Introduction
Gliomas are the most prevalent malignant tumors of
the central nervous system (CNS), particularly the brain. The
histological appearance of these cells resembles that of normal
glial cells whose origin remains uncertain (1). Traditionally, gliomas were classified
based on tumor cellular structure with tumor histology and location
guiding treatment strategies and predicting clinical outcomes
(2). The fifth edition of the World
Health Organization's (WHO) CNS classification in 2021 (3) implemented significant updates based on
the molecular characteristics of gliomas. As such, histological
features are combined with molecular and genetic variations that,
together, define glioma molecular pathology and aid in patient
stratification, as well as targeted therapies (4).
Over time, the classification of gliomas has
evolved, employing tools such as immunohistochemistry and electron
microscopy for lineage identification. The histologically based
classification has numerous limitations, one of which is the
‘interobserver variability’ or the concordance among multiple
independent observers in clinical environments (5). This results in a decreased accuracy in
patient prognoses under the conventional classification system.
Diagnostic differences remained unaddressed, even when considering
variables such as patient age, response to treatment and surgical
resection. Consequently, a comprehensive understanding of gliomas
at the molecular level became necessary. Such an understanding is
vital for improving diagnostic criteria and identifying prognostic
biomarkers, which are key to the development of effective targeted
therapeutics (6).
The recent incorporation of molecular surrogates has
enabled bioinformatics tools to offer valuable prognostic,
diagnostic and predictive assistance in classifying gliomas. In
adult patients, the majority of gliomas are of the diffuse type.
Diffuse gliomas can be classified as astrocytic, oligodendroglial
or a combination of both cell types. The categorization of these
subtypes is determined by comparing normal, non-cancerous glial
cells and tumor cells (7,8).
Gliomas are now categorized based on genetic
etiology and prognostic factors, according to the WHO CNS5 2021.
Diffuse gliomas in adults are now classified into three categories:
Astrocytoma [with mutations in isocitrate dehydrogenase (IDH),
classified as grade II, III or IV], oligodendroglioma
(characterized by IDH mutation and co-deletion of 1p/19q, either
grade II or III) and glioblastoma multiforme (GBM) [IDH wild-type
(wt), grade IV] (3). Several
studies describe the chromosomal and molecular heterogeneity of
glioma, which opens the door for a new classification system for
this disease (8–10). Genetic heterogeneity in GBM promotes
treatment resistance and disease progression by enabling cellular
plasticity and diverse expression states. Despite this diversity,
malignant cells converge into neural development- and cell
cycle-related states driven by alterations in genes such as EGFR
and PDGFRA, enhancing adaptability and limiting single-target
therapies (9,11). Personalized treatments integrating
genetic, epigenetic and microenvironmental data are essential
(12).
Since the mapping of the human genome sequence,
advances in bioinformatics and sequencing technology have
substantially improved the investigation of genomic sequences in
human malignancies. Single-cell RNA sequencing has revealed
distinct cell lineages in GBM, including astrocytic, neuronal,
oligodendrocytic, mesenchymal and GBM stem cells (GSCs), which
drive tumor growth and heterogeneity. It has uncovered a
neurodevelopmental hierarchy led by progenitor-like GSCs and has
highlighted tumorigenic, rapidly cycling GSCs as key drivers of
growth. Notably, neuronal lineages lack HLA expression, suggesting
immunotherapy resistance (13,14).
Microsatellite instability is observed in 5.5–25% of GBM cases,
particularly in recurrent tumors, due to mismatch repair defects.
Its clinical implications remain elusive, with studies showing
mixed outcomes in terms of prognosis and potential as a predictor
of immune checkpoint inhibitor efficacy (15,16).
One key feature driving the rapid proliferation of
GBM cells is altered glucose metabolism, characterized by
significant metabolic reprogramming. Altered glucose metabolism
supports GBM growth by increasing glucose uptake via upregulated
glucose transporter 1 (GLUT1) and GLUT3 and enhancing glycolysis
through enzymes such as phosphofructokinase platelet-type (PFKP)
and pyruvate kinase M2 (PKM2). This metabolic shift, known as the
Warburg effect, prioritizes glycolysis for ATP and biosynthetic
precursor production, even in oxygen-rich conditions, fueling rapid
proliferation (17,18). Also, GBM exhibits altered lipid
biosynthesis, including increased fatty acid synthesis via fatty
acid synthase (FASN) and ATP citrate lyase (ACLY), lipid droplet
accumulation for energy storage and reliance on exogenous
cholesterol through low-density lipoprotein (LDL) uptake. Abnormal
sphingolipid metabolism, with reduced ceramide and elevated
sphingosine-1-phosphate (S1P) levels, promotes tumor growth,
survival and therapy resistance (19,20).
