Introduction
Androgen deprivation therapy remains an important
therapy in the treatment of advanced prostate cancer; however,
despite its initial effectiveness, therapeutic resistance often
arises, thus resulting in the development of castration-resistant
prostate cancer (CRPC) (1,2). CRPC has a limited response to current
therapies, such as docetaxel with or without enzalutamide
(MDV3100), and is therefore considered a clinical challenge
(3–5). It is crucial to promptly identify new
therapeutic or chemosensitizing targets for prostate cancer
treatment.
Altered transcriptional programs confer survival
advantages to cancer cells by enhancing their overall biological
fitness across a range of different selective environments
(6). Dysregulation of transcription
factor (TF) expression drives carcinogenesis and determines the
therapeutic responsiveness of cancer cells by modulating cancer
hallmark signaling pathways (7–9).
Consequently, translational efforts targeting cancer-associated TFs
are crucial for advancing therapeutic strategies against various
types of human cancer, including prostate cancer.
Assay for Transposase-Accessible Chromatin with
sequencing (ATAC-seq) identifies accessible chromatin regions that
are closely associated with gene expression in the genome, thus
enabling the assessment of the gene regulatory landscape in human
cancer (10,11). However, the relationship between
chromatin accessibility and therapeutic response in prostate cancer
remains poorly understood. The present study used ATAC-seq data
from The Cancer Genome Atlas (TCGA) to examine the mechanisms
underlying differential therapeutic responses in patients with
prostate cancer. The results revealed the chromatin accessibility
patterns associated with therapeutic resistance in prostate cancer,
providing insights into strategies for overcoming therapeutic
challenges.
Materials and methods
Data collection and preprocessing
Raw BAM ATAC-seq files of TCGA-prostate
adenocarcinoma (PRAD) (n=26) were obtained via the NCI GDC Data
Portal (https://portal.gdc.cancer.gov/). The peak calls for
ATAC-seq profiles in prostate cancer were downloaded from TCGA
Publication Page (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG),
as previously described (10).
RNA-seq data and clinical information for TCGA-PRAD were retrieved
from the cBioPortal for Cancer Genomics (https://www.cbioportal.org/study/summary?id=prad_tcga_pan_can_atlas_2018).
ATAC-seq peak clustering
DESeq2 (v1.34.0; http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html)
was employed to construct multifactorial models for ATAC-seq read
counts in peaks. Subsequently, a variance stabilizing
transformation was computed based on the DESeq2 model, and
principal component analysis (PCA) was conducted to illustrate the
distinction between the drug response (Remission) and drug
resistance (Disease) groups (Fig.
S1). The principal component (PC) 1 coefficient was set to 0 as
the threshold to identify ambiguous samples in the PCA plot. A PC1
coefficient >0 indicates a positive correlation with the new PCA
variable, whereas a coefficient <0 indicates a negative
correlation. A total of 13 out of 20 complete response (CR)/partial
response (PR) samples had a PC1 coefficient >0, whereas 7 CR
samples had a PC1 coefficient <0 where progressive disease
(PD)/stable disease (SD) samples dominated. These 7 CR samples
cannot be clustered with the PD/SD group and were thus excluded
from further analyses.
Differential peak accessibility
Using the countOverlaps function of the R packages
GenomicAlignments (v1.30.0; http://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html)
and GenomicRanges (v1.46.1; http://bioconductor.org/packages/release/bioc/html/GenomicRanges.html),
reads aligning to global peak regions were tallied. To mitigate the
bias stemming from low-count peaks, 4,778 peaks with mean counts
<50 across all samples were removed. DESeq2 (v1.34.0) was
employed to evaluate differential peak accessibility. The
significant peaks were displayed in a hierarchical clustering
heatmap using the DESeq size-factor normalized read counts and the
complete distance metric for clustering. The R packages ChiPseeker
(v1.36.0; http://bioconductor.org/packages/release/bioc/html/ChIPseeker.html)
and clusterProfiler (v4.8.1; https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
were used to visualize peak coverage across chromosomes and
annotate peak regions. To analyze the relationship between
chromatin accessibility and transcriptional output, log2 fold
change was calculated by comparing the average peak calling for
each gene from ATAC-seq data or mean expression of genes from the
bulk RNA-seq data between Remission and Disease groups. Spearman
correlation coefficient was used to measure the strength and
direction of association. P<0.01 was considered to indicate a
statistically significant difference.
De novo TF motif analysis
The Hypergeometric Optimization of Motif EnRichment
(HOMER; v.4.11.1; http://homer.ucsd.edu/homer/motif/) utility
findMotifsGenome.pl was employed to identify the top 10 TF motifs
that were enriched in differentially accessible (DA) peaks. The
analysis focused on 100 bp-wide regions around the DA peak summits,
with hg19 serving as the reference genome. Additionally, custom
background regions were generated, spanning a 150 bp-wide range
around the peak summits. The top motifs were cross-referenced with
the known motifs from the HOMER database and then manually curated
to include only TFs that exhibited expression according to RNA-seq
data, grouping similar motifs from TFs of the same family.
Association between TFs and
therapeutic response groups
All accessible cis-regulatory elements (CREs) in the
entire genome were analyzed using Cis-Regulatory Element Motif
Activities (CREMA; http://crema.unibas.ch/crumara/). This tool can
determine the intensity signals of each CRE, calculate TF motif
activities, and identify the binding sites for hundreds of TFs
within the CREs in each sample. The vector of mean TF activities
was compared, and the association between TFs and therapeutic
response groups was evaluated, using the Wilcoxon rank-sum test.
Subsequently, the resulting P-values (one for each TF) underwent
adjustment for multiple hypothesis testing using the false
discovery rate (FDR) method. The analytical results were depicted
as a scatterplot, with the x-axis representing the mean TF activity
difference and the y-axis indicating FDR q-value. Significant TF
motifs were selected based on an absolute mean TF activity
difference >0.05 and an FDR q-value <0.05. The correlation of
their activities across patients was calculated using Pearson
correlation coefficient and was presented visually using a heatmap
to assess the consistency of the significant TFs in both the
Remission and Disease groups.
Pathway enrichment analysis
The Genomic Regions Enrichment of Annotations Tool
(v1.26; http://great.stanford.edu/public/html/) was used to
associate the sub-cluster of the DA peaks with genes. Pathway
analysis was then performed to discover the functional significance
of the DA peaks from the associated genes and to identify
overrepresented pathways (12).
