurrent
NCOA1-3 rearrangement. Am J Surg Pathol. 44:30–42. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hagenbuchner J and Ausserlechner MJ:
Targeting transcription factors by small compounds-current
strategies and future implications. Biochem Pharmacol. 107:1–13.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu J and Li Q: Review of the in vivo
functions of the p160 steroid receptor coactivator family. Mol
Endocrinol. 17:1681–1692. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu J and O'Malley BW: Molecular mechanisms
and cellular biology of the steroid receptor coactivator (SRC)
family in steroid receptor function. Rev Endocr Metab Disord.
3:185–192. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hultqvist G, Åberg E, Camilloni C, Sundell
GN, Andersson E, Dogan J, Chi CN, Vendruscolo M and Jemth P:
Emergence and evolution of an interaction between intrinsically
disordered proteins. Elife. 6:e160592017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heery DM, Kalkhoven E, Hoare S and Parker
MG: A signature motif in transcriptional co-activators mediates
binding to nuclear receptors. Nature. 387:733–736. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lodrini M, Münz T, Coudevylle N,
Griesinger C, Becker S and Pfitzner E: P160/SRC/NCoA coactivators
form complexes via specific interaction of their PAS-B domain with
the CID/AD1 domain. Nucleic Acids Res. 36:1847–1860. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szwarc MM, Kommagani R, Lessey BA and
Lydon JP: The p160/steroid receptor coactivator family: Potent
arbiters of uterine physiology and dysfunction. Biol Reprod.
91:1222014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Yi X, Sun X, Yin N, Shi B, Wu H,
Wang D, Wu G and Shang Y: Differential gene regulation by the SRC
family of coactivators. Genes Dev. 18:1753–1765. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Litterst CM and Pfitzner E:
Transcriptional activation by STAT6 requires the direct interaction
with NCoA-1. J Biol Chem. 276:45713–45721. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karlsson E, Lindberg A, Andersson E and
Jemth P: High affinity between CREBBP/p300 and NCOA evolved in
vertebrates. Protein Sci. 29:1687–1691. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Na SY, Lee SK, Han SJ, Choi HS, Im SY and
Lee JW: Steroid receptor coactivator-1 interacts with the p50
subunit and coactivates nuclear factor kappaB-mediated
transactivations. J Biol Chem. 273:10831–10834. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beischlag TV, Wang S, Rose DW, Torchia J,
Reisz-Porszasz S, Muhammad K, Nelson WE, Probst MR, Rosenfeld MG
and Hankinson O: Recruitment of the NCoA/SRC-1/p160 family of
transcriptional coactivators by the aryl hydrocarbon receptor/aryl
hydrocarbon receptor nuclear translocator complex. Mol Cell Biol.
22:4319–4333. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rohira AD and Lonard DM: Steroid receptor
coactivators present a unique opportunity for drug development in
hormone-dependent cancers. Biochem Pharmacol. 140:1–7. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koh SS, Chen D, Lee YH and Stallcup MR:
Synergistic enhancement of nuclear receptor function by p160
coactivators and two coactivators with protein methyltransferase
activities. J Biol Chem. 276:1089–1098. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Spencer TE, Jenster G, Burcin MM, Allis
CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ and
O'Malley BW: Steroid receptor coactivator-1 is a histone
acetyltransferase. Nature. 389:194–198. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Drazic A, Myklebust LM, Ree R and Arnesen
T: The world of protein acetylation. Biochim Biophys Acta.
1864:1372–1401. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding XF, Anderson CM, Ma H, Hong H, Uht
RM, Kushner PJ and Stallcup MR: Nuclear receptor-binding sites of
coactivators glucocorticoid receptor interacting protein 1 (GRIP1)
and steroid receptor coactivator 1 (SRC-1): Multiple motifs with
different binding specificities. Mol Endocrinol. 12:302–313. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kalkhoven E, Valentine JE, Heery DM and
Parker MG: Isoforms of steroid receptor co-activator 1 differ in
their ability to potentiate transcription by the oestrogen
receptor. EMBO J. 17:232–243. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumar MB and Perdew GH: Nuclear receptor
coactivator SRC-1 interacts with the Q-rich subdomain of the AhR
and modulates its transactivation potential. Gene Expr. 8:273–286.