Tumor-associated macrophages and microglia (TAMs)
and hypoxia significantly contribute to GBM progression and
therapeutic resistance. M2-like TAMs secrete factors like epidermal
growth factor (EGF), transforming growth factor-beta (TGF-β),
vascular endothelial growth factor (VEGF) and matrix
metalloproteinases (MMPs), promoting tumor growth, invasion,
angiogenesis and immune evasion, while tumor cells polarize TAMs
into a pro-tumor phenotype, amplifying malignancy (21). Hypoxia activates the
hypoxia-inducible factor pathway, upregulating genes such as VEGF,
ZEB1 and CXCR4, which drive metabolism, angiogenesis and invasion.
It further promotes glioma stem cells, enhances glycolysis,
suppresses immune responses and contributes to therapy resistance,
highlighting key therapeutic challenges (22).
In the present study, the bioinformatic analysis of
key genes that are altered in GBM according to the new WHO
classification were reviewed. Such understanding is crucial for
analyzing genes and pathways involved in the development and
progression of GBM, allowing for the development of targeted
therapies.
Overview of the genetic alterations in
GBM
GBM, a grade IV tumor, resembles the most prevalent
and deadliest form of brain cancer. Key factors influencing the
prognosis of GBM include the extent of tumor removal, the patient's
age at the time of diagnosis and their Karnofsky performance status
(1,23). Survival rates for patients differ
based on the glioma type, with pilocytic astrocytoma (grade I)
showing the highest survival rates and GBM having the lowest. The
five-year survival rate for individuals diagnosed with GBM ranges
from 0.05 to 4.7% (1).
GBM is defined by a wide range of genetic or
epigenetic changes. The extensive range of modifications results in
various mutation subgroups, each necessitating distinct
therapeutics and each associated with unique patient survival
outcomes. Therefore, the designation ‘multiforme’ was adopted to
reflect the genotypic and phenotypic diversity of this tumor type
(24). In 2008, The Cancer Genome
Atlas (TCGA) released a classification for GBM that was centered
around genetic mutations and molecular indicators, and consisting
of the classical, mesenchymal, neural and pro-neural categories
(25,26). EGFR amplification, cyclin-dependent
kinase inhibitor 2A (CDKN2A) deletion and p53 mutations are
characteristics of the classical subtype. The mesenchymal subtype
displays changes in neurofibromatosis type 1 (NF1) and phosphatase
and tensin homolog (PTEN), as well as expression of the MET and
CD44 mesenchymal genes, while the pro-neural subtype is
characterized by mutations in the p53, platelet-derived growth
factor receptor alpha, phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit alpha (PIK3CA) and IDH1 genes, along
with heightened expression of the homeobox protein NKX2-2 and the
oligodendrocyte transcription factor 2 (OLIG2) genes. Finally, the
neural subtype, which accounts for 16% of GBM cases, is
characterized by its expression of neural markers (27). Chromosomal gains (e.g., EGFR on
chromosome 7) and losses (e.g., PTEN on chromosome 10, CDKN2A/B on
chromosome 9 disrupt pathways such as RTK/PI3K, p53 and
retinoblastoma protein (RB), driving GBM growth, survival and
therapy resistance (28).
In GBM, micro (mi)RNA dysregulation impacts tumor
suppressors and oncogenes. Overexpressed miRNAs, such as miR-21 and
the miR-17-92 cluster, act as oncogenes by promoting proliferation,
invasion and therapy resistance, while downregulated miRNAs, such
as miR-7, miR-34a and miR-137, suppress tumor growth. Altered
miRNAs also regulate metabolism, angiogenesis and immune responses,
highlighting their role in GBM progression and potential as
therapeutic targets (29–31).
LncRNAs regulate GBM cell proliferation and invasion
by acting as oncogenes or tumor suppressors. Pro-tumorigenic long
non-coding (lnc)RNAs such as H19 and MIR31HG promote proliferation
and invasion via Wnt/β-catenin and NF-κB signaling, while
tumor-suppressive lncRNAs such as GAS5 and LINC-PINTinhibit these
processes by targeting pathways such as STAT5. These lncRNAs hold
potential as biomarkers and therapeutic targets (32–34).