PANTHER knowledgebase (http://www.pantherdb.org/) was used for the pathway
analysis (hypergeometric test, adjusted P-value <0.05).
Cell culture and reagents
Prostate cancer cell lines PC-3 (cat. no. CRL-1435),
LNCaP (clone FGC; cat. no. CRL-1740), DU145 (cat. no. HTB-81) and
22Rv1 (cat. no. CRL-2505) were purchased from the American Type
Culture Collection. The normal prostate epithelial cell line PNT1A
(cat. no. 95012614) was obtained from the European Collection of
Authenticated Cell Cultures. Cells were cultured in RPMI1640 (PC-3,
LNCaP, 22Rv1, PNT1A) or DMEM (DU145) containing 10% fetal bovine
serum, penicillin (100 U/ml) and streptomycin (100 mg/ml). Cell
culture reagents were obtained from Welgene, Inc. All cell lines
tested negative for mycoplasma contamination using the Mycoplasma
PCR Detection kit (Intron Biotechnology, Inc.). The cell lines were
authenticated using short tandem repeat analysis. Cabazitaxel (cat.
no. S3022), docetaxel (cat. no. S1148) and MDV3100 (cat. no. S1250)
were purchased from Selleck Chemicals.
Small interfering RNA (siRNA)
transfection
A siRNA against forkhead box (FOX)M1 (siFOXM1;
sense, 5′-GCUCAUACCUGGUACCUAU-3′; antisense,
5′-AUAGGUACCAGGUAUGAGC-3′) (13)
was obtained from Genolution, Inc. A control siRNA (ON-TARGETplus
Non-targeting Pool; cat. no. D-001810-10), which was used as a
negative control, was purchased from Horizon Discovery; Revvity,
Inc. PC-3 (2.5×105 cells/well), LNCaP
(2.5×105 cells/well), DU145 (2.5×105
cells/well), 22Rv1 (2.5×105 cells/well) and PNT1A
(2.5×105 cells/well) cells were seeded into 6-well
plates. Each cell line was transfected with 50 nM control siRNA or
siFOXM1 for the indicated times at 37°C using
Lipofectamine® RNAiMAX reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). For the MTT assay, cells were transfected
with either siControl or siFOXM1 for 72 h. For the colony formation
assay, cells were transfected with siControl or siFOXM1 once every
3 days for 9 days. To assess the effect of FOXM1 on drug response,
cells were treated with each drug 24 h after transfection with
siControl or siFOXM1. After 72 h of transfection, FOXM1 knockdown
was confirmed by western blotting.
MTT assay
After 24 h of siFOXM1 transfection, PC-3, LNCaP,
DU145, 22Rv1 and PNT1A cells were treated with 50 nM cabazitaxel or
0.5 nM docetaxel for 48 h, or 10 µM MDV3100 for 72 h at 37°C. The
MTT assay (MilliporeSigma) was performed to assess cell viability,
according to the manufacturer's instructions. The purple formazan
was dissolved in DMSO and was quantitated by determining the
absorbance at 570 nm on a BioTek SynergyMx microplate reader
(BioTek; Agilent Technologies, Inc.).
Colony formation assay
PC-3 (3×103 cells/well), LNCaP
(8×103 cells/well), DU145 (1×103 cells/well),
22Rv1 (3×103 cells/well) and PNT1A (3×103
cells/well) cells were plated into 6-well plates. Cells were
transfected with 50 nM siControl or siFOXM1 once every 3 days for 9
days at 37°C. After the cells were fixed with 3.7% paraformaldehyde
(MilliporeSigma) for 10 min at room temperature, they were stained
with 0.5% crystal violet (MilliporeSigma) for 15 min at room
temperature. Next, the number of colonies, defined as >50
cells/colony, was counted using ImageJ (1.8.0 172; National
Institutes of Health).
Western blotting
The crude extracts from PC-3, LNCaP, DU145, 22Rv1
and PNT1A cells were prepared with RIPA buffer [50 mM Tris-HCl (pH
7.4), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1%
SDS and 0.5 EDTA] including protease and phosphatase inhibitor
cocktails (MilliporeSigma). The total protein concentration was
quantified using the BCA assay kit (Thermo Fisher Scientific Inc.).
Subsequently, the samples were separated by SDS-PAGE on 6% (FOXM1,
30 µg), 10% (β-tubulin, 10 µg) or 15% (caspase-3, 10 µg; cleaved
caspase-3, 80 µg) gels and transferred onto nitrocellulose
membranes (Bio-Rad Laboratories, Inc.). The membranes were blocked
with 5% skim milk in Tris-buffered saline-0.1% Tween 20 for 1 h at
room temperature and were then incubated with anti-FOXM1 (1:1,000;
cat. no. 5436S; Cell Signaling Technology, Inc.), anti-caspase-3
(1:1,000; cat. no. 9662S; Cell Signaling Technology, Inc.),
anti-cleaved caspase-3 (1:1,000; cat. no. 9661S; Cell Signaling
Technology, Inc.) and anti-β-tubulin (1:5,000; cat. no. T4026;
MilliporeSigma) overnight at 4°C. β-tubulin was used as the loading
control. After primary antibody incubation, the membranes were
incubated with a goat anti-rabbit IgG-HRP antibody (1:5,000; cat.
no. A120-101P; Bethyl Laboratories, Inc.) or a goat anti-mouse
antibody (1:5,000, cat. no. A90-116P; Bethyl Laboratories, Inc.)
for 1 h at room temperature. Subsequently, the signals were
determined using Amersham ECL reagent (Cytiva). X-ray films were
scanned on an EPSON scanner. The data are representative of at
least three independent experiments.
Statistical analysis and data
visualization
R version 4.1.1 (R Foundation for Statistical
Computing) was used for all statistical analyses. The R package
ggplot2 (v3.3.5; http://cran.r-project.org/web/packages/ggplot2/index.html)
was used to create graphs, whereas the R package ComplexHeatmap
(v2.10.0; http://www.bioconductor.org/packages/ComplexHeatmap/)
was utilized to produce heatmaps. The P-values for multiple
comparisons were adjusted using the Benjamini-Hochberg method. The
Kaplan-Meier survival curve and log-rank test were employed to
estimate overall survival (14–16).
