Cells represent the fundamental units and building
blocks of life, serving a pivotal role in various aspects of
existence through their proliferation, differentiation, functional
characteristics and eventual death. Cell death was initially
perceived to be passive and unregulated. However, by the 1970s,
apoptosis was discovered to be is a form of programmed cell death,
executed through developmentally programmed pathways (1). The broader classification of regulated
cell death refers to death programs that are molecularly regulated
but not always associated with developmental programming (2). One such example includes ferroptosis,
a term that was first introduced in 2012 (3), and which has had a profound impact on
research into metabolic reprogramming in cancer. Ferroptosis is
driven by the iron-dependent peroxidation of phospholipids and is
influenced by a variety of cellular metabolic events, including
redox balance, iron handling, mitochondrial activity and the
metabolism of amino acids, lipids and sugars, as well as a number
of disease-related signaling pathways (3).
Copper-induced death, also known as cuproptosis, is
another type of regulated cell death, which results from toxic
stress inflicted on proteins associated with the tricarboxylic acid
(TCA) cycle within the mitochondria due to lipid acylation
(4). By contrast, disulfide-induced
cell death, also known as disulfide death or disulfidptosis, arises
from the accumulation of abnormal disulfide bonds in cells
expressing high levels of solute carrier family 7 member 11
(SLC7A11) when starved of glucose. This leads to the formation of
disulfide bonds between actin cytoskeletal proteins, which disrupts
the F-actin network. The F-actin network comprises actin filaments
that interact with myosin to generate movement within the cell and
thereby contribute to processes such as cell migration and
deformation. Disruption of this network leads to structural damage
and ultimately triggers cell death (5). However, gaps remain in our
understanding of the field of disulfide death. Research in this
field has evolved through the exploration of trace elements, amino
acid metabolism, redox reactions and cell death, and has generated
a large volume of published research (5). Therefore, it is imperative to
critically examine and consolidate recent research progress. In the
present review, the causes, regulatory mechanisms and potential
physiological roles of disulfide death, as well as associated
therapeutic approaches and their effects on tumors are explored.
Additionally, new theoretical challenges and issues facing this
field are discussed, and recommendations are offered to guide
future research into disulfide death.
Sulfur is not only an essential element for living
organisms but is also one of the most abundant resources on Earth.
A number of sulfur compounds participate in the formation of
inorganic sulfate salts, and sulfur is also incorporated into
eukaryotes, e.g., via the amino acids methionine, cysteine and
cysteine (6). Methionine is
considered a key amino acid, as it plays a central role in
polyamine synthesis by serving as a methyl donor, and is involved
in the production of glutathione (GSH) (7). Methionine is a precursor for cysteine
in the synthesis of GSH; the latter primarily exists in the body in
its reduced thiol form and an oxidized disulfide form (8). The liver is the primary source of GSH,
where it plays a crucial role in antioxidative protection,
detoxification, cysteine storage and the regulation of immune
function (9). GSH deficiency or
changes in the GSH/GSSG ratio increase the vulnerability of cells
to oxidative stress, inflammation and tumor progression. By
contrast, elevated GSH levels increase antioxidant capacity and
resistance to oxidative stress, a phenomenon evident in various
tumors (10). Exogenous GSH has
been shown to inhibit inflammatory responses via the modulation of
reactive oxygen species (ROS), whereas endogenous GSH has been
shown to serve a role in the precise regulation of the innate
immune response to infection, thereby modulating inflammation
(10). Thus, GSH has a dual role in
the inflammatory response; it acts both as an antioxidant by
scavenging ROS during oxidative stress and as a signaling molecule
that regulates protein function through thiol-disulfide bond
exchange reactions, such as protein glutathionylation. Numerous
examples of this regulatory role in oncogenes including p53,
hypoxia-inducible factor-1 and, c-jun are included in a database
developed by Chen et al (11) and are also discussed in a previous
review (12). GSH-deficient
regulatory T cells (Tregs) exhibit increased serine uptake and
de novo synthesis, which enhances the one-carbon metabolic
pathway. GSH not only controls the redox state of Tregs, but also
serves as a negative feedback regulator to limit serine
input/synthesis. The inhibition of serine uptake restores the
suppressive function of GSH-deficient Tregs. According to relevant
experiments, using mice that specifically ablate the catalytic
subunit of Treg glutamate cysteine ligase (Gclc), the researchers
found that removing GSH from Treg alters serine introduction and
synthesis, and that the integrity of this feedback loop is critical
to Treg's ability to inhibit it (13). Thus, GSH not only controls the redox
state of Tregs, but also serves as a negative feedback regulator to
limit serine input/synthesis (13).
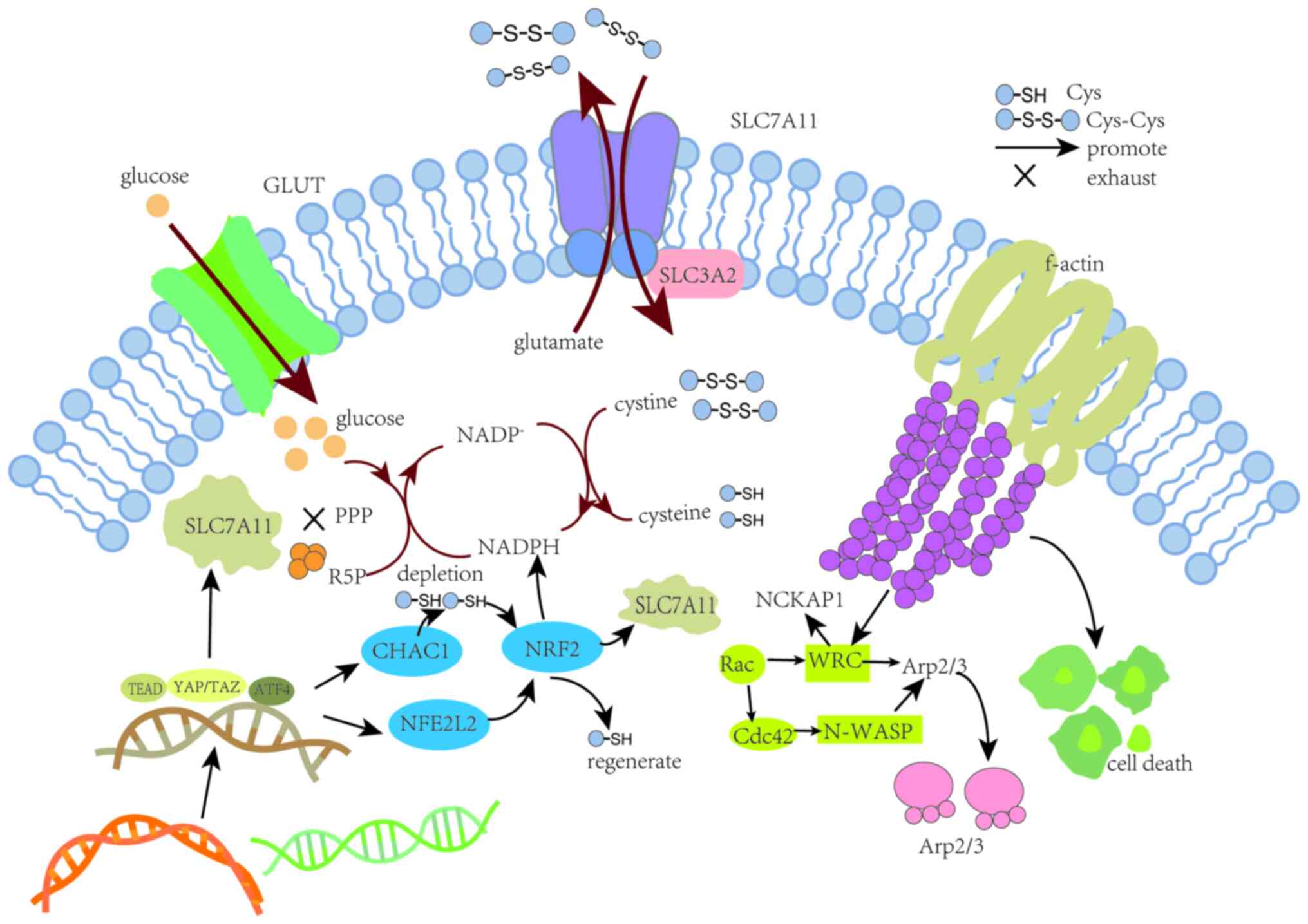
SLC7A11 is a cystine/glutamate antiporter, also
known as xCT, that plays a key role in numerous cancer types
(14–16), such as colorectal cancer (17), gastric cancer (18), breast cancer (19) and non-small cell lung cancer
(20). SLC7A11 facilitates the
cellular import of cystine and export of glutamate. Once inside the
cell, cystine is converted to cysteine with the consumption of
nicotinamide adenine dinucleotide phosphate (NADPH) generated via
the pentose phosphate pathway (PPP). Thus, SLC7A11 facilitates the
biosynthesis of GSH from cysteine and thereby promotes the
antioxidative defense (21). The
cystine/glutamate antiporter system Xc− is
primarily a dimeric structure comprising SLC7A11 as a light chain
subunit and SLC3A2 as a heavy chain subunit. The former regulates
the biological activity of Xc− while the
latter anchors the SLC7A11 protein to the cell membrane (22) (Fig.