A new stratification for GBM was established by the
2021 WHO CNS5. In adult cases of IDH-wt diffuse astrocytic gliomas,
the presence of any one of three genetic factors is sufficient to
identify the most severe grade of glioma. These parameters are: i)
Telomerase reverse transcriptase (TERT) promoter mutation, ii) EGFR
gene amplification and iii) combined gain of the entire chromosome
7 and loss of the entire chromosome 10 (+7/-10 chromosome copy
number changes) (35,36). Consequently, these three genetic
parameters have been incorporated into the diagnostic criteria for
GBM. Histone acetylation and methylation regulate gene expression
in GBM as well, promoting tumor progression. Acetylation by histone
acetyltransferases such as p300/CBP enhances gene activity but
drives gliomagenesis, while methylation by lysine
methyltransferases and protein arginine methyltransferase 2 is
linked to aggression and oncogenesis. The H3K27M mutation and
epigenetic modifications in GSCs offer potential therapeutic
targets (37–39).
ATP-binding cassette (ABC) transporters, such as
ABCB1, ABCC1 and ABCG2, contribute to chemotherapy resistance in
GBM by actively effluxing chemotherapeutic drugs like temozolomide,
reducing their intracellular concentration and rendering them less
effective. Genetic variations, including single nucleotide
polymorphisms, further influence the chemotherapy response,
complicating the prediction of treatment outcomes, as tumors with
higher expression of ABC transporters may be less responsive to
treatment (40,41).
Studying the molecular and genetic changes
underlying the development of GBM is crucial for gaining a
comprehensive understanding of this disease and developing targeted
treatment plans for patients. Given the above, a one-size-fits-all
approach does not exist for treating all cases of GBM. While
surgery, radiation and chemotherapy remain the cornerstone
therapeutic approaches for cancer, progress in understanding the
molecular processes driving tumor development is leading to novel
targeted strategies for the diagnosis and treatment of cancer. Over
the last decade, there has been a surge in biological data on
genes, proteins, CpG islands and a variety of other topics, which
led to the introduction of numerous bioinformatics tools for
analyzing biological data. Eventually, new databases emerged, such
as The Cancer Genome Atlas (TCGA); http://www.cancer.gov/tcga), Therapeutically
Applicable Research to Generate Effective Treatments (TARGET;
http://www.cancer.gov/ccg/research/genome–sequencing/target)
and the Cancer Genome Characterization Initiative (CGCI)
(https://www.cancer.gov/ccg/research/genome-sequencing/cgci),
leading to groundbreaking changes in the cancer field (42). In this review, a genomic evaluation
of key genes involved in GBM tumorigenesis and progression was
carried out, combining results of laboratory research and
bioinformatics tools.
Overview of bioinformatics tools used to
identify genetic alterations
cBioPortal
Given the nature of GBM and the multitude of genes
implicated in its tumorigenesis, it is essential to employ a tool
for the comprehensive analysis and comparison of the genetic
alterations associated with this specific type of cancer.
cBioPortal, an open-access web resource used in cancer research and
molecular profiling, effectively meets this need and provides a
platform for cancer researchers to explore, visualize and analyze
multidimensional cancer genomics data compiled from extensive
cancer genomics projects such as the International Cancer Genome
Consortium and TCGA. This portal facilitates access to DNA copy
number alterations, somatic mutations, DNA methylation, mRNA and
miRNA expression, as well as protein and phosphoprotein abundance
data (43). Scientists can then
analyze the datasets to uncover patterns and biomarkers associated
with cancer progression, or novel therapeutic targets that aid in
the development of personalized GBM treatment strategies.
cBioportal offers an oncoprint feature that allows
users to generate graphical representations of genomic alterations
across specific genes within a sample where every row corresponds
to a gene and every column represents a sample. This representation
uses colors and symbols to depict amplifications, mutations,
deletions and alterations in gene expression (43,44).
This allows researchers to easily identify patterns and
relationships between genetic alterations and specific cancer
types.
cBioPortal also provides an analysis of mutual
exclusivity, using Fisher's exact test to ascertain whether certain
genetic alterations occur together or are mutually exclusive within
the same tumor type. In mutual exclusivity, one genetic alteration
replaces another within a tumor, suggesting that each tumor may
harbor only one of these alterations. Alternatively, Co-occurring
genetic alterations can arise concomitantly in the same tumor type
(43,44).
The mutations tab is another feature that provides
both a graphical and a tubular summary of the non-synonymous
mutations detected within the queried gene. The ‘lollipop model’,
or visual representation, illustrates the frequency and location of
mutations within the protein domains as encoded by the standard
gene isoform. Notably, all DNA mutations are adapted to the
standard RefSeq isoform. The tabular summary offers additional
details regarding every mutation within each queried gene, such as
the sample case ID, amino acid change, annotations, type (missense,
nonsense, splice site, frameshift insertion or deletion, in-frame
insertion or deletion, nonstop, nonstart), copy number, number of
mutations in a sample, number of mutations at a particular position
in the Catalogue of Somatic Mutations in Cancer and frequency
(43).