The samples were divided into FOXM1 high-expression (n=79) or
low-expression (n=299) groups from TCGA transcriptomic data. To
divide samples into high- and low-expression groups, the
bifurcation point approach (17)
was used because the cohort might have biased expression pattern of
a specific gene, and the median or quantile is not the most natural
cut-point for gene expression profiles and survival prediction.
Subsequently, the Cox proportional hazards model was applied to
calculate hazard ratios (HRs) and 95% confidence intervals. A
one-way analysis of variance (ANOVA) was employed to compare means
among the experimental groups, followed by Bonferroni's multiple
comparisons test. A two-way ANOVA followed by Bonferroni's test was
used for pairwise comparisons between siControl and siFOXM1, or
siFOXM1 and its combinations with drugs (docetaxel, cabazitaxel, or
MDV3100). P- or FDR q-values <0.05 were considered to indicate a
statistically significant difference.
Results
Chromatin accessibility differences
between the Remission and Disease groups
To examine the association between chromatin
accessibility and therapeutic response in prostate cancer, ATAC-seq
data consisting of 98,905 peaks from 26 TCGA-PRAD samples were
analyzed (10) (Fig. S1). Therapeutic response was
categorized into CR, PR, PD and SD based on response criteria
(18). PCA of peak read count
showed a clear separation between CR/PR samples (n=13) and PD/SD
samples (n=6) when the 7 CR samples were excluded (Fig. S2A). In addition, it was confirmed
that the 7 excluded CR samples exhibited a gene expression pattern
more similar to PD/SD samples than to CR/PR samples (Fig. S2B). Excluding these samples allowed
for more distinct CR/PR and PD/SD groups, despite the reduction in
sample size. Henceforth, these two groups were designated as the
Remission (n=13) and Disease (n=6) groups (Fig. 1A). The present study then
investigated the chromatin accessibility differences between the
Remission and Disease groups. A total of 20,610 DA peaks (absolute
log2 fold change >1.0 and adjusted P<0.05) were
identified, which accounted for 20.84% of the total ATAC-seq peaks
(Fig. 1B and C). Among these,
11,385 peaks (55.24%) exhibited increased accessibility in the
Remission group, whereas 9,225 peaks (44.76%) showed increased
accessibility in the Disease group (Fig. 1B and C). The DA ATAC-seq peaks were
distributed across various chromatin regions, including promoter,
exon, intron and distal intergenic regions, for both the Remission
and Disease groups (Fig. 1D). These