1).
The inward and outward transport of GSH, including
its disulfide-bonded form, proceeds via the
Xc− system (23). Under normal physiological
conditions, the reducing cellular environment promotes the
formation of disulfide bonds in cytoplasmic proteins. Disulfide
bond activation has been suggested to depend on three key features:
i) High expression of SLC7A11, since high levels of extracellular
cystine uptake and intracellular cysteine accumulation contribute
to the occurrence of disulfide bond stress in cellular metabolic
processes. Furthermore, the high expression of SLC7A11 stimulates
tumor growth but also inhibits tumor metastasis (21,22,24–27).
ii) Glucose is critical to metabolic processes, and its metabolism
through the PPP generates NADPH in its reduced form (28). The deprivation of glucose and
administration of anti-glycolytic drugs can cause tumor cells to
die. This death may be due to necrosis, mitochondrial- or
caspase-8-mediated apoptosis, or the formation of abnormal
disulfide bonds between actin cytoskeletal proteins (29). iii) The excessive accumulation of
disulfides promotes the formation of disulfide bonds between actin
cytoskeletal proteins. This results in the contraction of actin and
its detachment from the plasma membrane, ultimately leading to cell
contraction and death (30)
(Fig. 1). When the expression of
SLC7A11 is low and combined with a state of glucose deprivation or
the interruption of glucose intake, intracellular glucose levels
decrease. This inhibits the production of glucose 6-phosphate via
the inhibition of hexokinase, and also suppresses the generation of
NADPH and pyruvate through the PPP and glycolysis (28). As a result, pyruvate levels are
effectively reduced via the TCA cycle and mitochondrial oxidative
phosphorylation. These physiological processes trigger oxidative
stress and deplete ATP levels, which in turn increases the
expression levels of Bax and Bak. Cytochrome c is then
released from the mitochondria, which activates caspase-3 and
further promotes membrane blebbing and cell apoptosis mediated by
poly (ADP-ribose) polymerase (31).
By contrast, the high expression of SLC7A11 combined with glucose
starvation or reduced intake leads to a marked intake of cystine,
decreased cysteine levels, reduced consumption of NADPH, and the
accumulation of intracellular disulfides, ultimately triggering a
disulfide bond stress response. This stress activates the Rac-WAVE
regulatory complex (WRC)-actin related protein 2/3 (Arp2/3)
signaling pathway, thereby producing abnormal disulfide bonds and
sulfidation in the actin cytoskeleton of muscle cells (32).
The primary characteristic of the tumor
microenvironment is hypoxia, which is closely associated with the
rapid progression and metastasis of cancer (33). Hypoxia inhibits the formation of
disulfide bonds and impairs protein folding in the endoplasmic
reticulum (ER), thereby disrupting ER homeostasis (34). In most types of cancer, hypoxia
causes the cellular energy supply to be derived primarily from
glycolysis and the PPP. A key factor in triggering disulfide death
is the interruption of glucose metabolism during glucose
starvation, as this impairs the PPP pathway and thereby reduces the
generation of NADPH. However, cancer cells have the ability to
exploit the tumor microenvironment to grow rapidly in nutrient-poor
conditions (35). Glucose
metabolism is inextricably associated with lipid and amino acid
metabolism, and the regulation of one aspect may cause alterations
in other metabolic pathways that can lead to disulfide death
(36). In addition to the Warburg
effect, increased glutamine catabolism and de novo fatty
acid synthesis are key hallmarks of cancer. Glutamine catabolism
provides nutrients for the TCA cycle, and supplies nitrogen and
carbon for the synthesis of nucleotides and amino acids (36). Furthermore, rapidly dividing cancer
cells require increased de novo synthesis of fatty acids
from acetyl coenzyme A using the reducing power of NADPH for
membrane biosynthesis (36). When
cysteine availability is limited, some cells utilize the
transsulfuration pathway to biosynthesize cysteine from methionine.
Specifically, cystathionine β-synthase converts methionine-derived
homocysteine to cystathionine, which is then converted to cysteine
by cystathionine γ-lyase (37).
Activation of the transsulfuration pathway inhibits the disulfide
death-dependent pathway by producing cysteine or GSH, acting in
conjunction with system Xc−.
High expression of SLC7A11 is one of the key
conditions for disulfide bond formation; its expression is
influenced by multiple mechanisms, potentially involving
transcriptional regulation by transcription factors and epigenetic
regulators. These mechanisms aim to control the mRNA levels,
protein stability and subcellular localization and transport
protein activity of SLC7A11 (21).
The expression of SLC7A11 can be induced under various stress
conditions, including oxidative stress, amino acid starvation,
metabolic stress and genotoxic stress, likely as an adaptive
response enabling cells to restore redox homeostasis and maintain
survival under stress condition (22) (Fig.
1).
Activating transcription factor 4 (ATF4) and nuclear
factor erythroid 2-related factor 2 (NRF2) are considered to be the
main transcription factors that mediate the stress-induced
transcription of SLC7A11 (38).
ATF4 is a member of the ATF/cAMP response element-binding family
that not only serves as a transcription activator but also as a
repressor protein, capable of responding to various types of
cellular stress including ER stress, oxidative stress, amino acid
depletion and the integrated stress response (38). The association of ATF4 and NRF2 has
been reported to induce the expression of cyclooxygenase-2 in
melanoma, and to regulate amino acid homeostasis via the production
of asparagine and serine in non-small cell lung cancer (39–41).
In addition, ATF4 promotes NRF2 expression through two pathways.
The first pathway involves the depletion of GSH in ChaC
glutathione-specific γ-glutamylcyclotransferase 1-dependent cells,
and the second pathway directly promotes the expression of NFE2
like the transcriptional induction of NFE2L2 expression (42). In-depth analyses reveal that NRF2
supports ATF4-induced cells by increasing cystine uptake via the
glutamate-cystine antiporter xCT. In addition, NRF2 upregulates
genes mediating thioredoxin usage and regeneration, thus balancing
the glutathione decrease (42).
Ultimately, both pathways promote the transcription of SLC7A11 and
the generation of GSH. A study has shown that the expression level
of ATF4 in gastric cancer is significantly increased, and that the
transcriptional activity of ATF4 accelerates the growth and spread
of gastric cancer (43). The
antitumor activity of salbutamycin has been demonstrated to proceed
via induction of the autophagic degradation of protein disulfide
bond isomerase family A member 4 (PDIA4) and attenuation of the
eukaryotic translation initiation factor 2 α kinase 3-ATF4-SLC7A11
signaling pathway (44), suggesting
that the promotion of PDIA4 and upregulation of SLC7A11 promote the
occurrence of disulfide death events. Studies have shown that
PDIA4, an enzyme in the ER, is involved in the formation, breakage
and rearrangement of protein disulfide bonds, thereby mediating
oxidative protein folding (45,46).