Additionally, the portal has a survival analysis
section enabling researchers to compare overall survival and
disease-free survival rates, represented in Kaplan-Meier plots,
between tumor samples with and without alterations in the genes
under investigation (43). These
methods provided by cBioportal aid in unraveling the complex
genetic dynamics underlying cancers such as GBM.
cBioPortal in GBM research enables the
identification of key genomic alterations such as EGFR, PTEN and
IDH1 mutations, as well as disrupted pathways such as PI3K/AKT and
p53. It stratifies patients with GBM into molecular subtypes,
supports personalized treatment strategies and integrates clinical
outcomes with genomic data to identify prognostic biomarkers and
therapeutic responses. Tools such as oncoprints and survival plots
facilitate the discovery of actionable targets and advancements in
GBM diagnostics and treatment (45).
In a study published in Nature, cBioPortal
was used for the analysis of large-scale genomic and transcriptomic
datasets to uncover GBM-specific alterations such as TP53 mutations
and EGFR amplifications. It revealed molecular drivers, therapeutic
targets and distinct subtypes, correlating genomic changes with
prognosis and treatment outcomes, making it crucial for
translational GBM research (46).
cBioPortal also has several limitations. Its
retrospective design introduces the possibility of convenience
sampling and it lacks the ability to perform multivariate analyses
to identify and adjust for confounding factors. However, metadata
can be downloaded and analyzed using external statistical software
like R or SPSS (47). While
cBioPortal supports correlation analysis between query gene
alterations, tools such as Regulome Explorer (https://www.regulomedb.org) and Oncomine (https://www.oncomine.com) are needed for more complex
gene correlations, including mRNA expression (43). Currently, this tool is for research
use only and not approved for patient diagnosis or treatment.
Future developments aim to ensure compliance with Medical
Diagnostic Regulation (https://eur–lex.europa.eu/eli/reg/2017/745/oj)
and In Vitro Diagnostic Regulation (https://eur-lex.europa.eu/eli/reg/2017/746/oj)
(48).
Cytoscape
The recent development of high-throughput ‘omics’
research areas, including transcriptomics, proteomics and
metabolomics, has led to the generation of extensive datasets,
which considerably increased the current understanding of
biological processes at the molecular and physiological levels,
particularly in the context of GBM. However, interpreting these
large databases remains a challenge. As a result, there is an
increased demand for computer-based support in visualizing and
analyzing biological data, which plays an essential role in
illustrating the numerous biological interactions as organized and
logical pathways (49).
Cytoscape is an open-source software platform
developed for visualizing molecular interaction networks and
combining them with annotations, gene expression profiles and other
high-throughput expression data (50). While it can be applied to various
molecular systems, Cytoscape is most effective when utilized
alongside large databases of molecular interactions, for both
humans and model organisms, particularly those involving
protein-protein and protein-DNA interactions as well as numerous
genetic interactions (50,51). The core functionality of Cytoscape
includes network layouts, data integration with molecular states
and linking to functional annotation databases (50). The software's extensibility through
plugins allows the rapid development of additional computational
analyses. Overall, Cytoscape is an effective and adaptable
framework for depicting and evaluating pathways at the biomolecular
level (49).
Using the Cytoscape tool in GBM research
facilitates the analysis and visualization of protein-protein
interaction (PPI) networks and molecular pathways involved in the
development of this disease. It enables researchers to identify key
hub proteins and deregulated pathways, such as PI3K/AKT/mTOR and
EGFR signaling, which are critical to GBM progression and
resistance mechanisms (52).
Integrating genomics, transcriptomics, proteomics, epigenomics and
metabolomics provides a comprehensive view of GBM by identifying
molecular interactions, tumor subtypes and biomarkers. Techniques
like single-cell multi-omics and machine learning uncover cellular
heterogeneity, disrupted pathways and therapeutic targets (53–55).
By integrating multi-omics data, Cytoscape provides graphical
representations of molecular interactions, aiding in the
identification of therapeutic targets and enhancing the
understanding of GBM's molecular biology.
In specific studies, Cytoscape was used to construct
and analyze PPI networks for GBM using publicly available datasets.
Plug-ins such as Molecular Complex Detection (MCODE; http://mcode.readthedocs.io) were employed to identify
highly connected clusters or modules in the network that are
crucial to GBM pathology. Topological analysis revealed hub genes
and key nodes involved in tumor progression and cellular processes
such as cell cycle regulation (56).