results identified a distinct difference in chromatin accessibility
between the two groups.
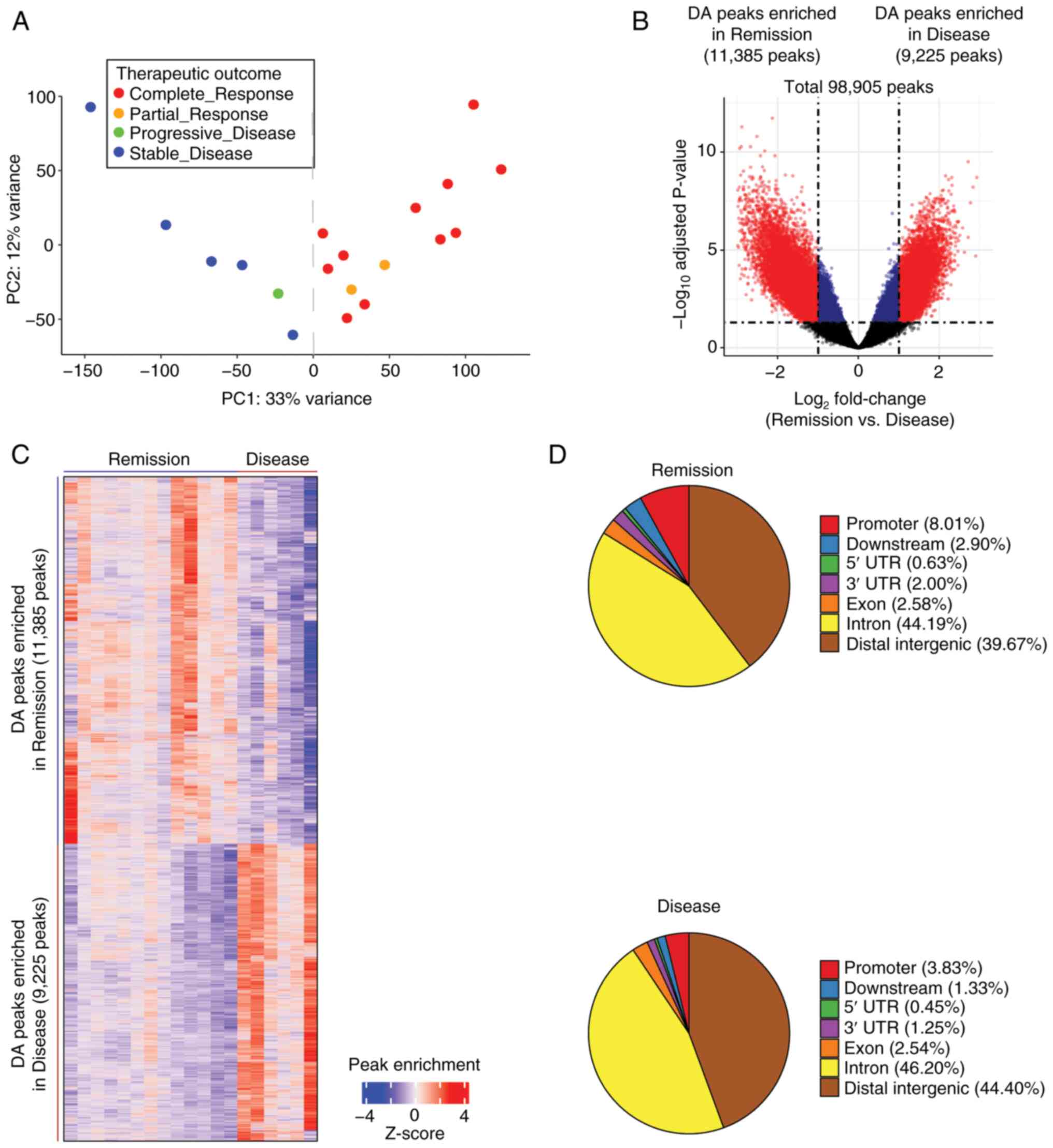 | Figure 1.Chromatin accessibility differences
between the Remission and Disease groups. (A) Principal component
analysis plot of the ATAC-seq signal showed distinct clusters
between the complete response/partial response (n=13) and
progressive disease/stable disease (n=6) groups, effectively
reclassifying them as the Remission and Disease groups. (B) Volcano
plot of ATAC-seq peaks exhibited discrete separation between the
Remission and Disease groups. The adjusted P-values were obtained
using DESeq2. The vertical dotted lines indicate absolute
log2 fold change of 1.0, whereas the horizontal dotted
line represents an adjusted P-value of 0.05. Among the total 98,905
peaks, significant peaks with log2 fold change >1.0
are highlighted in red, whereas those with log2 fold
change <1.0 are depicted in blue. Notably, 20,610 significant DA
peaks (highlighted in red) were respectively enriched in the
Remission (n=11,385) and Disease (n=9,225) groups. (C) Hierarchical
clustering of the 20,610 DA peaks in the Remission and Disease
groups. The color indicator represents log2-transformed
peak count data, which has been normalized by z-score row. (D) Pie
charts of the distribution of DA ATAC-seq peaks (false discovery
rate q-value <0.05) across various chromatin regions for the
Remission and Disease groups. ATAC-seq, Assay for
Transposase-Accessible Chromatin with sequencing. |
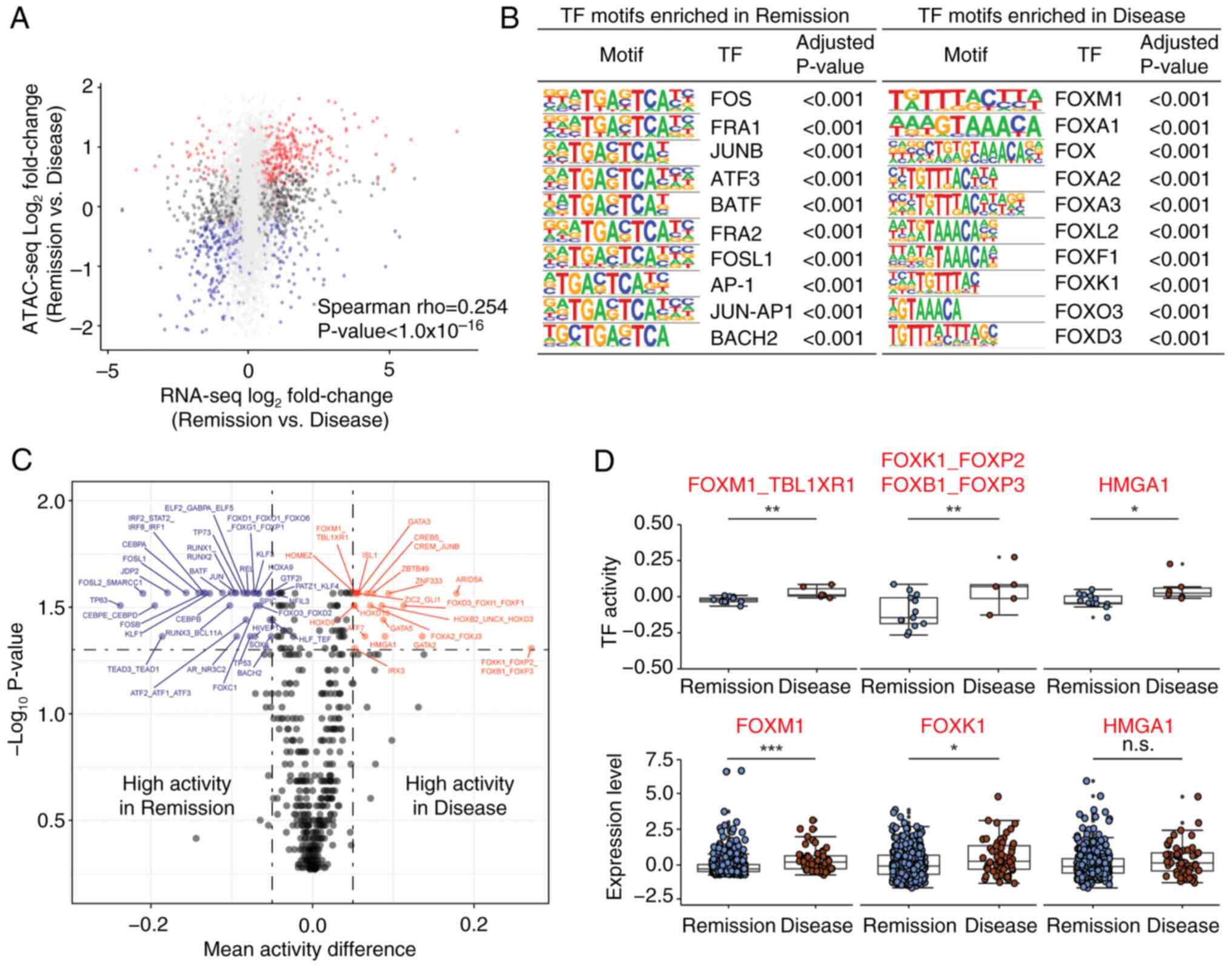
Chromatin accessibility is associated
with transcriptional output and TF activity between the Remission
and Disease groups
To explore the relationship between chromatin
accessibility and transcriptional output, ATAC-seq data were
compared with RNA-seq data. The chromatin accessibility of
individual genes showed a moderate association with the
corresponding gene expression between the Remission and Disease
groups (Fig. 2A). Subsequently, to
identify key TFs that drive the transcription program difference
between the Remission and Disease groups, HOMER motif analysis of
DA ATAC-seq peaks was performed. Several proto-oncogenes, such as
FOS, FRA1 and JUNB, were enriched in the Remission group (Fig. 2B left), whereas FOX family genes,
such as FOXM1, FOXA1 and FOXA2, were enriched in the Disease group
(Fig. 2B right).
Sample-specific TF binding motif activities were
also inferred using CREMA, which enables the mapping of chromatin
accessibility profiles to a lower-dimensional inferred TF activity.