ATF4 is known to regulate SLC7A11 via a number of
other methods. For example, in hepatocellular carcinoma,
Yes1-associated transcriptional regulator (YAP)/transcription
coactivator with PDZ-binding motif (TAZ) are key effectors of the
Hippo signaling pathway that play a crucial role in the maintenance
of the intracellular GSH balance (47). These transcription factors
coordinate the ATF4-dependent induction of SLC7A11 expression
through interaction with TEA domain transcription factors (TEADs)
(47). Research by Gao et al
(47) indicated that the knockdown
of YAP and TAZ resulted in a significant reduction in SLC7A11
expression at both the mRNA and protein levels in hepatocellular
carcinoma cells. In the same study, chromatin immunoprecipitation
using specific antibodies against YAP or TAZ followed by
quantitative PCR (qPCR) showed that YAP and TAZ bind to DNA
fragments containing TEAD motifs from the SLC7A11 gene promoter.
These findings demonstrate that SLC7A11 is a direct transcriptional
target of YAP/TAZ. Further mechanistic experiments showed that
YAP/TAZ interacts with ATF4, forming a complex that binds to TEAD
motifs, thereby enhancing the nuclear localization of ATF4, further
increasing transcriptional activity, and preventing proteasomal
degradation and ubiquitination in the cytoplasm. In addition, the
study demonstrated that within the nucleus, ATF4 binds to DNA
fragments in the amino acid responsive element region of the
SLC7A11 promoter, thereby coordinating the expression of SLC7A11
(47). Thus, these findings
indicate that a feedback loop between Hippo signaling and ATF4
activation regulates SLC7A11 expression. ATF4 and NRF2 are also
involved in SLC7A11 transcription in melanoma, where enzyme
activation is predominantly associated with thiol metabolism and
oxidative stress (48,49). As aforementioned, NRF2 supports
ATF4-induced cells by increasing cystine uptake via system
Xc− component SLC7A11 (42). Mechanistically, the high uptake of
cystine by cancer cells, high SLC7A11 expression and reduced
glucose metabolism leads to the toxic accumulation of cystine and
other disulfide bond-containing molecules in the cell, NADPH
depletion and collapse of the redox system. SLC7A11 plays a key
role in the synthesis of GSH and negatively regulates ferroptosis,
yet is upregulated in disulfide death (50). Promoting the upregulation of SLC7A11
through alternative pathways may also promote the disulfide death
process (Fig. 1).
Rac has been demonstrated to activate the WRC to
promote the formation of lamellipodial protrusions from cells
(51,52). Cell migration typically involves the
formation of lamellipodia, which are induced by Rac GTPase, and
peripheral cell protrusions, which are driven by actin networks
formed by the branching activity of the Arp2/3 complex (53,54).
NCK-associated protein 1 (NCKAP1) within the WRC is responsible for
activating the Arp2/3 complex. This activation drives the formation
of branched actin filaments and also promotes the reorganization of
actin and the formation of lamellipodia, resulting in the
construction of a branched cortical actin network beneath the
plasma membrane (55,56). The absence of NCKAP1 has been shown
to attenuate the glucose starvation-induced formation of disulfide
bonds, contraction of F-actin and detachment of F-actin from the
plasma membrane in UMRC6 cells (57). The WAVE-2 catalytic subunit of the
WRC facilitates Arp2/3-mediated actin polymerization and
lamellipodia formation. The branched network of actin filaments
that form the lamellipodia is a critical environment in which
disulfide linkages can form between actin cytoskeletal proteins. In
a study of melanoma, it was established that WAVE pathway typically
involves two key mechanisms, the first being the direct activation
of neuronal-Wiskott-Aldrich syndrome protein (N-WASP)-Arp2/3 by the
Rac-independent activator cell division control protein 42 to form
a plate-like structure, and the second being the WRC-Rac-dependent
formation of lamellipodia (58).
The deletion of either of these two pathways can disrupt F-actin
filament formation and subsequently lead to cellular death
(Fig. 1).
GLUT inhibitors can induce the dimerization of
SLC7A11 in GSH-abundant cells and synergize with immune therapies,
such as programmed cell death protein 1/programmed cell death
ligand 1 antibodies, resulting in enhancing tumor-killing efficacy
(59). MutS homolog 3 (MSH3) has
been demonstrated to induce disulfide bond deposition in renal cell
carcinoma (RCC) cell lines under glucose starvation conditions
(60). MSH3 is a DNA mismatch
repair protein that binds to MSH2 to form the MutSβ heterodimer,
which repairs incorrect base insertions and deletions (61). MSH3 is closely associated with the
tumor microenvironment, immune checkpoint genes and
immunotherapeutic susceptibility (60). In the aforementioned study, a
predictive model of disulfide-related genes was constructed and
MSH3 was identified as a key gene, the expression of which was
negatively correlated with the number of Tregs, which contribute to
the promotion of tumor progression and immune escape (60). The GLUT1 inhibitors BAY-876 and
KL-11743 have been utilized to inhibit glucose uptake (62,63),
consequently reducing NADPH production and increasing the
NADP/NADPH ratio. This inhibition induced the formation of
disulfide bonds in the actin cytoskeleton and induced its collapse
in highly SLC7A11-expressing UMRC6 RCC cells, efficiently leading
to cell death (30). In addition,
these GLUT inhibitors also significantly reduced colorectal tumor
volume in a mouse xenograft model, suggesting that they have
potential as a treatment for colorectal cancer (64). Glucose uptake is significantly
increased in cancer cells (65),
and GLUT inhibition has been shown to be effective in reducing the
proliferation and metabolism of human lung and cervical cancer
cells (66). In addition, a study
demonstrated that GLUT upregulation is associated with
5-fluorouracil resistance, and the inhibition of GLUT significantly
improved the outcome in a mouse model of colorectal cancer
(67). The upregulation of SLC7A11
expression in colon cancer cells, which may be induced by mutations
in tumor suppressor genes such as BRCA1-associated deubiquitinase 1
or kelch-like ECH-associated protein 1, has been shown to promote
the sensitivity of the cells to GLUT inhibitors (68,69).
In a study by Zhao et al (70) in which HCT-116 colorectal cancer
cells were treated with the GLUT1 selective inhibitor BAY-876 and
the GLUT1/GLUT3 inhibitor KL-11743, sensitivity to these inhibitors
was reduced in estrogen-related receptor α knockdown cells compared
with control cells, indicating that estrogen-related receptor α
sensitizes the cells to GLUT inhibition and ultimately to cell
death. Overall, research suggests that the occurrence of
disulfide-related death events is promoted by the inhibition of
GLUT.
Metabolic reprogramming is often regarded as a
notable hallmark of cancer, with disulfide death becoming a focal
point of research. Studies performed using public databases have
detected the expression and mutations of disulfide death-related
genes in cancers including lung adenocarcinoma (LUAD) (71), breast cancer (72) and hepatocellular carcinoma (73), as shown in Table I. Our group went from the study of
iron death to copper death to the current study of disulfide death,
which is an ongoing process of exploration in the field of cancer.
Centered on disufidptosis-associated genes, the studies performed
cluster subtyping and analyzed the differentially expressed genes
associated with disulfide death. Following this, multiple
differentially expressed genes were employed to construct
prognostic risk models, and the association of these genes with
prognosis were analyzed through immune infiltration, immune
checkpoint and drug sensitivity analyses, with the aim of
determining precise individualized treatments according to
different disease types (71–73).
In the study of LUAD, qPCR was utilized to assess
the expression of seven core differentially expressed genes in the
A549 lung cancer cell line and the BEAS-2B normal bronchial
epithelial cell line. Glucose-6-phosphate dehydrogenase (G6PD) had
the highest risk coefficient as a disulfide death factor in LUAD
among these genes, and western blotting experiments confirmed the
elevated expression of G6PD in LUAD cells. Treatment with a G6PD
inhibitor effectively reduced the expression of G6PD in the A549
cells, and the results of Ki67 staining and colony formation assays
showed that the inhibition of G6PD significantly inhibited the
proliferation of these cells (71).