Additionally, pathway enrichment analysis using
Cytoscape tools such as ClueGO and CluePedia mapped identified
genes to biological pathways, uncovering disruptions in cell
proliferation, apoptosis and DNA repair mechanisms (56). Enhanced DNA repair mechanisms,
including homologous recombination, non-homologous end joining and
base excision repair, enable GBM cells to resist
radiotherapy-induced DNA damage. Key factors like ataxia
telangiectasia mutated (ATM), ATM- and Rad3-related (ATR),
DNA-dependent protein kinase (DNA-PK) and RAD51, along with cancer
stem cells' efficient repair capacity, further enhance resistance
(57,58). In addition, Notch pathway activation
sustains GBM stem cells by promoting self-renewal, inhibiting
differentiation and driving tumor progression and therapy
resistance (59,60). Targeting these pathways could
improve radiotherapy outcomes.
In order to overcome the limitations of Cytoscape,
it is important to connect directly with databases such as Database
of Interacting Proteins (DIP) (http://dip.doe-mbi.ucla.edu), Gene Expression Omnibus
(GEO; http://www.ncbi.nlm.nih.gov/geo) and Gene Ontology
(GO; http://geneontology.org), eliminating
the need for manual data parsing into annotations or attributes. A
longer-term goal is to integrate high-level interaction networks
with detailed physico-chemical models of biological processes,
which are addressed by tools such as Ecell (https://www.e–cell.org), VirtualCell (https://vcell.org), Gepasi (http://www.gepasi.org) and Systems Biology (https://sbw.sourceforge.net) (50).
MutationTaster
As molecular biology progresses and its
methodologies become more cost-effective, the use of DNA sequencing
tools helps investigate potential biological markers and identify
novel genetic alterations involved in the progression of GBM.
MutationTaster is one of several web-based tools utilized for
predicting the effects of DNA variants. It was developed in 2014 by
Cardiff University and Universitätsmedizin Berlin to assess
intronic, synonymous and short indel mutations using an annotation
software on its website (mutationtaster.org).
To predict variant effect, the program uses
pathogenic variants found in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) and the Human
Gene Mutation Database (https://www.hgmd.cf.ac.uk) (61), as well as, among others, single
nucleotide polymorphisms and deletions from the 1,000 Genomes
Project (62). Pathogenic
variations in ClinVar are considered disease-causing, while
variants that appear >4 times in 1,000 Genomes (https://www.internationalgenome.org)/HapMap
(https://ftp.ncbi.nlm.nih.gov/hapmap)
are considered neutral (63).
A significant advantage of MutationTaster2 (a new
version of the tool) compared to other mutation-predicting tools is
the ability to provide high-accuracy predictions, as evidenced by
various benchmarking studies. In addition, MutationTaster2 is
capable of processing large-scale datasets, which is particularly
useful for analyzing data from high-throughput sequencing. However,
a notable drawback of MutationTaster2 is its dependence on
predefined rules and algorithms, which may not fully account for
the intricate and situation-specific impacts of genetic variations
(64).
The latest release of MutationTaster21, aimed at
boosting whole-exome sequencing success rates by subjecting each
variation to a battery of in silico tests and replacing
Bayes classifier with Random Forest models, is suited for several
kinds of variation. The models were built on balanced accuracy,
ensuring equal predictive performance for both benign and malignant
variations (64).
Several studies have demonstrated the advantages of
using MutationTaster in GBM by predicting the functional impact of
genetic mutations in key GBM-associated genes such as TP53,
EGFR, PTEN and IDH1. MutationTaster analyzes these
mutations to determine whether they are disease-causing by
evaluating protein function, splice sites and regulatory elements.
Hence, it helps prioritize mutations for further validation and
identifies their role in tumor progression, therapy resistance and
altered cellular processes, supporting the development of
personalized therapies for GBM (65).
MutationTaster is limited by the fact that it can
only predict the deleteriousness of variants within protein-coding
genes and cannot assess variants in RNA genes or non-genic regions
(64).
Analysis of genes involved in GBM survival
pathways
EGFR
GBM is often associated with the amplification and
overexpression and EGFR, a tyrosine kinase receptor. EGFR signaling
is essential for GBM cell growth and survival, and targeting EGFR
has been a major focus of GBM research. One of the most prevalent
mutations in GBM is the EGFRvIII mutation, which is caused by an
in-frame deletion of exons 2–7 of the EGFR gene. EGFRvIII has been
associated with enhanced GBM cell proliferation, invasion and
angiogenesis. It is expressed in ~30% of patients with GBM.