A total of 54 TF motif activities were significantly associated
with therapeutic response, as determined by FDR q-value <0.05
and absolute mean activity difference >0.05 (Figs. 2C and S3A). TF activities from the same
therapeutic response group (either the Remission or Disease) were
also correlated across samples by Spearman correlation (Fig. S3B). Notably, the Wilcoxon rank-sum
test showed that several TFs, including FOXM1, FOXK1 and HMGA1,
consistently exhibited higher activity and mRNA expression in the
Disease group compared with in the Remission group (Fig. 2D). Taken together, these results
indicated that chromatin accessibility determines differential
transcriptional output and TF activity between the two groups.
Experiments involving FOXM1 exemplify
the usefulness of chromatin accessibility analysis for determining
therapeutic response
Notably, FOXM1 was frequently identified among the
TFs highlighted in the present analyses (Fig. 2B and D). Therefore, to validate the
results of computational analysis, TCGA-PRAD data were analyzed.
The expression levels of FOXM1 were markedly upregulated in
metastasis samples compared with those in benign or primary cancer
samples (Fig. 3A), providing a
potential underlying mechanism of elevated FOXM1 TF activity in
metastasis (Fig. 2B and D).
Kaplan-Meier analysis revealed that high expression levels of FOXM1
were associated with a worse prognosis in patients with prostate
cancer (HR=3.0, P=1.18×10−3) (Fig. 3B). These results suggested that
FOXM1 may serve a crucial role in determining therapeutic response
and could be considered a useful prognostic marker for prostate
cancer.
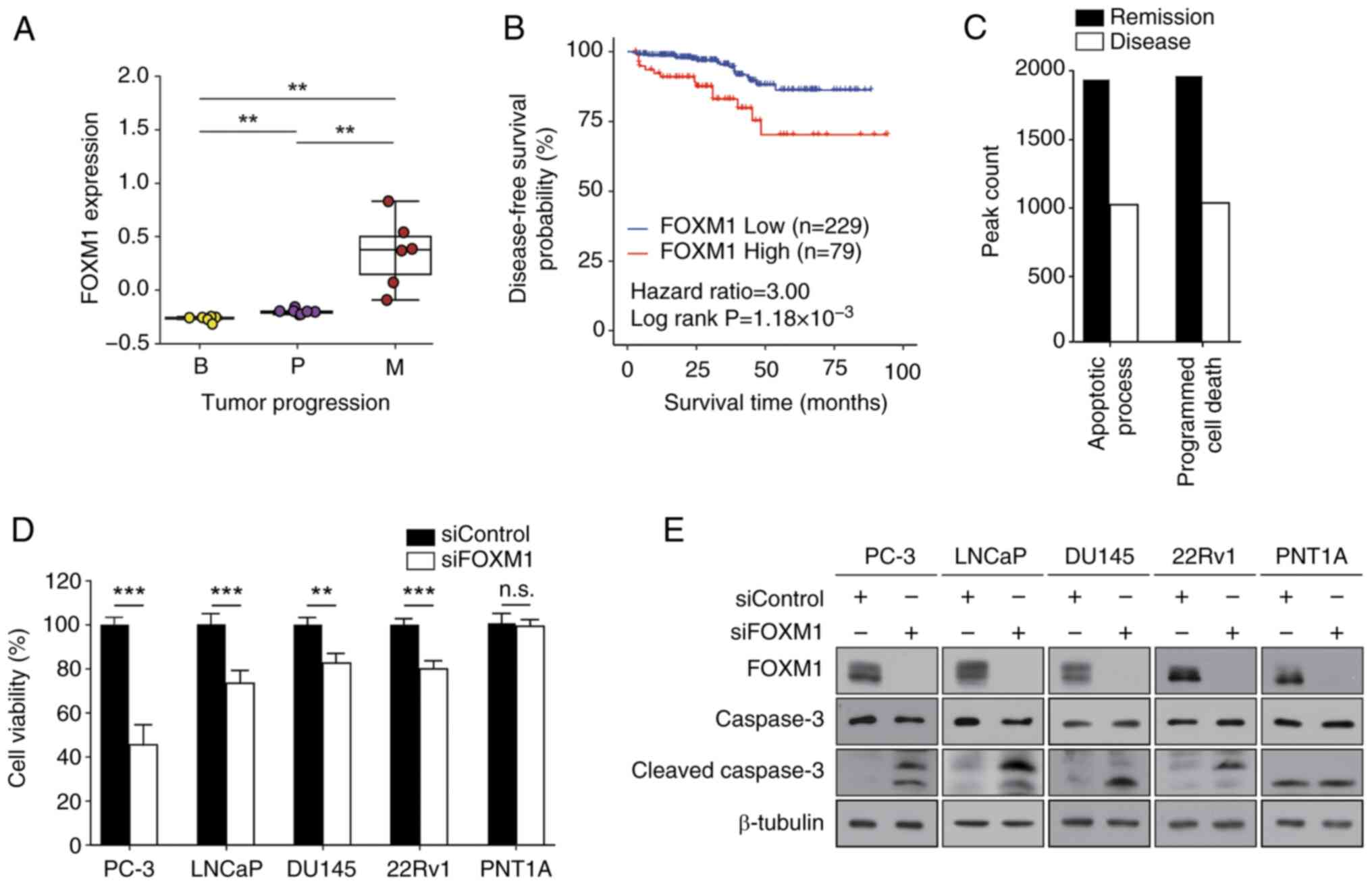 | Figure 3.Effects of FOXM1 on prostate cancer.
(A) FOXM1 expression levels in patients with prostate cancer are
represented in the box plots. The x-axis indicates three different
stages of prostate cancer, and the y-axis depicts the normalized
expression levels. (B) Disease-free survival curve for patients
with prostate cancer based on the expression levels of FOXM1 from
The Cancer Genome Atlas data. (C) Enrichment of PANTHER pathways of
differential accessibilities in the Remission or Disease groups.
The bar represents the number of peak region hits associated with
the pathway. (D and E) PC-3, LNCaP, DU145, 22Rv1 and PNT1A cells
were transfected with 50 nM siRNAs for 72 h prior to the MTT assay
and western blot analysis. (D) Cell viability was expressed as a
relative value compared with that of the siControl, which was set
to 100%. Data are presented as the mean ± SEM (n=4). (E) Caspase-3
expression was assessed using anti-cleaved caspase-3 antibody. A
representative image from three independent experiments is shown.