G6PD acts as a catalyst in the metabolism of glucose via the PPP,
and also contributes to maintenance of the biological redox balance
and biosynthesis processes (74).
In tumors and other proliferating or developing cells, the rate of
glucose uptake significantly increases, leading to the production
of lactate, even in the presence of oxygen and fully functioning
mitochondria. This process, known as the Warburg effect, has been
studied extensively, as summarized in a previous review (75). In addition to meeting the
biosynthetic needs of the cell during synthetic metabolic
processes, the PPP also provides an antioxidative defense mechanism
and generates NADPH (76). G6PD has
a rate-limiting role in the PPP and is dynamically modified by
O-linked β-N-acetylglucosamine, via a process termed
O-GlcNAcylation, under hypoxia (77). This glycosylation activates G6PD and
enhances the flux of glucose through the PPP, providing necessary
precursors for the biosynthesis of nucleotides and lipids, and
reducing agents such as NADPH for antioxidative defense (78). It has been shown that the inhibition
of G6PD glycosylation reduces the growth of A549 lung cancer cells
in vitro and tumor growth in vivo, while G6PD
O-GlcNAcylation promotes the proliferation and tumor growth of
these cells. This highlights that the upregulation of G6PD in LUAD
may serve as a new target for LUAD treatment (79). G6PD has also been indicated to
regulate ferroptosis through an NADPH-dependent mechanism, while
NADPH also affects the disulfide bond formation process (71). A reduction in NADPH levels triggers
the formation of disulfide bonds between actin and the cytoskeleton
proteins and the contraction of F-actin, which are key steps
leading to disulfide death (80).
The classical molecular subtypes of breast cancer
include luminal, HER2 and basal subtypes, which have different
prognostic characteristics and drug sensitivities (81). A study identified that
NADH:ubiquinone oxidoreductase core unit S1 (NDUFS1), leucine rich
pentatricopeptide repeat containing (LRPPRC) and SLC7A11 are
differentially expressed and have prognostic value for disulfide
death in breast cancer by screening The Cancer Genome Atlas (TCGA)
data and the GSE86166 dataset (72). NDUFS1 is the initiating enzyme of
the mitochondrial respiratory chain, which is responsible for
electron transfer from NADH to coenzyme Q10 and for the
translocation coupling of protons from the matrix to the
intermembrane space (82). It is
the largest core subunit encoded by nuclear genes, and is part of
the eight iron-sulfur clusters that are responsible for the
oxidation of NADH (83).
LRPPRC is a leucine-rich PPR motif protein that
regulates gene expression at the transcriptional and
post-transcriptional levels by binding to target RNAs. It
contributes to RNA stabilization, regulation, processing, splicing,
translation and editing (84). As a
multifunctional protein, LRPPRC regulates energy metabolism and is
involved the maturation and stability of nuclear-encoded mRNA, as
well as cellular oxidative phosphorylation. It also plays a role in
the regulation of signaling pathways and mitochondrial function
(84). Mitochondria are important
for NADPH synthesis and the regulation of disulfide bond stress;
thus, the functional state of mitochondria may be closely
associated with the onset and progression of disulfide toxicity.
LRPPRC deficiency reduces the stability of mitochondrial mRNAs,
resulting in the loss of polyadenylation of these mRNAs and
abnormalities in mitochondrial translational (85).
NDUFS1 plays a key role in the mitochondrial fusion
pathway. Mitochondrial fusion is an endogenous protective process
for mitochondrial quality control and the maintenance of
mitochondrial homeostasis and function (86,87).
NDUFS1 is the largest core subunit in respiratory chain complex I,
and the reduced expression of this subunit leads to reduced
homeostasis of respiratory chain complex I and mitochondrial
dysfunction (88). It has been
demonstrated that significantly reduced levels of LRPPRC and NDUFS1
in ulcerative colitis may disrupt the homeostasis of the relevant
subunits from respiratory chain complexes IV and I, causing the
respiratory electron transport chain to become dysfunctional and
ultimately inhibiting NADPH production and the reduction of
disulfide bonds, thereby promoting disulfide death (85).
The LRPPRC gene has been found to be upregulated in
a variety of human malignancies, and this upregulation is closely
associated with a poor prognosis (89,90).
LRPPRC has also been demonstrated to serve as an independent
prognostic factor in hepatocellular carcinoma (91). The downregulation of LRPPRC induces
apoptosis in cancer cells and reduces their invasive ability,
suggesting that LRPPRC may be a promising biomarker and potential
molecular target for cancer treatment (84). It has been shown that LRPPRC and
NDUFS1 are resistant to disulfide death and that their inactivation
synergizes with glucose starvation and regulation of the energy
metabolism of NADPH to induce cell death (30). LRPPRC and NDUFS1 have both been
observed to be significantly downregulated in breast cancer, and it
has been suggested that they may have a notable synergistic role in
the induction of disulfide death in breast cancer (92).
Studies of genes associated with disulfide death in
gastric cancer have identified that apolipoprotein D (APOD) and
NCKAP1 are potential therapeutic targets for this disease (93,94).
The NCKAP1 protein is a component of the WAVE complex, along with
Abl interactor 1 and 2, BRICK1, cytoplasmic FMR1 interacting
proteins 1 and 2, and WASP family members 1 and 2. NCKAP1 deletion
has been shown to affect actin nucleation in fibroblast laminar
pseudopod formation by affecting the spreading and focal adhesion
dynamics of cells, suggesting a role for NCKAP1 in cell migration
(95). In addition, NCKAP1 is
involved in the formation of the actin network, and has been shown
to be a key gene in disulfide death, which regulates a variety of
processes such as apoptosis, migration and invasion, and serves a
crucial role in pathogenesis (94).
APOD is one of the 22 members of the human APO
family, encoded by a gene located on human chromosome 3 (96). APOD is involved in the inflammatory
response and lipid metabolism, and plays an important role in a
number of cancers (97). For
instance, high APOD expression has been shown to be associated with
shorter overall survival and recurrence-free survival in patients
with non-small cell lung cancer (98). Studies have also shown that elevated
expression of APOD in cancer of the breast and central nervous
system is closely associated with highly differentiated,
non-invasive and non-metastatic cancers, and that APOD reduces the
osteopontin-mediated adhesion, invasiveness and proliferation of
Rama37 breast cancer cells in vitro (99–101).
However, APOD has been suggested to be an independent predictor of
metastasis-free survival and overall survival in patients with
invasive breast cancer, with increased APOD expression being
associated with a poor prognosis (102). In gastric cancer, bioinformatics
analyses have been used to indicate that APOD may be included as a
component of genetic risk models for disulfide death, and is
associated with an increased tumor mutational load and immune cell
infiltration (103–105). In addition, positive correlations
have been identified for APOD with macrophage infiltration and the
Wnt signaling pathway in stomach cancer as evaluated by the
analysis of CD68 and Wnt family member 2B, respectively (106). In addition to its involvement in
the immune response, APOD is also a marker of hypoxia, which has a
fundamental role in the immune microenvironment of tumors (107,108). Bioinformatics analysis has also
indicated that APOD expression is positively associated with
cancer-associated fibroblasts, endothelial cells and hematopoietic
stem cells in most cancers, but negatively associated with
myeloid-derived suppressor cells. It is hypothesized that APOD may
affect formation of the actin cytoskeleton, subsequently
influencing the collapse of the cytoskeleton during disulfide death
(109).