Furthermore, EGFRvIII has been linked to radiation- and
chemotherapy resistance (26,66). A
cBioPortal analysis of 592 GBM samples, based on the TCGA database,
indicated that EGFR shows the highest mutation percentage (47%)
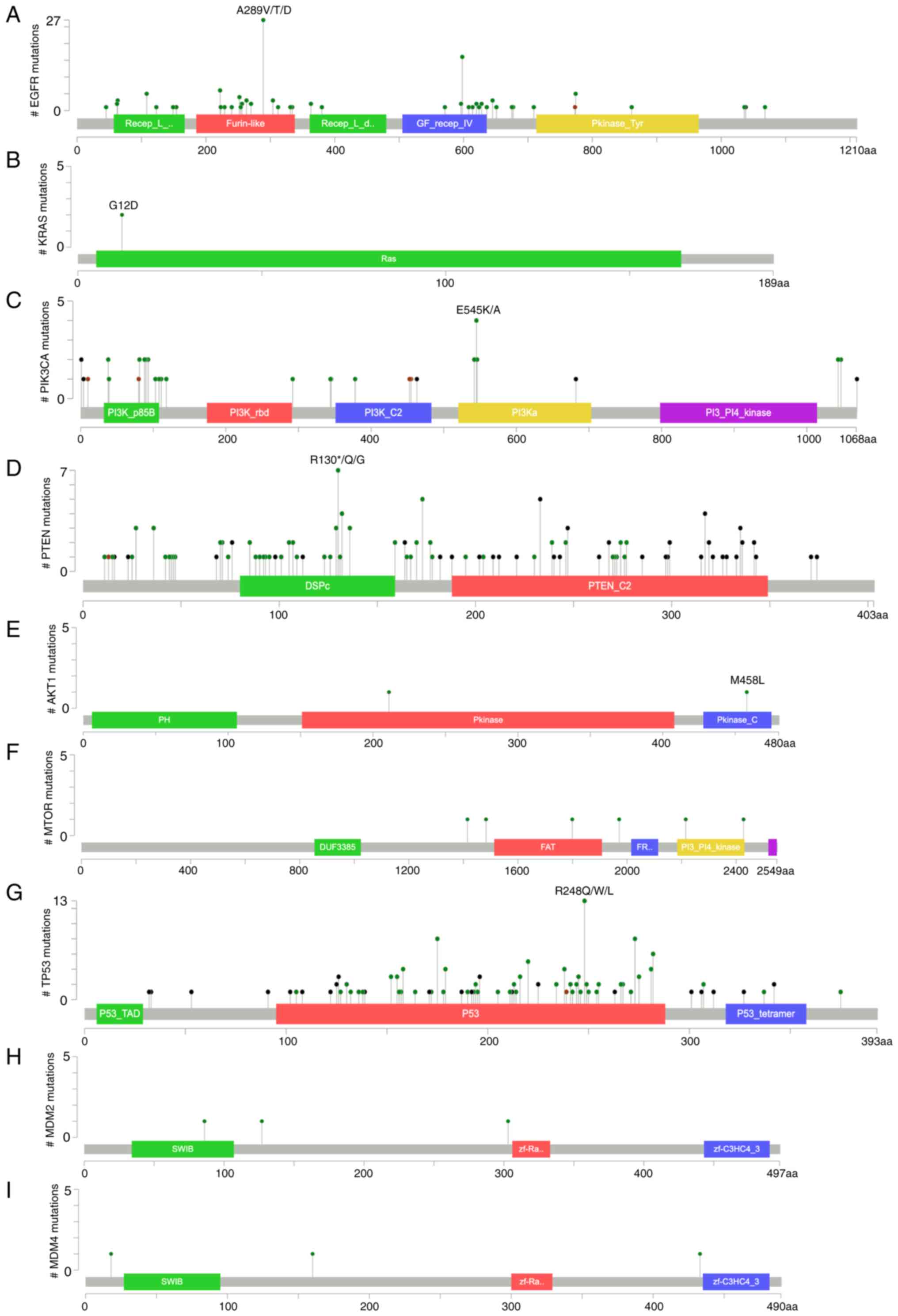
among the genes evaluated in this review (Figs. 1A and B and 2). Brennan et al (26) examined the genomic alterations in
GBM and discovered that EGFR was amplified in ~45% of cases.
Bioinformatics analysis of the 592 GBM samples (TCGA, Pan
Atlas-GBM) for mutual exclusivity and co-occurrence using
cBioPortal demonstrated that EGFR mutations co-occur with
modification of AKT1 and MAP3K1 but are mutually exclusive with
IDH1 and TP53 mutations (P<0.01) (Fig. 1C and D). Additional research
confirmed this and showed that EGFR amplification is associated
with both higher gene mRNA expression and increased activity of the
PI3K/AKT/mTOR signaling pathway, an important regulator of cell
growth and survival. The lollipop model developed by cBioPortal
indicated that 26 out of 126 mutations are the highly oncogenic
A289V/T/D and 16 mutations are G598 V/A (67). The pathogenicity of missense
mutations may be predicted using the Rare Exome Variant Ensemble
Learner (REVEL) for better understanding of their effect. Variants
have scores that range between 0 and 1, and the greater their
value, the more likely they are pathogenic. The A to V amino acid
substitution is expected to have the highest REVEL score among the
three A289 substitutions (A289V/T/D), suggesting that it is more
oncogenic than the other two. Likewise, the amino acid shift from G
to V in G598V/A is more oncogenic than the switch from G to A.