**P<0.01, ***P<0.005. B, benign; FOX, forkhead box; M,
metastatic; P, primary; si, small interfering. |
To assess the role of FOXM1 in prostate cancer,
pathway enrichment analysis was performed. The apoptotic process
and programmed cell death pathways were distinct in the DA ATAC-seq
peaks of Disease samples (Fig. 3C).
To confirm the computational prediction results, MTT and colony
formation assays were then performed. FOXM1 expression was knocked
down and successful transfection was confirmed (Fig. 3E). FOXMI knockdown suppressed cell
viability (Fig. 3D) and colony
formation (Table I; Fig. S4) of prostate cancer cells (PC-3,
LNCaP, DU145 and 22Rv1), but not of normal prostate epithelial
cells (PNT1A). In addition, FOXM1 knockdown elevated the expression
levels of cleaved caspase-3 in prostate cancer cells (Figs. 3E and S5). Taken together, these results
indicated that FOXM1 may have a crucial role in regulating cell
survival and proliferation in prostate cancer.
 | Table I.Effect of FOXM1 knockdown on colony
formation in prostate cancer cells. |
Table I.
Effect of FOXM1 knockdown on colony
formation in prostate cancer cells.
|
| Number of
colonies |
|---|
|
|
|
|---|
| Cell line | siControl | siFOXM1 |
|---|
| PC-3 | 310.70±6.49 |
10.67±1.45a |
| LNCaP | 332.00±20.42 |
12.33±3.48a |
| DU145 | 157.00±6.43 |
24.00±3.06a |
| 22Rv1 | 227.70±2.33 |
76.33±16.60a |
| PNT1A | 0 | 0 |
The present study also examined whether FOXM1
expression could influence the therapeutic response of prostate
cancer cells to several therapeutic agents. Transfection with
siFOXM1 increased the sensitivity of PC-3 [androgen receptor
(AR)-negative], LNCaP [AR-positive, AR splice variant-7
(AR-V7)-negative], DU145 (AR-negative) and 22Rv1 (AR-positive,
AR-V7-positive) cells to docetaxel or cabazitaxel compared with
each drug alone (Fig. 4A and B). In
addition, FOXM1 knockdown enhanced the therapeutic activity of the
AR antagonist MDV3100 only in LNCaP cells (Fig. 4C). By contrast, siFOXM1 did not
enhance chemotherapeutic effects in PNT1A cells (Fig. 4A-C). These results suggested that
FOXM1 inhibition may be a potential strategy for improving the
efficacy of nonselective chemotherapy or androgen blockade therapy.
Notably, there was a weak, but significant, positive
co-expressional correlation between FOXM1 and AR in TCGA
transcriptomic data (Fig. S6),
underscoring the clinical significance of the combined inhibition
of FOXM1 and AR. These findings indicated that FOXM1 represents a
promising therapeutic target for prostate cancer.
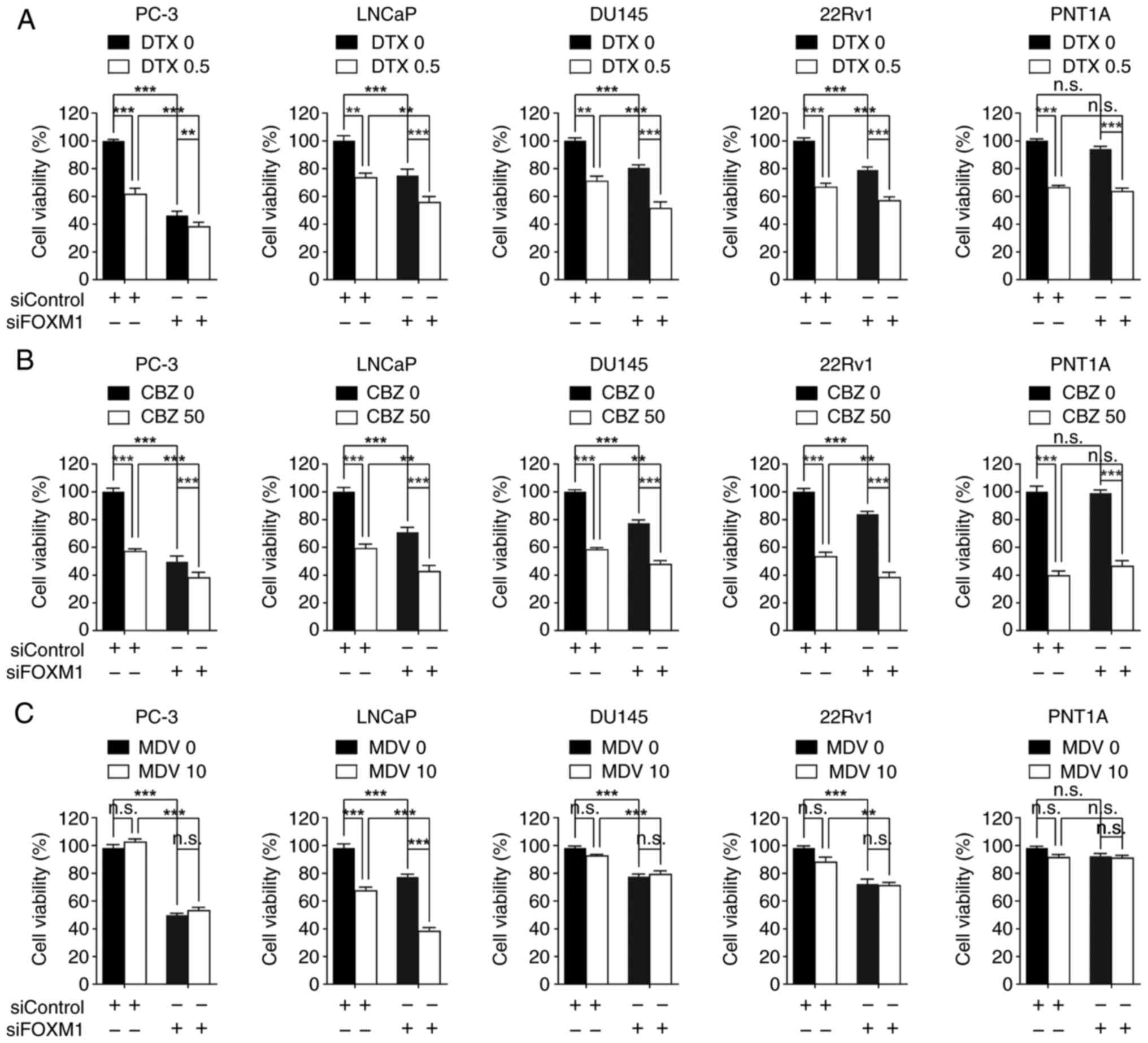 | Figure 4.FOXM1 knockdown increases therapeutic
response in prostate cancer. Each cell line was transfected with 50
nM siRNAs for 24 h, and then cells were further treated with (A)
0.5 nM DTX, (B) 50 nM CBZ or (C) 10 µM MDV for 48 h prior to MTT
assay. Cell viability was expressed as a relative value compared
with that of the siControl, which was set to 100%. Data are
presented as the mean ± SEM (n=4). **P<0.01, ***P<0.005. CBZ,
cabazitaxel; DTX, docetaxel; FOX, forkhead box; MDV, MDV3100; n.s.,
not significant; si, small interfering. |
Discussion
The present study described three main findings: i)
Chromatin accessibility was associated with distinct therapeutic
responses in prostate cancer; ii) chromatin accessibility was
associated with distinct transcriptional output and TF activity in
prostate cancer; and iii) FOXM1 exemplified the usefulness of
chromatin accessibility analysis in assessing therapeutic
response.