1999.PubMed/NCBI
|
|
35
|
Bevan CL, Hoare S, Claessens F, Heery DM
and Parker MG: The AF1 and AF2 domains of the androgen receptor
interact with distinct regions of SRC1. Mol Cell Biol.
19:8383–8392. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Varadi M, Anyango S, Deshpande M, Nair S,
Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al:
AlphaFold protein structure database: Massively expanding the
structural coverage of protein-sequence space with high-accuracy
models. Nucleic Acids Res. 50:D439–D444. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Razeto A, Ramakrishnan V, Litterst CM,
Giller K, Griesinger C, Carlomagno T, Lakomek N, Heimburg T,
Lodrini M, Pfitzner E and Becker S: Structure of the NCoA-1/SRC-1
PAS-B domain bound to the LXXLL motif of the STAT6 transactivation
domain. J Mol Biol. 336:319–329. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Russo L, Giller K, Pfitzner E, Griesinger
C and Becker S: Insight into the molecular recognition mechanism of
the coactivator NCoA1 by STAT6. Sci Rep. 7:168452017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li S and Shang Y: Regulation of SRC family
coactivators by post-translational modifications. Cell Signal.
19:1101–1112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han SJ, Lonard B and O'Malley W:
Multi-modulation of nuclear receptor coactivators through
posttranslational modifications. Trends Endocrinol Metab. 20:8–15.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rowan BG, Garrison N, Weigel NL and
O'Malley BW: 8-Bromo-cyclic AMP induces phosphorylation of two
sites in SRC-1 that facilitate ligand-independent activation of the
chicken progesterone receptor and are critical for functional
cooperation between SRC-1 and CREB binding protein. Mol Cell Biol.
20:8720–8730. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Narayanan R, Adigun AA, Edwards DP and
Weigel NL: Cyclin-dependent kinase activity is required for
progesterone receptor function: Novel role for cyclin A/Cdk2 as a
progesterone receptor coactivator. Mol Cell Biol. 25:264–277. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ueda T, Mawji NR, Bruchovsky N and Sadar
MD: Ligand-independent activation of the androgen receptor by
interleukin-6 and the role of steroid receptor coactivator-1 in
prostate cancer cells. J Biol Chem. 277:38087–38094. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rowan BG, Weigel NL and O'Malley BW:
Phosphorylation of steroid receptor coactivator-1: Identification
of the phosphorylation sites and phosphorylation through the
mitogen-activated protein kinase pathway. J Biol Chem.
275:4475–4483. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hoang T, Fenne IS, Cook C, Børud B, Bakke
M, Lien EA and Mellgren G: cAMP-dependent protein kinase regulates
ubiquitin-proteasome-mediated degradation and subcellular
localization of the nuclear receptor coactivator GRIP1. J Biol
Chem. 279:49120–49130. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Oh AS, Lahusen JT, Chien CD, Fereshteh MP,
Zhang X, Dakshanamurthy S, Xu J, Kagan BL, Wellstein A and Riegel
AT: Tyrosine phosphorylation of the nuclear receptor coactivator
AIB1/SRC-3 is enhanced by Abl kinase and is required for its
activity in cancer cells. Mol Cell Biol. 28:6580–6593. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Baumann CT, Ma H, Wolford R, Reyes JC,
Maruvada P, Lim C, Yen PM, Stallcup MR and Hager GL: The
glucocorticoid receptor interacting protein 1 (GRIP1) localizes in
discrete nuclear foci that associate with ND10 bodies and are
enriched in components of the 26S proteasome. Mol Endocrinol.