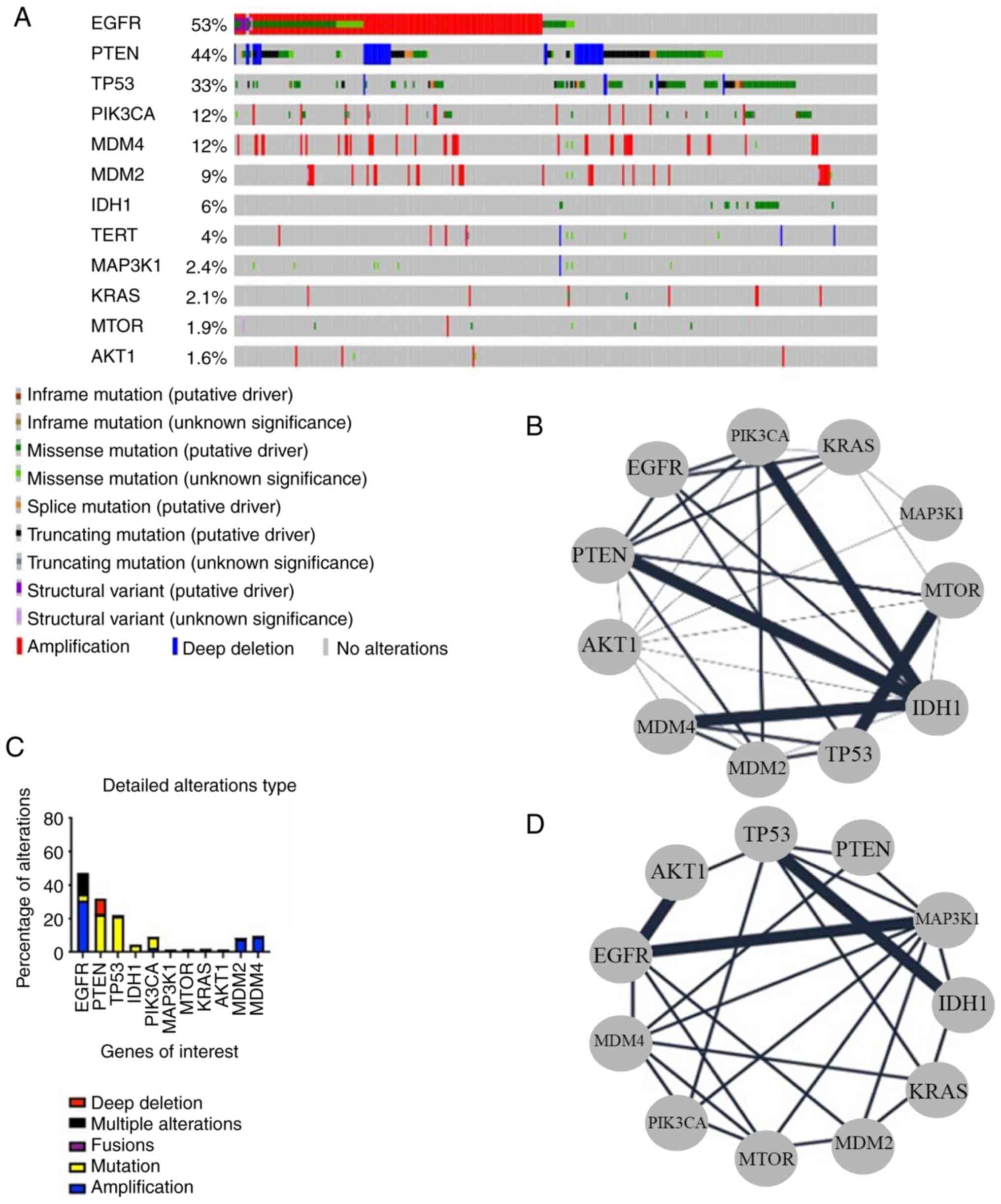 | Figure 1.Bioinformatics analysis of genetic
alterations in GBM. (A) Oncoprint of EGFR, PTEN, TP53, PIK3CA,
MDM2, MDM4, IDH1, TERT, MAPK3K1, KRAS, mTOR and AKT1 in 592 samples
of GBM showing the alteration type. (B) Network of the genes of
interest showing their mutual exclusivity relationship. The network
was generated by Cytoscape. (C) Percentage of alteration in each of
the genes of interest according to the oncoprint. (D) Network of
the genes of interest showing their co-occurrence relationship. The
network was generated by cystoscape. GBM, glioblastoma. |
Additional bioinformatics studies have been carried
out to better elucidate the role of EGFR in GBM. Lu et al
(68) used gene expression
profiling to determine which gene expression patterns were
different in GBM tumors with and without EGFR amplification.