Balancing sample cluster integrity with sample size
is a common challenge in bioinformatics, particularly when
excluding outliers. The small sample size of 26 in the PRAD
ATAC-seq study raised concerns. To address this, an initial PCA was
conducted to identify 7 CR samples with gene expression patterns
similar to PD/SD samples. These outliers likely represent technical
or biological confounders, complicating the interpretation of their
response status. Although excluding these 7 CR samples reduced the
sample size, it improved the robustness of the findings, minimized
data skew, and allowed for more accurate associations between
molecular profiles and clinical outcomes. This step was critical
for understanding chromatin accessibility differences underlying
treatment response.
The prediction of therapeutic response remains a
significant challenge in treating prostate cancer, with epigenetic
changes serving a crucial role in its development (19). Chromatin accessibility profiling
using ATAC-seq is an important tool for assessing epigenetic
changes in cancer tissues (20);
however, only a few studies have applied the ATAC-seq approach
specifically to prostate cancer. Most research has analyzed the
ATAC-seq data from prostate cancer cell lines (21), patient-derived xenografts (11) or murine organoids (22), rather than from clinical samples.
While some studies have utilized ATAC-seq data from patient
samples, they primarily focused on molecular classification, cell
heterogeneity, or alterations in AR signaling of CRPC (23,24).
By contrast, the present study integrated ATAC-seq data from
primary human prostate cancer samples with individual patient
therapeutic response data from TCGA. This approach revealed
distinct chromatin accessibility signatures, regulatory pathways
and TF activities linked to therapeutic response in prostate
cancer. Notably, the present analysis uniquely identified FOXM1 as
a modulator of therapeutic response; to the best of our knowledge,
this has not been reported in other studies with ATAC-seq data. The
present study provides a pioneering genome-wide analysis, revealing
significant epigenetic differences between responsive and resistant
cases, thus providing valuable insights into the mechanisms
underlying therapeutic response in prostate cancer.
Chromatin profiling provides a valuable approach for
classifying prostate cancer and discovering potential drug targets
(11). In the present study,
ATAC-seq data analysis was employed to identify distinct chromatin
accessibility and specific TF activities associated with
therapeutic response in prostate cancer. The analysis revealed that
various FOX TF family genes were enriched in the Disease group.
Dysregulation of FOX genes is frequently observed in human cancer
(25), where they drive tumor
development, progression and drug resistance (26,27).
The present study demonstrated that FOXM1 was associated with poor
clinical outcomes in patients with prostate cancer and served as a
key TF governing transcriptional programs in therapeutic resistance
in prostate cancer, which is consistent with previous findings that
FOXM1 is functionally required not only for tumorigenesis, tumor
proliferation, metastasis and invasion, but also serves as a poor
prognostic marker and induces docetaxel resistance in CRPC
(28).
Although the present study primarily focused on
FOXM1, further investigation is needed to identify which other FOX
isotypes may have a critical role in mediating therapeutic
resistance. Additionally, future studies are required to
investigate functional interaction among FOX family genes in
therapeutic resistance and to assess the impact of their
co-inhibition on cancer resistance. Moreover, the molecular
mechanisms underlying how FOXM1 induces therapeutic resistance in
prostate cancer are still unclear and require elucidation.
In conclusion, the present study demonstrated that
chromatin accessibility may be associated with therapeutic response
in prostate cancer. These findings provide valuable insights into
the understanding of prostate cancer biology and could inform the
development of therapeutic approaches for this disease.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from the National Research
Foundation of Korea grant funded by the Korea government (The
Ministry of Science and ICT) (grant nos. 2018R1A4A1023822 and
2020R1A2C1102574), a grant from the Seoul National University
Hospital Research Fund (grant no. 0320230140), and the Education
and Research Encouragement Fund of Seoul National University
Hospital.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SL, DYL, IS, JNC and JHJ designed the research and
wrote the main manuscript text. SL analyzed the ATAC-seq data and
made the figures. DYL and JNC designed and performed the validation
experiment using prostate cancer cell lines. DYL, JNC and JHJ
confirm the authenticity of all the raw data. IS contributed to
data analysis and interpretation. All authors read and approved the
final version of the manuscript, and consented to its
publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chandrasekar T, Yang JC, Gao AC and Evans
CP: Mechanisms of resistance in castration-resistant prostate
cancer (CRPC). Transl Androl Urol. 4:365–380. 2015.
|
|
2
|
Dai C, Dehm SM and Sharifi N: Targeting
the androgen signaling axis in prostate cancer. J Clin Oncol.
41:4267–4278. 2023. View Article : Google Scholar
|
|
3
|
Park S, Kim YS, Kim DY, So I and Jeon JH:
PI3K pathway in prostate cancer: All resistant roads lead to PI3K.
Biochim Biophys Acta Rev Cancer. 1870:198–206. 2018. View Article : Google Scholar
|
|
4
|
Cai M, Song XL, Li XA, Chen M, Guo J, Yang
DH, Chen Z and Zhao SC: Current therapy and drug resistance in
metastatic castration-resistant prostate cancer. Drug Resist Updat.