A study of glioblastoma identified three genes that
were significantly associated with disulfide death in this type of
cancer, namely LRPPRC, ribophorin I (RPN1) and glycogen synthase 1
(GYS1). These three genes were demonstrated to be located in the
mitochondria, cytosol and microtubules, respectively, of U-251MG
glioblastoma cells (110). TCGA
glioma dataset analysis revealed that LRPPRC is downregulated in
glioma, whereas RPN1 and GYS1 are upregulated. In addition, a
Kaplan-Meier survival analysis showed that high LRPPRC expression
was positively associated with a good prognosis while the high
expression of RPN1 and GYS1 was positively associated with a poor
prognosis in patients with glioma (110). RPN1 contributes to protein
synthesis and glycoprotein formation in the ER (111,112). It acts as a receptor and regulator
of protein translocation in the ER, helping to direct and anchor
nascent proteins to the ER membrane, thereby facilitating their
proper folding and glycan modification (113). In a study aiming to evaluate RPN1
as a pan-cancer marker, wild-type (WT) and RPN1 knockout (KO)
MDA-MB-231 breast cancer and A549 lung cancer cells cultured in
glucose-containing or -deficient medium were analyzed by flow
cytometry to evaluate cell death. In addition, F-actin in the cells
was immunofluorescently labeled to visualize changes in the actin
cytoskeleton under different glucose conditions. The results
indicated that cell death and cytoskeletal breakdown occurred under
glucose deprivation. This cytoskeletal breakdown was inhibited in
RPN1 KO cells, suggesting that RPN1 mediates cell death through the
degradation of cytoskeletal proteins. In addition, protein
extraction and western blotting analyses performed using
non-reducing and reducing methods indicated that when these cells
are deprived of glucose, RPN1 KO contributes to the stabilization
of cytoskeletal proteins. This suggests that RPN1 deletion promotes
cytoskeletal integrity. These findings suggest that RPN1 plays a
role in the mediation of disulfide death in breast and lung cancer,
possibly through a mechanism involving the induction of
cytoskeletal protein degradation (114). It is possible that this pathway
may also be associated with disulfide death in glioblastoma. GYS1
is the most important rate-limiting enzyme in the last step of
glycogen synthesis (115). A lack
of GYS1 has been shown to cause type 0 myoglycogen storage disease
that may lead to death (116,117). GYS1 is rapidly induced under
hypoxic conditions and is positively associated with the
accumulation of glycogen in glioblastoma (118). A hypoxic environment is favorable
for the growth, development and infiltration of tumors, and the
upregulation of hypoxia-induced factors has been verified as a
basic pathway in most tumors (119). Notably, glycogen metabolism is a
metabolic pathway that is upregulated by hypoxia (120). Carbon and sulfur from glycogen
breakdown can enter the PPP and contribute to the production of
NADPH, which serves as a reducing agent to scavenge ROS and
maintain immune cell survival (121). GSH and thioredoxin are two major
cellular antioxidants that scavenge intracellular ROS and rely on
NADPH, produced via the PPP, as an electron donor. Therefore, it
may be hypothesized that GYS1 regulates disulfide death by
influencing the production of NADPH by the PPP.
In summary, the current literature supports that
disulfide death occurs in tumors, and G6PD, LRPPRC, SLC7A11, APOD,
NCKAP1, RPN1 and GYS1 have been demonstrated to serve as predictive
target genes in different tumor types. Among these, SLC7A11 serves
as a key link to disulfide death and is also a promising target for
research in a variety of cancer types.
In summary, cell death is known to progress via
various processes, including apoptosis and necrosis, as well as
ferroptosis, copper-induced death and disulfide death. While
cellular respiration in the form of oxidative phosphorylation is
not essential for all cells, tumor cells commonly rely on the
Warburg effect, an alternative pathway. In disulfide death,
blocking the PPP prevents the supply of NADPH, resulting in the
abnormal accumulation of disulfide bonds, which ultimately leads to
cell death. Tumor cells require the glycolysis pathway to supply
energy for survival, which can be inhibited by GLUT inhibitors to
impede tumor growth. Among the solid tumors studied to date, most
of the genes on disulfide death are associated with mitochondrial
regulation, and respiratory chain 1 occupies an important role in
mitochondria. SLC7A11 is a key channel protein associated with
disulfide death, and also serves as a therapeutic target for tumor
cells. The expression of SLC7A11 has been found to be downregulated
in ferroptosis and upregulated in disulfide death, suggesting a
certain association between the two mechanisms. Potential means of
regulating the relationship between these mechanisms and
identifying a suitable target in tumor cells merit exploration in
future studies.
Not applicable.
Funding: No funding was received.
Not applicable.
XL conceptualized and designed the article,
conducted the literature search and chart preparation and wrote the
manuscript. DZ reviewed the manuscript and supervised the study.
Data authentication is not applicable. Both authors read and
approved the final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the Nomenclature Committee on Cell Death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu X, Zhuang L and Gan B: Disulfidptosis:
Disulfide stress-induced cell death. Trends Cell Biol. 34:327–337.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ravilious GE and Jez JM: Structural
biology of plant sulfur metabolism: from assimilation to
biosynthesis. Nat Prod Rep. 29:1138–1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wanders D, Hobson K and Ji X: Methionine
restriction and cancer biology. Nutrients. 12:6842020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaplowitz N, Aw TY and Ookhtens M: The
regulation of hepatic glutathione. Annu Rev Pharmacol Toxicol.
25:715–744. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu SC: Glutathione synthesis. Biochim
Biophys Acta. 1830:3143–3153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Diotallevi M, Checconi P, Palamara AT,
Celestino I, Coppo L, Holmgren A, Abbas K, Peyrot F, Mengozzi M and
Ghezzi P: Glutathione Fine-Tunes the Innate Immune Response toward
Antiviral Pathways in a Macrophage Cell Line Independently of Its
Antioxidant Properties. Front Immunol. 8:12392017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen YJ, Lu CT, Lee TY and Chen YJ: dbGSH:
A database of S-glutathionylation. Bioinformatics. 30:2386–2388.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lermant A and Murdoch CE: Cysteine
Glutathionylation Acts as a Redox Switch in Endothelial Cells.
Antioxidants (Basel). 8:3152019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kurniawan H, Franchina DG, Guerra L,
Bonetti L, Baguet LS, Grusdat M, Schlicker L, Hunewald O, Dostert
C, Merz MP, et al: Glutathione Restricts Serine Metabolism to
Preserve Regulatory T Cell Function. Cell Metab. 31:920–936.e7.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gai X, Liu Y, Lan X, Chen L, Yuan T, Xu J,
Li Y, Zheng Y, Yan Y, Yang L, et al: Oncogenic KRAS induces
arginine auxotrophy and confers a therapeutic vulnerability to
SLC7A1 inhibition in non-small cell lung cancer. Cancer Res.
84:1963–1977. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
You S, Zhu X, Yang Y, Du X, Song K, Zheng
Q, Zeng P and Yao Q: SLC7A1 overexpression is involved in energy
metabolism reprogramming to induce tumor progression in epithelial
ovarian cancer and is associated with immune-infiltrating cells. J
Oncol. 2022:58648262022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shen C and Wang Y: Ferroptosis biomarkers
for predicting prognosis and immunotherapy efficacy in
adrenocortical carcinoma. Arch Med Res. 54:45–55. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lei S, Chen C, Han F, Deng J, Huang D,
Qian L, Zhu M, Ma X, Lai M, Xu E and Zhang H: AMER1 deficiency
promotes the distant metastasis of colorectal cancer by inhibiting
SLC7A11- and FTL-mediated ferroptosis. Cell Rep. 42:1131102023.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ouyang S, Li H, Lou L, Huang Q, Zhang Z,
Mo J, Li M, Lu J, Zhu K, Chu Y, et al: Inhibition of
STAT3-ferroptosis negative regulatory axis suppresses tumor growth
and alleviates chemoresistance in gastric cancer. Redox Biol.