15:485–500. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chauchereau A, Amazit L, Quesne M,
Guiochon-Mantel A and Milgrom E: Sumoylation of the progesterone
receptor and of the steroid receptor coactivator SRC-1. J Biol
Chem. 278:12335–12343. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kotaja N, Karvonen U, Jänne OA and Palvimo
JJ: The nuclear receptor interaction domain of GRIP1 is modulated
by covalent attachment of SUMO-1. J Biol Chem. 277:30283–30288.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu H, Sun L, Zhang Y, Chen Y, Shi B, Li R,
Wang Y, Liang J, Fan D, Wu G, et al: Coordinated regulation of AIB1
transcriptional activity by sumoylation and phosphorylation. J Biol
Chem. 281:21848–21856. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen H, Lin RJ, Xie W, Wilpitz D and Evans
RM: Regulation of hormone-induced histone hyperacetylation and gene
activation via acetylation of an acetylase. Cell. 98:675–686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Naeem H, Cheng D, Zhao Q, Underhill C,
Tini M, Bedford MT and Torchia J: The activity and stability of the
transcriptional coactivator p/CIP/SRC-3 are regulated by
CARM1-dependent methylation. Mol Cell Biol. 27:120–134. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
McKenna NJ and O'Malley BW: Combinatorial
control of gene expression by nuclear receptors and coregulators.
Cell. 108:465–474. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Voegel JJ, Heine MJ, Tini M, Vivat V,
Chambon P and Gronemeyer H: The coactivator TIF2 contains three
nuclear receptor-binding motifs and mediates transactivation
through CBP binding-dependent and -independent pathways. EMBO J.
17:507–519. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Johnson AB and Barton MC: Hypoxia-induced
and stress-specific changes in chromatin structure and function.
Mutat Res. 618:149–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang L, Motoi T, Khanin R, Olshen A,
Mertens F, Bridge J, Cin PD, Antonescu CR, Singer S, Hameed M, et
al: Identification of a novel, recurrent HEY1-NCOA2 fusion in
mesenchymal chondrosarcoma based on a genome-wide screen of
exon-level expression data. Genes Chromosomes Cancer. 51:127–139.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sumegi J, Streblow R, Frayer RW, Cin PD,
Rosenberg A, Meloni-Ehrig A and Bridge JA: Recurrent t(2;2) and
t(2;8) translocations in rhabdomyosarcoma without the canonical
PAX-FOXO1 fuse PAX3 to members of the nuclear receptor
transcriptional coactivator family. Genes Chromosomes Cancer.
49:224–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bekers EM, Groenen PJTA, Verdijk MAJ,
Raaijmakers-van Geloof WL, Roepman P, Vink R, Gilhuijs NDB, van
Gorp JM, Bovée JVMG, Creytens DH, et al: Soft tissue angiofibroma:
Clinicopathologic, immunohistochemical and molecular analysis of 14
cases. Genes Chromosomes Cancer. 56:750–757. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Argani P, Reuter VE, Kapur P, Brown JE,
Sung YS, Zhang L, Williamson R, Francis G, Sommerville S, Swanson
D, et al: Novel MEIS1-NCOA2 gene fusions define a distinct
primitive spindle cell sarcoma of the kidney. Am J Surg Pathol.