68:1009622023. View Article : Google Scholar
|
|
5
|
Gebrael G, Fortuna GG, Sayegh N, Swami U
and Agarwal N: Advances in the treatment of metastatic prostate
cancer. Trends Cancer. 9:840–854. 2023. View Article : Google Scholar
|
|
6
|
Bradner JE, Hnisz D and Young RA:
Transcriptional Addiction in Cancer. Cell. 168:629–643. 2017.
View Article : Google Scholar
|
|
7
|
Bhagwat AS and Vakoc CR: Targeting
transcription factors in cancer. Trends Cancer. 1:53–65. 2015.
View Article : Google Scholar
|
|
8
|
Bushweller JH: Targeting transcription
factors in cancer-From undruggable to reality. Nat Rev Cancer.
19:611–624. 2019. View Article : Google Scholar
|
|
9
|
Shiah JV, Johnson DE and Grandis JR:
Transcription factors and cancer: Approaches to Targeting. Cancer
J. 29:38–46. 2023. View Article : Google Scholar
|
|
10
|
Corces MR, Granja JM, Shams S, Louie BH,
Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al:
The chromatin accessibility landscape of primary human cancers.
Science. 362:eaav18982018. View Article : Google Scholar
|
|
11
|
Tang F, Xu D, Wang S, Wong CK,
Martinez-Fundichely A, Lee CJ, Cohen S, Park J, Hill CE, Eng K, et
al: Chromatin profiles classify castration-resistant prostate
cancers suggesting therapeutic targets. Science. 376:eabe15052022.
View Article : Google Scholar
|
|
12
|
McLean CY, Bristor D, Hiller M, Clarke SL,
Schaar BT, Lowe CB, Wenger AM and Bejerano G: GREAT improves
functional interpretation of cis-regulatory regions. Nat
Biotechnol. 28:495–501. 2010. View
Article : Google Scholar
|
|
13
|
Shan L, Zhao M, Lu Y, Ning H, Yang S, Song
Y, Chai W and Shi X: CENPE promotes lung adenocarcinoma
proliferation and is directly regulated by FOXM1. Int J Oncol.
55:257–266. 2019.
|
|
14
|
Park S, Lim JM, Chun JN, Lee S, Kim TM,
Kim DW, Kim SY, Bae DJ, Bae SM, So I, et al: Altered expression of
fucosylation pathway genes is associated with poor prognosis and
tumor metastasis in non-small cell lung cancer. Int J Oncol.
56:559–567. 2020.
|
|
15
|
Lee DY, Lee S, Kim YS, Park S, Bae SM, Cho
EA, Park EJ, Park HH, Kim SY, So I, et al: Cyclosporin A inhibits
prostate cancer growth through suppression of E2F8 transcription
factor in a MELK-dependent manner. Oncol Rep. 50:2182023.
View Article : Google Scholar
|
|
16
|
Lee S, Park YR, Kim SH, Park EJ, Kang MJ,
So I, Chun JN and Jeon JH: Geraniol suppresses prostate cancer
growth through down-regulation of E2F8. Cancer Med. 5:2899–2908.
2016. View
Article : Google Scholar
|
|
17
|
Marco E, Karp RL, Guo G, Robson P, Hart
AH, Trippa L and Yuan GC: Bifurcation analysis of single-cell gene
expression data reveals epigenetic landscape. Proc Natl Acad Sci
USA. 111:E5643–5650. 2014. View Article : Google Scholar
|
|
18
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S,
Mooney M, et al: New response evaluation criteria in solid tumours:
Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247.
2009. View Article : Google Scholar
|
|
19
|
Conteduca V, Hess J, Yamada Y, Ku SY and
Beltran H: Epigenetics in prostate cancer: Clinical implications.
Transl Androl Urol. 10:3104–3116. 2021. View Article : Google Scholar
|
|
20
|
Grandi FC, Modi H, Kampman L and Corces
MR: Chromatin accessibility profiling by ATAC-seq. Nat Protoc.
17:1518–1552. 2022. View Article : Google Scholar
|
|
21
|
Taavitsainen S, Engedal N, Cao S, Handle
F, Erickson A, Prekovic S, Wetterskog D, Tolonen T, Vuorinen EM,
Kiviaho A, et al: Single-cell ATAC and RNA sequencing reveal
pre-existing and persistent cells associated with prostate cancer
relapse. Nat Commun. 12:53072021. View Article : Google Scholar
|
|
22
|
Grbesa I, Augello MA, Liu D, McNally DR,
Gaffney CD, Huang D, Lin K, Ivenitsky D, Goueli R, Robinson BD, et
al: Reshaping of the androgen-driven chromatin landscape in normal
prostate cells by early cancer drivers and effect on therapeutic
sensitivity. Cell Rep. 36:1096252021. View Article : Google Scholar
|
|
23
|
Shrestha R, Chesner LN, Zhang M, Zhou S,
Foye A, Lundberg A, Weinstein AS, Sjöström M, Zhu X,
Moreno-Rodriguez T, et al: An atlas of accessible chromatin in
advanced prostate cancer reveals the epigenetic evolution during
tumor progression. Cancer Res. 84:3086–3100. 2024. View Article : Google Scholar
|
|
24
|
Bian X, Wang W, Abudurexiti M, Zhang X, Ma
W, Shi G, Du L, Xu M, Wang X, Tan C, et al: Integration analysis of
single-cell multi-omics reveals prostate cancer heterogeneity. Adv
Sci (Weinh). 11:e23057242024. View Article : Google Scholar
|
|
25
|
Katoh M, Igarashi M, Fukuda H, Nakagama H
and Katoh M: Cancer genetics and genomics of human FOX family
genes. Cancer Lett. 328:198–206. 2013. View Article : Google Scholar
|
|
26
|
Myatt SS and Lam EW: The emerging roles of
forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 7:847–859.
2007. View
Article : Google Scholar
|
|
27
|
Castaneda M, Hollander PD and Mani SA:
Forkhead box transcription factors: Double-edged swords in cancer.
Cancer Res. 82:2057–2065. 2022. View Article : Google Scholar
|
|
28
|
Lee DY, Chun JN, So I and Jeon JH:
Oncogenic role of FOXM1 in human prostate cancer (Review). Oncol
Rep. 51:152024. View Article : Google Scholar
|