52:1023172022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Zhou Y, Xie S, Wang J, Li Z, Chen
L, Mao M, Chen C, Huang A, Chen Y, et al: Metformin induces
Ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J
Exp Clin Cancer Res. 40:2062021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Shen N, Wang Z, Yu L, Yang S, Wang
Y, Liu Y, Han G and Zhang Q: TRIM3 facilitates ferroptosis in
non-small cell lung cancer through promoting SLC7A11/xCT K11-linked
ubiquitination and degradation. Cell Death Differ. 31:53–64. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koppula P, Zhang Y, Zhuang L and Gan B:
Amino acid transporter SLC7A11/xCT at the crossroads of regulating
redox homeostasis and nutrient dependency of cancer. Cancer Commun
(Lond). 38:122018.PubMed/NCBI
|
|
23
|
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M,
Ding HF, Zhang J, Wang H, Chen X and Yan C: ATF3 promotes
erastin-induced ferroptosis by suppressing system Xc. Cell Death
Differ. 27:662–675. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao H, Jedrychowski MP, Schweppe DK,
Huttlin EL, Yu Q, Heppner DE, Li J, Long J, Mills EL, Szpyt J, et
al: A Quantitative Tissue-Specific Landscape of Protein Redox
Regulation during Aging. Cell. 180:968–983.e4. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Musaogullari A and Chai YC: Redox
Regulation by Protein S-Glutathionylation: From Molecular
Mechanisms to Implications in Health and Disease. Int J Mol Sci.
21:81132020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Machesky LM: Deadly actin collapse by
disulfidptosis. Nat Cell Biol. 25:375–376. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan Y, Teng H, Hang Q, Kondiparthi L, Lei
G, Horbath A, Liu X, Mao C, Wu S, Zhuang L, et al: SLC7A11
expression level dictates differential responses to oxidative
stress in cancer cells. Nat Commun. 14:36732023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia N, Guo X, Guo Q, Gupta N, Ji N, Shen
B, Xiao L and Feng Y: Metabolic flexibilities and vulnerabilities
in the pentose phosphate pathway of the zoonotic pathogen
Toxoplasma gondii. PLoS Pathog. 18:e10108642022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
El Mjiyad N, Caro-Maldonado A,
Ramírez-Peinado S and Muñoz-Pinedo C: Sugar-free approaches to
cancer cell killing. Oncogene. 30:253–264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu X, Nie L, Zhang Y, Yan Y, Wang C,
Colic M, Olszewski K, Horbath A, Chen X, Lei G, et al: Actin
cytoskeleton vulnerability to disulfide stress mediates
disulfidptosis. Nat Cell Biol. 25:404–414. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen J, Ma B, Yang Y, Wang B, Hao J and
Zhou X: Disulfidptosis decoded: A journey through cell death
mysteries, regulatory networks, disease paradigms and future
directions. Biomark Res. 12:452024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fregoso FE, van Eeuwen T, Simanov G,
Rebowski G, Boczkowska M, Zimmet A, Gautreau AM and Dominguez R:
Molecular mechanism of Arp2/3 complex inhibition by Arpin. Nat
Commun. 13:6282022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wicks EE and Semenza GL: Hypoxia-inducible
factors: Cancer progression and clinical translation. J Clin
Invest. 132:e1598392022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rozpedek W, Pytel D, Mucha B, Leszczynska
H, Diehl JA and Majsterek I: The Role of the PERK/eIF2α/ATF4/CHOP
signaling pathway in tumor progression during endoplasmic reticulum
stress. Curr Mol Med. 16:533–544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Y, Lin C, Liu Z, Sun Y, Chen M, Guo
Y, Liu W, Zhang C, Chen W, Sun J, et al: Cancer cells co-opt
nociceptive nerves to thrive in nutrient-poor environments and upon
nutrient-starvation therapies. Cell Metab. 34:1999–2017.e10. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang
W, Wang XY and Fang X: Fatty acid oxidation: An emerging facet of
metabolic transformation in cancer. Cancer Lett. 435:92–100. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang L, Cai H, Hu Y, Liu F, Huang S, Zhou
Y, Yu J, Xu J and Wu F: A pharmacological probe identifies
cystathionine β-synthase as a new negative regulator for
ferroptosis. Cell Death Dis. 9:10052018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tang H, Kang R, Liu J and Tang D: ATF4 in
cellular stress, ferroptosis, and cancer. Arch Toxicol.
98:1025–1041. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jessen C, Kreß JKC, Baluapuri A, Hufnagel
A, Schmitz W, Kneitz S, Roth S, Marquardt A, Appenzeller S, Ade CP,
et al: The transcription factor NRF2 enhances melanoma malignancy
by blocking differentiation and inducing COX2 expression. Oncogene.
39:6841–6855. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
DeNicola GM, Chen PH, Mullarky E, Sudderth
JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al: NRF2
regulates serine biosynthesis in non-small cell lung cancer. Nat
Genet. 47:1475–1481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gwinn DM, Lee AG, Briones-Martin-Del-Campo
M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D and
Sweet-Cordero EA: Oncogenic KRAS Regulates Amino Acid Homeostasis
and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to
L-Asparaginase. Cancer Cell. 33:91–107.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kreß JKC, Jessen C, Hufnagel A, Schmitz W,
Xavier da Silva TN, Ferreira Dos Santos A, Mosteo L, Goding CR,
Friedmann Angeli JP and Meierjohann S: The integrated stress
response effector ATF4 is an obligatory metabolic activator of
NRF2. Cell Rep. 42:1127242023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Y, Ali M, Zhang Q, Sun Q, Ren J, Wang
W, Tang D and Wang D: ATF4 transcriptionally activates SHH to
promote proliferation, invasion, and migration of gastric cancer
cells. Cancers (Basel). 15:14292023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kang L, Wang D, Shen T, Liu X, Dai B, Zhou
D, Shen H, Gong J, Li G, Hu Y, et al: PDIA4 confers resistance to
ferroptosis via induction of ATF4/SLC7A11 in renal cell carcinoma.
Cell Death Dis. 14:1932023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Z, Zhang H and Cheng Q: PDIA4: The
basic characteristics, functions and its potential connection with
cancer. Biomed Pharmacother. 122:1096882020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee CH, Chiang CF, Lin FH, Kuo FC, Su SC,
Huang CL, Li PF, Liu JS, Lu CH, Hsieh CH, et al: PDIA4, a new
endoplasmic reticulum stress protein, modulates insulin resistance
and inflammation in skeletal muscle. Front Endocrinol (Lausanne).
13:10538822022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao R, Kalathur RKR, Coto-Llerena M, Ercan
C, Buechel D, Shuang S, Piscuoglio S, Dill MT, Camargo FD,
Christofori G and Tang F: YAP/TAZ and ATF4 drive resistance to
Sorafenib in hepatocellular carcinoma by preventing ferroptosis.
EMBO Mol Med. 13:e143512021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen D, Fan Z, Rauh M, Buchfelder M,
Eyupoglu IY and Savaskan N: ATF4 promotes angiogenesis and neuronal
cell death and confers ferroptosis in a xCT-dependent manner.
Oncogene. 36:5593–5608. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sasaki H, Sato H, Kuriyama-Matsumura K,
Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M and Bannai
S: Electrophile response element-mediated induction of the
cystine/glutamate exchange transporter gene expression. J Biol
Chem. 277:44765–44771. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Miki H, Suetsugu S and Takenawa T: WAVE, a
novel WASP-family protein involved in actin reorganization induced
by Rac. EMBO J. 17:6932–6941. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Steffen A, Rottner K, Ehinger J, Innocenti
M, Scita G, Wehland J and Stradal TE: Sra-1 and Nap1 link Rac to
actin assembly driving lamellipodia formation. EMBO J. 23:749–759.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kage F, Döring H, Mietkowska M, Schaks M,
Grüner F, Stahnke S, Steffen A, Müsken M, Stradal TEB and Rottner
K: Lamellipodia-like actin networks in cells lacking WAVE
regulatory complex. J Cell Sci. 135:jcs2603642022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rotty JD, Wu C and Bear JE: New insights
into the regulation and cellular functions of the ARP2/3 complex.
Nat Rev Mol Cell Biol. 14:7–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Alekhina O, Burstein E and Billadeau DD:
Cellular functions of WASP family proteins at a glance. J Cell Sci.