42:1562–1570. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Piscuoglio S, Burke KA, Ng CK,
Papanastasiou AD, Geyer FC, Macedo GS, Martelotto LG, de Bruijn I,
De Filippo MR, Schultheis AM, et al: Uterine adenosarcomas are
mesenchymal neoplasms. J Pathol. 238:381–388. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dickson BC, Childs TJ, Colgan TJ, Sung YS,
Swanson D, Zhang L and Antonescu CR: Uterine tumor resembling
ovarian sex cord tumor: A distinct entity characterized by
recurrent NCOA2/3 gene fusions. Am J Surg Pathol. 43:178–186. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Le Loarer F, Laffont S, Lesluyes T, Tirode
F, Antonescu C, Baglin AC, Delespaul L, Soubeyran I, Hostein I,
Pérot G, et al: Clinicopathologic and molecular features of a
series of 41 biphenotypic sinonasal sarcomas expanding their
molecular spectrum. Am J Surg Pathol. 43:747–754. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lacambra MD, Weinreb I, Demicco EG, Chow
C, Sung YS, Swanson D, To KF, Wong KC, Antonescu CR and Dickson BC:
PRRX-NCOA1/2 rearrangement characterizes a distinctive fibroblastic
neoplasm. Genes Chromosomes Cancer. 58:705–712. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wachtel M, Dettling M, Koscielniak E,
Stegmaier S, Treuner J, Simon-Klingenstein K, Bühlmann P, Niggli FK
and Schäfer BW: Gene expression signatures identify
rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23)
translocation fusing PAX3 to NCOA1. Cancer Res. 64:5539–5545. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bean GR, Anderson J, Sangoi AR, Krings G
and Garg K: DICER1 mutations are frequent in mullerian
adenosarcomas and are independent of rhabdomyosarcomatous
differentiation. Mod Pathol. 32:280–289. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
El Beaino M, Roszik J, Livingston JA, Wang
WL, Lazar AJ, Amini B, Subbiah V, Lewis V and Conley AP:
Mesenchymal chondrosarcoma: A review with emphasis on its
fusion-driven biology. Curr Oncol Rep. 20:372018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schneiderman BA, Kliethermes SA and
Nystrom LM: Survival in mesenchymal chondrosarcoma varies based on
age and tumor location: A survival analysis of the SEER database.
Clin Orthop Relat Res. 475:799–805. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Brown RE and Boyle JL: Mesenchymal
chondrosarcoma: Molecular characterization by a proteomic approach,
with morphogenic and therapeutic implications. Ann Clin Lab Sci.
33:131–141. 2003.PubMed/NCBI
|
|
69
|
Fischer A and Gessler M: Delta-Notch-and
then? Protein interactions and proposed modes of repression by Hes
and Hey bHLH factors. Nucleic Acids Res. 35:4583–4596. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Swanson PE, Lillemoe TJ, Manivel JC and
Wick MR: Mesenchymal chondrosarcoma. An immunohistochemical study.
Arch Pathol Lab Med. 114:943–948. 1990.PubMed/NCBI
|
|
71
|
Qi W, Rosikiewicz W, Yin Z, Xu B, Jiang H,
Wan S, Fan Y, Wu G and Wang L: Genomic profiling identifies genes
and pathways dysregulated by HEY1-NCOA2 fusion and shines a light
on mesenchymal chondrosarcoma tumorigenesis. J Pathol. 257:579–592.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tepes PS, Segovia D, Jevtic S, Ramirez D,
Lyons SK and Sordella R: Patient-derived xenografts and in vitro
model show rationale for imatinib mesylate repurposing in
HEY1-NCoA2-driven mesenchymal chondrosarcoma. Lab Invest.
102:1038–1049. 2021. View Article : Google Scholar
|
|
73
|
de Jong Y, van Maldegem AM,
Marino-Enriquez A, de Jong D, Suijker J, Briaire-de Bruijn IH,
Kruisselbrink AB, Cleton-Jansen AM, Szuhai K, Gelderblom H, et al:
Inhibition of Bcl-2 family members sensitizes mesenchymal
chondrosarcoma to conventional chemotherapy: Report on a novel
mesenchymal chondrosarcoma cell line. Lab Invest. 96:1128–1137.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tanaka M, Homme M, Teramura Y, Kumegawa K,
Yamazaki Y, Yamashita K, Osato M, Maruyama R and Nakamura T:
HEY1-NCOA2 expression modulates chondrogenic differentiation and
induces mesenchymal chondrosarcoma in mice. JCI Insight.