130:2235–2241. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ibarra N, Pollitt A and Insall RH:
Regulation of actin assembly by SCAR/WAVE proteins. Biochem Soc
Trans. 33:1243–1246. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhu A, Zong Y, Wei S, Li Y, Fan Y, Liu S
and Gao X: Pan-cancer Analysis of the Disulfidptosis-related Gene
NCKAP1 and Its Prognostic Value for Lung Adenocarcinoma. J Cancer.
14:3351–3367. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tang H, Li A, Bi J, Veltman DM, Zech T,
Spence HJ, Yu X, Timpson P, Insall RH, Frame MC and Machesky LM:
Loss of Scar/WAVE complex promotes N-WASP- and FAK-dependent
invasion. Curr Biol. 23:107–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Koppula P, Olszewski K, Zhang Y,
Kondiparthi L, Liu X, Lei G, Das M, Fang B, Poyurovsky MV and Gan
B: KEAP1 deficiency drives glucose dependency and sensitizes lung
cancer cells and tumors to GLUT inhibition. iScience.
24:1026492021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu K, Zhang Y, Yan Z, Wang Y, Li Y, Qiu Q,
Du Y, Chen Z and Liu X: Identification of disulfidptosis related
subtypes, characterization of tumor microenvironment infiltration,
and development of DRG prognostic prediction model in RCC, in which
MSH3 is a key gene during disulfidptosis. Front Immunol.
14:12052502023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sakellariou D, Bak ST, Isik E, Barroso SI,
Porro A, Aguilera A, Bartek J, Janscak P and Peña-Diaz J: MutSβ
regulates G4-associated telomeric R-loops to maintain telomere
integrity in ALT cancer cells. Cell Rep. 39:1106022022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Siebeneicher H, Cleve A, Rehwinkel H,
Neuhaus R, Heisler I, Müller T, Bauser M and Buchmann B:
Identification and optimization of the first highly selective GLUT1
Inhibitor BAY-876. ChemMedChem. 11:2261–2271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Olszewski K, Barsotti A, Feng XJ,
Momcilovic M, Liu KG, Kim JI, Morris K, Lamarque C, Gaffney J, Yu
X, et al: Inhibition of glucose transport synergizes with chemical
or genetic disruption of mitochondrial metabolism and suppresses
TCA cycle-deficient tumors. Cell Chem Biol. 29:423–435.e10. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li Y, Tang M, Dang W, Zhu S and Wang Y:
Identification of disulfidptosis-related subtypes, characterization
of tumor microenvironment infiltration, and development of a
prognosis model in colorectal cancer. J Cancer Res Clin Oncol.
149:13995–14014. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shriwas P, Roberts D, Li Y, Wang L, Qian
Y, Bergmeier S, Hines J, Adhicary S, Nielsen C and Chen X: A
small-molecule pan-class I glucose transporter inhibitor reduces
cancer cell proliferation in vitro and tumor growth in vivo by
targeting glucose-based metabolism. Cancer Metab. 9:142021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chang CK, Chiu PF, Yang HY, Juang YP, Lai
YH, Lin TS, Hsu LC, Yu LC and Liang PH: Targeting colorectal cancer
with conjugates of a glucose transporter inhibitor and
5-fluorouracil. J Med Chem. 64:4450–4461. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang Y, Koppula P and Gan B: Regulation
of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell
Cycle. 18:773–783. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang Y, Shi J, Liu X, Feng L, Gong Z,
Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al: BAP1 links
metabolic regulation of ferroptosis to tumour suppression. Nat Cell
Biol. 20:1181–1192. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhao M, Xu C and Zhu H: Efficacy of
glucose transporter inhibitors for the treatment of
ERRα-overexpressed colorectal cancer. Acta Biochim Pol. 69:567–572.
2022.PubMed/NCBI
|
|
71
|
Qi C, Ma J, Sun J, Wu X and Ding J: The
role of molecular subtypes and immune infiltration characteristics
based on disulfidptosis-associated genes in lung adenocarcinoma.
Aging (Albany NY). 15:5075–5095. 2023.PubMed/NCBI
|
|
72
|
Xia Q, Yan Q, Wang Z, Huang Q, Zheng X,
Shen J, Du L, Li H and Duan S: Disulfidptosis-associated lncRNAs
predict breast cancer subtypes. Sci Rep. 13:162682023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhao J, Luo Z, Fu R, Zhou J, Chen S, Wang
J, Chen D and Xie X: Disulfidptosis-related signatures for
prognostic and immunotherapy reactivity evaluation in
hepatocellular carcinoma. Eur J Med Res. 28:5712023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL,
Stern A and Chiu DT: The Redox Role of G6PD in Cell Growth, Cell
Death, and Cancer. Cells. 8:10552019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liberti MV and Locasale JW: The Warburg
Effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hart GW, Housley MP and Slawson C: Cycling
of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins.
Nature. 446:1017–1022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chen L, Zhang Z, Hoshino A, Zheng HD,
Morley M, Arany Z and Rabinowitz JD: NADPH production by the
oxidative pentose-phosphate pathway supports folate metabolism. Nat
Metab. 1:404–415. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Rao X, Duan X, Mao W, Li X, Li Z, Li Q,
Zheng Z, Xu H, Chen M, Wang PG, et al: O-GlcNAcylation of G6PD
promotes the pentose phosphate pathway and tumor growth. Nat
Commun. 6:84682015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Liu X, Zhang Y, Zhuang L, Olszewski K and
Gan B: NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated
cystine uptake as a double-edged sword in cellular redox
regulation. Genes Dis. 8:731–745. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Holm J, Eriksson L, Ploner A, Eriksson M,
Rantalainen M, Li J, Hall P and Czene K: Assessment of breast
cancer risk factors reveals subtype heterogeneity. Cancer Res.
77:3708–3717. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ni Y, Hagras MA, Konstantopoulou V, Mayr
JA, Stuchebrukhov AA and Meierhofer D: Mutations in ndufs1 cause
metabolic reprogramming and disruption of the electron transfer.
Cells. 8:11492019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhu J, Vinothkumar KR and Hirst J:
Structure of mammalian respiratory complex I. Nature. 536:354–358.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cui J, Wang L, Ren X, Zhang Y and Zhang H:
LRPPRC: A multifunctional protein involved in energy metabolism and
human disease. Front Physiol. 10:5952019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Song H, Zhang F, Bai X, Liang H, Niu J and
Miao Y: Comprehensive analysis of disulfidptosis-related genes
reveals the effect of disulfidptosis in ulcerative colitis. Sci
Rep. 14:157052024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wan S, Maitiabula G, Wang P, Zhang Y, Gao
X, Zhang L, Gao T and Wang X: Down regulation of NDUFS1 is involved
in the progression of parenteral-nutrition-associated liver disease
by increasing Oxidative stress. J Nutr Biochem. 112:1092212023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yang Y, Yuan H, Zhao L, Guo S, Hu S, Tian
M, Nie Y, Yu J, Zhou C, Niu J, et al: Targeting the
miR-34a/LRPPRC/MDR1 axis collapse the chemoresistance in P53
inactive colorectal cancer. Cell Death Differ. 29:2177–2189. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wei WS, Wang N, Deng MH, Dong P, Liu JY,
Xiang Z, Li XD, Li ZY, Liu ZH, Peng YL, et al: LRPPRC regulates
redox homeostasis via the circANKHD1/FOXM1 axis to enhance bladder
urothelial carcinoma tumorigenesis. Redox Biol. 48:1022012021.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yang L, Zhang W and Yan Y: Identification
and characterization of a novel molecular classification based on
disulfidptosis-related genes to predict prognosis and immunotherapy
efficacy in hepatocellular carcinoma. Aging (Albany NY).
15:6135–6151. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zeng M, Wu B, Wei W, Jiang Z, Li P, Quan Y
and Hu X: Disulfiram: A novel repurposed drug for cancer therapy.
Chin Med J (Engl). 137:1389–1398. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li Q and Yin LK: Comprehensive analysis of
disulfidptosis related genes and prognosis of gastric cancer. World
J Clin Oncol. 14:373–399. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Yan J, Fang Z, Shi M, Tu C, Zhang S, Jiang
C, Li Q and Shao Y: Clinical Significance of Disulfidptosis-related
Genes and Functional Analysis in Gastric Cancer. J Cancer.
15:1053–1066. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Whitelaw JA, Swaminathan K, Kage F and
Machesky LM: The WAVE Regulatory Complex Is Required to Balance
Protrusion and Adhesion in Migration. Cells. 9:16352020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Drayna DT, McLean JW, Wion KL, Trent JM,
Drabkin HA and Lawn RM: Human apolipoprotein D gene: Gene sequence,
chromosome localization, and homology to the alpha 2u-globulin
superfamily. DNA. 6:199–204. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ren L, Yi J, Li W, Zheng X, Liu J, Wang J
and Du G: Apolipoproteins and cancer. Cancer Med. 8:7032–7043.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Cury SS, de Moraes D, Freire PP, de
Oliveira G, Marques DVP, Fernandez GJ, Dal-Pai-Silva M, Hasimoto
ÉN, Dos Reis PP, Rogatto SR and Carvalho RF: Tumor Transcriptome
Reveals High Expression of IL-8 in Non-Small Cell Lung Cancer
Patients with Low Pectoralis Muscle Area and Reduced Survival.
Cancers (Basel). 11:12512019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hunter S, Young A, Olson J, Brat DJ,
Bowers G, Wilcox JN, Jaye D, Mendrinos S and Neish A: Differential
expression between pilocytic and anaplastic astrocytomas:
Identification of apolipoprotein D as a marker for low-grade,
non-infiltrating primary CNS neoplasms. J Neuropathol Exp Neurol.
61:275–281. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Porter D, Lahti-Domenici J, Keshaviah A,
Bae YK, Argani P, Marks J, Richardson A, Cooper A, Strausberg R,
Riggins GJ, et al: Molecular markers in ductal carcinoma in situ of
the breast. Mol Cancer Res. 1:362–375. 2003.PubMed/NCBI
|
|
101
|
Jin D, El-Tanani M and Campbell FC:
Identification of apolipoprotein D as a novel inhibitor of
osteopontin-induced neoplastic transformation. Int J Oncol.
29:1591–1599. 2006.PubMed/NCBI
|
|
102
|
Jankovic-Karasoulos T, Bianco-Miotto T,
Butler MS, Butler LM, McNeil CM, O'Toole SA, Millar EKA, Sakko AJ,
Ruiz AI, Birrell SN, et al: Elevated levels of tumour
apolipoprotein D independently predict poor outcome in breast
cancer patients. Histopathology. 76:976–987. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yu J, Zhang Q, Wang M, Liang S, Huang H,
Xie L, Cui C and Yu J: Comprehensive analysis of tumor mutation
burden and immune microenvironment in gastric cancer. Biosci Rep.
41:BSR202033362021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Guo X, Liang X, Wang Y, Cheng A, Zhang H,
Qin C and Wang Z: Significance of Tumor Mutation Burden Combined
With Immune Infiltrates in the Progression and Prognosis of
Advanced Gastric Cancer. Front Genet. 12:6426082021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Huo J, Wu L and Zang Y: Construction and
Validation of a Universal Applicable Prognostic Signature for
Gastric Cancer Based on Seven Immune-Related Gene Correlated With
Tumor Associated Macrophages. Front Oncol. 11:6353242021.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Khan M, Lin J, Wang B, Chen C, Huang Z,
Tian Y, Yuan Y and Bu J: A novel necroptosis-related gene index for
predicting prognosis and a cold tumor immune microenvironment in
stomach adenocarcinoma. Front Immunol. 13:9681652022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Beemelmanns A, Zanuzzo FS, Xue X,
Sandrelli RM, Rise ML and Gamperl AK: The transcriptomic responses
of Atlantic salmon (Salmo salar) to high temperature stress alone,
and in combination with moderate hypoxia. BMC Genomics. 22:2612021.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Shida M, Kitajima Y, Nakamura J,
Yanagihara K, Baba K, Wakiyama K and Noshiro H: Impaired mitophagy
activates mtROS/HIF-1α interplay and increases cancer
aggressiveness in gastric cancer cells under hypoxia. Int J Oncol.
48:1379–1390. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang Z, Chen H, Sun L, Wang X, Xu Y, Tian
S and Liu X: Uncovering the potential of APOD as a biomarker in
gastric cancer: A retrospective and multi-center study. Comput
Struct Biotechnol J. 23:1051–1064. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang X, Yang J, Yang F and Mu K: The
disulfidptosis-related signature predicts prognosis and immune
features in glioma patients. Sci Rep. 13:179882023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ding J, Xu J, Deng Q, Ma W, Zhang R, He X,
Liu S and Zhang L: Knockdown of Oligosaccharyltransferase Subunit
Ribophorin 1 Induces Endoplasmic-Reticulum-Stress-Dependent Cell
Apoptosis in Breast Cancer. Front Oncol. 11:7226242021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Qin SY, Hu D, Matsumoto K, Takeda K,
Matsumoto N, Yamaguchi Y and Yamamoto K: Malectin forms a complex
with ribophorin I for enhanced association with misfolded
glycoproteins. J Biol Chem. 287:38080–38089. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wilson CM, Roebuck Q and High S:
Ribophorin I regulates substrate delivery to the
oligosaccharyltransferase core. Proc Natl Acad Sci USA.
105:9534–9539. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wang X, Zhu HQ, Lin SM, Xia BY and Xu B:
RPN1: A pan-cancer biomarker and disulfidptosis regulator. Transl
Cancer Res. 13:2518–2534. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
López-Ramos JC, Duran J, Gruart A,
Guinovart JJ and Delgado-García JM: Role of brain glycogen in the
response to hypoxia and in susceptibility to epilepsy. Front Cell
Neurosci. 9:4312015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Cameron JM, Levandovskiy V, MacKay N,
Utgikar R, Ackerley C, Chiasson D, Halliday W, Raiman J and
Robinson BH: Identification of a novel mutation in GYS1
(muscle-specific glycogen synthase) resulting in sudden cardiac
death, that is diagnosable from skin fibroblasts. Mol Genet Metab.
98:378–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Pederson BA, Chen H, Schroeder JM, Shou W,
DePaoli-Roach AA and Roach PJ: Abnormal cardiac development in the
absence of heart glycogen. Mol Cell Biol. 24:7179–7187. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Favaro E, Bensaad K, Chong MG, Tennant DA,
Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Günther UL, et al:
Glucose utilization via glycogen phosphorylase sustains
proliferation and prevents premature senescence in cancer cells.
Cell Metab. 16:751–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wigerup C, Påhlman S and Bexell D:
Therapeutic targeting of hypoxia and hypoxia-inducible factors in
cancer. Pharmacol Ther. 164:152–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
de Heer EC, Zois CE, Bridges E, van der
Vegt B, Sheldon H, Veldman WA, Zwager MC, van der Sluis T, Haider
S, Morita T, et al: Glycogen synthase 1 targeting reveals a
metabolic vulnerability in triple-negative breast cancer. J Exp
Clin Cancer Res. 42:1432023. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ma R, Ji T, Zhang H, Dong W, Chen X, Xu P,
Chen D, Liang X, Yin X, Liu Y, et al: A Pck1-directed glycogen
metabolic program regulates formation and maintenance of memory
CD8(+) T cells. Nat Cell Biol. 20:21–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Chen H, Yang W, Li Y, Ma L and Ji Z:
Leveraging a disulfidptosis-based signature to improve the survival
and drug sensitivity of bladder cancer patients. Front Immunol.
14:11988782023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhang D, Zhang X, Liu Z, Han T, Zhao K, Xu
X, Zhang X, Ren X and Qin C: An integrative multi-omics analysis
based on disulfidptosis-related prognostic signature and distinct
subtypes of clear cell renal cell carcinoma. Front Oncol.
13:12070682023. View Article : Google Scholar : PubMed/NCBI
|