8:e1602792023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nakayama S, Nishio J, Aoki M, Koga K,
Nabeshima K and Yamamoto T: Angiofibroma of soft tissue: Current
status of pathology and genetics. Histol Histopathol. 37:717–722.
2022.PubMed/NCBI
|
|
76
|
Sugita S, Aoyama T, Kondo K, Keira Y,
Ogino J, Nakanishi K, Kaya M, Emori M, Tsukahara T and Nakajima H:
Diagnostic utility of NCOA2 fluorescence in situ hybridization and
Stat6 immunohistochemistry staining for soft tissue angiofibroma
and morphologically similar fibrovascular tumors. Hum Pathol.
45:1588–1596. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jin Y, Möller E, Nord KH, Mandahl N, Von
Steyern FV, Domanski HA, Mariño-Enríquez A, Magnusson L, Nilsson J,
Sciot R, et al: Fusion of the AHRR and NCOA2 genes through a
recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma
results in upregulation of aryl hydrocarbon receptor target genes.
Genes Chromosomes Cancer. 51:510–520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Uemura K, Komatsu M, Hara S, Kawamoto T,
Bitoh Y, Itoh T and Hirose T: CYP1A1 is a useful diagnostic marker
for angiofibroma of soft tissue. Am J Surg Pathol. 47:547–557.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yamashita K, Baba S, Togashi Y, Dobashi A,
Ae K, Matsumoto S, Tanaka M, Nakamura T and Takeuchi K:
Clinicopathologic and genetic characterization of angiofibroma of
soft tissue: A study of 12 cases including two cases with
AHRR::NCOA3 gene fusion. Histopathology. 83:57–66. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Deguchi K, Ayton PM, Carapeti M, Kutok JL,
Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML and Gilliland
DG: MOZ-TIF2-induced acute myeloid leukemia requires the MOZ
nucleosome binding motif and TIF2-mediated recruitment of CBP.
Cancer Cell. 3:259–271. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Carapeti M, Aguiar RC, Goldman JM and
Cross NC: A novel fusion between MOZ and the nuclear receptor
coactivator TIF2 in acute myeloid leukemia. Blood. 91:3127–3133.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Huntly BJ, Shigematsu H, Deguchi K, Lee
BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR,
et al: MOZ-TIF2, but not BCR-ABL, confers properties of leukemic
stem cells to committed murine hematopoietic progenitors. Cancer
Cell. 6:587–596. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Largeot A, Perez-Campo FM, Marinopoulou E,
Lie-a-Ling M, Kouskoff V and Lacaud G: Expression of the MOZ-TIF2
oncoprotein in mice represses senescence. Exp Hematol.
44:231–237.e234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Shima H, Yamagata K, Aikawa Y, Shino M,
Koseki H, Shimada H and Kitabayashi I: Bromodomain-PHD finger
protein 1 is critical for leukemogenesis associated with MOZ-TIF2
fusion. Int J Hematol. 99:21–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tam WF, Hähnel PS, Schüler A, Lee BH,
Okabe R, Zhu N, Pante SV, Raffel G, Mercher T, Wernig G, et al:
STAT5 is crucial to maintain leukemic stem cells in acute
myelogenous leukemias induced by MOZ-TIF2. Cancer Res. 73:373–384.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Aikawa Y, Katsumoto T, Zhang P, Shima H,
Shino M, Terui K, Ito E, Ohno H, Stanley ER, Singh H, et al:
PU.1-mediated upregulation of CSF1R is crucial for leukemia stem
cell potential induced by MOZ-TIF2. Nat Med. 16:580–585. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Miyamoto R, Okuda H, Kanai A, Takahashi S,
Kawamura T, Matsui H, Kitamura T, Kitabayashi I, Inaba T and
Yokoyama A: Activation of CpG-rich promoters mediated by MLL drives
MOZ-rearranged leukemia. Cell Rep. 32:1082002020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Shima H, Takamatsu-Ichihara E, Shino M,
Yamagata K, Katsumoto T, Aikawa Y, Fujita S, Koseki H and
Kitabayashi I: Ring1A and Ring1B inhibit expression of Glis2 to
maintain murine MOZ-TIF2 AML stem cells. Blood. 131:1833–1845.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cheung N, Fung TK, Zeisig BB, Holmes K,
Rane JK, Mowen KA, Finn MG, Lenhard B, Chan LC and So CW: Targeting
aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute
myeloid leukemia. Cancer Cell. 29:32–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Skapek SX, Ferrari A, Gupta AA, Lupo PJ,
Butler E, Shipley J, Barr FG and Hawkins DS: Rhabdomyosarcoma. Nat
Rev Dis Primers. 5:12019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sun X, Guo W, Shen JK, Mankin HJ, Hornicek
FJ and Duan Z: Rhabdomyosarcoma: Advances in molecular and cellular
biology. Sarcoma. 2015:2320102015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Alaggio R, Zhang L, Sung YS, Huang SC,
Chen CL, Bisogno G, Zin A, Agaram NP, LaQuaglia MP, Wexler LH and
Antonescu CR: A molecular study of pediatric spindle and sclerosing
rhabdomyosarcoma: Identification of novel and recurrent
VGLL2-related fusions in infantile cases. Am J Surg Pathol.
40:224–235. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mosquera JM, Sboner A, Zhang L,
Kitabayashi N, Chen CL, Sung YS, Wexler LH, LaQuaglia MP, Edelman
M, Sreekantaiah C, et al: Recurrent NCOA2 gene rearrangements in
congenital/infantile spindle cell rhabdomyosarcoma. Genes
Chromosomes Cancer. 52:538–550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Whittle S, Venkatramani R, Schönstein A,
Pack SD, Alaggio R, Vokuhl C, Rudzinski ER, Wulf AL, Zin A, Gruver
JR, et al: Congenital spindle cell rhabdomyosarcoma: An
international cooperative analysis. Eur J Cancer. 168:56–64. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Jia M, Sun PL and Gao H: Uterine lesions
with sex cord-like architectures: A systematic review. Diagn
Pathol. 14:1292019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Schraag SM, Caduff R, Dedes KJ, Fink D and
Schmidt AM: Uterine tumors resembling ovarian sex cord
tumors-treatment, recurrence, pregnancy and brief review. Gynecol
Oncol Rep. 19:53–56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Clement PB and Scully RE: Mullerian
adenosarcomas of the uterus with sex cord-like elements. A
clinicopathologic analysis of eight cases. Am J Clin Pathol.
91:664–672. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
McCluggage WG, Date A, Bharucha H and
Toner PG: Endometrial stromal sarcoma with sex cord-like areas and
focal rhabdoid differentiation. Histopathology. 29:369–374. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Boyraz B, Watkins JC, Young RH and Oliva
E: Uterine tumors resembling ovarian sex cord tumors: A
clinicopathologic study of 75 cases emphasizing features predicting
adverse outcome and differential diagnosis. Am J Surg Pathol.
47:234–247. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lee CH, Kao YC, Lee WR, Hsiao YW, Lu TP,
Chu CY, Lin YJ, Huang HY, Hsieh TH, Liu YR, et al:
Clinicopathologic characterization of GREB1-rearranged uterine
sarcomas with variable sex-cord differentiation. Am J Surg Pathol.
43:928–942. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Devereaux KA, Kertowidjojo E, Natale K,
Ewalt MD, Soslow RA and Hodgson A: GTF2A1-NCOA2-associated uterine
tumor resembling ovarian sex cord tumor (UTROSCT) shows focal
rhabdoid morphology and aggressive behavior. Am J Surg Pathol.
45:1725–1728. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bi R, Yao Q, Ji G, Bai Q, Li A, Liu Z,
Cheng Y, Tu X, Yu L, Chang B, et al: Uterine tumor resembling
ovarian sex cord tumors: 23 Cases indicating molecular
heterogeneity with variable biological behavior. Am J Surg Pathol.
47:739–755. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lu B, Xia Y, Chen J, Tang J, Shao Y and Yu
W: NCOA1/2/3 rearrangements in uterine tumor resembling ovarian sex
cord tumor: A clinicopathological and molecular study of 18 cases.
Hum Pathol. 135:65–75. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xiong SP, Luo RZ, Wang F, Yang X, Lai JP,
Zhang C and Liu LL: PD-L1 expression, morphology, and molecular
characteristic of a subset of aggressive uterine tumor resembling
ovarian sex cord tumor and a literature review. J Ovarian Res.
16:1022023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bernasconi M, Remppis A, Fredericks WJ,
Rauscher FJ III and Schafer BW: Induction of apoptosis in
rhabdomyosarcoma cells through down-regulation of PAX proteins.
Proc Natl Acad Sci USA. 93:13164–13169. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Oh AS, Lahusen JT, Chien CD, Fereshteh MP,
Zhang X, Dakshanamurthy S, Xu J, Kagan BL, Wellstein A and Riegel
AT: Tyrosine phosphorylation of the nuclear receptor coactivator
AIB1/SRC-3 is enhanced by Abl kinase and is required for its
activity in cancer cells. Mol Cell Biol. 28:6580–6593. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Crooke ST, Liang XH, Baker BF and Crooke
RM: Antisense technology: A review. J Biol Chem. 296:1004162021.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Quemener AM, Bachelot L, Forestier A,
Donnou-Fournet E, Gilot D and Galibert MD: The powerful world of
antisense oligonucleotides: From bench to bedside. Wiley
Interdiscip Rev RNA. 11:e15942020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Katti A, Diaz BJ, Caragine CM, Sanjana NE
and Dow LE: CRISPR in cancer biology and therapy. Nat Rev Cancer.
22:259–279. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Martinez-Lage M, Puig-Serra P, Menendez P,
Torres-Ruiz R and Rodriguez-Perales S: CRISPR/Cas9 for cancer
therapy: Hopes and challenges. Biomedicines. 6:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Martinez-Lage M, Torres-Ruiz R, Puig-Serra
P, Moreno-Gaona P, Martin MC, Moya FJ, Quintana-Bustamante O,
Garcia-Silva S, Carcaboso AM, Petazzi P, et al: In vivo CRISPR/Cas9
targeting of fusion oncogenes for selective elimination of cancer
cells. Nat Commun. 11:50602020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chen ZH, Yu YP, Zuo ZH, Nelson JB,
Michalopoulos GK, Monga S, Liu S, Tseng G and Luo JH: Targeting
genomic rearrangements in tumor cells through Cas9-mediated
insertion of a suicide gene. Nat Biotechnol. 35:543–550. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Sun X, Gao H, Yang Y, He M, Wu Y, Song Y,
Tong Y and Rao Y: PROTACs: Great opportunities for academia and
industry. Signal Transduct Target Ther. 4:642019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Bekes M, Langley DR and Crews CM: PROTAC
targeted protein degraders: The past is prologue. Nat Rev Drug
Discov. 21:181–200. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Lee Y, Heo J, Jeong H, Hong KT, Kwon DH,
Shin MH, Oh M, Sable GA, Ahn GO, Lee JS, et al: Targeted
degradation of transcription coactivator SRC-1 through the N-degron
pathway. Angew Chem Int Ed Engl. 59:17548–17555. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Tan GZL, Saminathan SN, Chang KTE, Odoño
EG, Kuick CH, Chen H and Lee VKM: A rare case of congenital spindle
cell rhabdomyosarcoma with TEAD1-NCOA2 fusion: A subset of spindle
cell rhabdomyosarcoma with indolent behavior. Pathol Int.
70:234–236. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Avenarius MR, Miller CR, Arnold MA, Koo S,
Roberts R, Hobby M, Grossman T, Moyer Y, Wilson RK, Mardis ER, et
al: Genetic characterization of pediatric sarcomas by targeted RNA
sequencing. J Mol Diagn. 22:1238–1245. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bennett JA, Lastra RR, Barroeta JE,
Parilla M, Galbo F, Wanjari P, Young RH, Krausz T and Oliva E:
Uterine tumor resembling ovarian sex cord stromal tumor (UTROSCT):
A series of 3 cases with extensive rhabdoid differentiation,
malignant behavior, and ESR1-NCOA2 fusions. Am J Surg Pathol.
44:1563–1572. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Panagopoulos I, Gorunova L, Viset T and
Heim S and Heim S: Gene fusions AHRR-NCOA2, NCOA2-ETV4, ETV4-AHRR,
P4HA2-TBCK, and TBCK-P4HA2 resulting from the translocations
t(5;8;17)(p15;q13;q21) and t(4;5)(q24;q31) in a soft tissue
angiofibroma. Oncol Rep. 36:2455–2462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Teramura Y, Tanaka M, Yamazaki Y,
Yamashita K, Takazawa Y, Ae K, Matsumoto S, Nakayama T, Kaneko T,
Musha Y and Nakamura T: Identification of novel fusion genes in
bone and soft tissue sarcoma and their implication in the
generation of a mouse model. Cancers (Basel). 12:23452020.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhou M, Gao L, Jing Y, Xu YY, Ding Y, Wang
N, Wang W, Li MY, Han XP, Sun JZ, et al: Detection of ETV6 gene
rearrangements in adult acute lymphoblastic leukemia. Ann Hematol.
91:1235–1243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhuravleva J, Paggetti J, Martin L,
Hammann A, Solary E, Bastie JN and Delva L: MYST3/NCOA2-induced
acute myeloid leukemia in transgenic fish. Blood. 112:53292008.
View Article : Google Scholar
|
|
123
|
Esteyries S, Perot C, Adelaide J, Imbert
M, Lagarde A, Pautas C, Olschwang S, Birnbaum D, Chaffanet M and
Mozziconacci MJ: NCOA3, a new fusion partner for MOZ/MYST3 in M5
acute myeloid leukemia. Leukemia. 22:663–665. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Chang B, Bai Q, Liang L, Ge H and Yao Q:
Recurrent uterine tumors resembling ovarian sex-cord tumors with
the growth regulation by estrogen in breast cancer 1-nuclear
receptor coactivator 2 fusion gene: A case report and literature
review. Diagn Pathol. 15:1102020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Yu J, Wu WK, Liang Q, Zhang N, He J, Li X,
Zhang X, Xu L, Chan MT, Ng SS and Sung JJ: Disruption of NCOA2 by
recurrent fusion with LACTB2 in colorectal cancer. Oncogene.
35:187–195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Cao Q, Liu Z, Huang Y, Qi C and Yin X:
NCOA1-ALK: A novel ALK rearrangement in one lung adenocarcinoma
patient responding to crizotinib treatment. Onco Targets Ther.
12:1071–1074. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yoshihara K, Wang Q, Torres-Garcia W,
Zheng S, Vegesna R, Kim H and Verhaak RG: The landscape and
therapeutic relevance of cancer-associated transcript fusions.
Oncogene. 34:4845–4854. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Robinson DR, Kalyana-Sundaram S, Wu YM,
Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso
CS, et al: Functionally recurrent rearrangements of the MAST kinase
and Notch gene families in breast cancer. Nat Med. 17:1646–1651.
2011. View Article : Google Scholar : PubMed/NCBI
|