Various genes that regulate the cell cycle and DNA replication have
been found to be upregulated in tumors with EGFR amplification
(26,69). Computational modelling was used in
another study by Jain et al (70) to find possible downstream effectors
of the EGFR signaling pathway in GBM. They discovered that one of
the major downstream pathways activated by EGFR in GBM was the
RAS/RAF/MEK/ERK pathway. Small molecule tyrosine kinase inhibitors
(TKIs) and monoclonal antibodies are two examples of targeted
therapeutics that have been developed to block EGFR signaling in
GBM (71,72). Clinical trials of these agents,
however, have had mixed results in terms of improving the overall
survival of patients with GBM (73). This can be the result of the complex
signaling network involving EGFR and its downstream effectors,
which can lead to other pathways being activated as a compensatory
mechanism in response to EGFR inhibition. Recent studies have
identified novel approaches to targeting EGFR in GBM, such as
combination therapies that target both EGFR and other signaling
pathways, or inhibitors that target specific downstream EGFR
effectors (73). Overall, EGFR
seems to be a potential therapeutic target in GBM. Traditional
tyrosine kinase inhibitors (TKIs), such as erlotinib and gefitinib,
are designed to target mutations in the intracellular tyrosine
kinase domain. However, since GBM mutations primarily occur in the
extracellular domain (ECD), these TKIs are less effective. By
contrast, lapatinib has a higher affinity for the ECD, which likely
accounts for its superior in vivo results (74). Additionally, GBM-associated EGFR
mutations enable active signaling despite an inactive receptor
conformation (75), further
limiting the effectiveness of traditional TKIs in targeting EGFR.
Moreover, erlotinib and gefitinib show limited efficacy in GBM
clinical trials, with no significant survival improvements, even
when used in combination therapies (73). Erlotinib's ineffectiveness is likely
due to its inability to target the EGFR extracellular domain. While
gefitinib slightly improves survival and efficiently
dephosphorylates EGFR, it fails to halt tumor growth signaling
(74,76). These findings underscore the need
for synergistic inhibition of downstream EGFR pathways and the
development of new TKIs that specifically target ECD variants in
GBM.
Monoclonal antibodies, which work by binding to EGFR
overexpressed on tumor cells and blocking signaling required for
cell growth, have also been investigated. However, antibodies
targeting EGFR in GBM, such as cetuximab, panitumumab and
nimotuzumab, show limited efficacy in clinical trials, with minimal
improvements in progression-free survival (PFS) and overall
survival (77–79). Cetuximab demonstrates potential in
combination therapies but causes side effects like rash and
gastrointestinal toxicity due to off-target effects on normal
tissue (80,81). These side effects are worsened by
the high doses required to penetrate the blood-brain barrier,
emphasizing the need for synergistic inhibition to overcome
resistance and the development of GBM-specific drugs or delivery
mechanisms to minimize off-target antibody accumulation (82,83).
Sym004, a combination of two recombinant human-mouse
chimeric monoclonal antibodies (futuximab and modotuximab), targets
non-overlapping EGFR epitopes and demonstrates activity against
various EGFR-expressing solid tumors in preclinical studies. It
shows superior tumor growth inhibition in xenograft models compared
to other anti-EGFR antibodies (84). However, in a phase II trial for
recurrent GBM, Sym004 shows limited efficacy, with PFS reaching a
maximum of 9.95 months (85). This
may be due to the complex signaling network involving EGFR and its
downstream effectors, which can activate compensatory pathways in
response to EGFR inhibition.
Similarly, although novel anti-EGFR antibody GC1118
demonstrates promising preclinical data (86), it fails to improve PFS in a phase II
trial for recurrent GBM patients with EGFR amplification, likely
due to tumor evolution and inadequate pathway suppression (87). Depatuxizumab mafodotin, an anti-EGFR
antibody-drug conjugate, also failed to improve overall survival in
EGFR-amplified GBM patients in a phase III trial, despite promising
preclinical data. While it improved PFS, particularly in patients
with the EGFRvIII variant, tumor resistance via alternative
pathways limits its efficacy (88).
Currently, novel approaches, including highly
CNS-penetrant small molecule EGFR inhibitors like ERAS-801 and
BDTX-1535, as well as dual-specific immunotoxins targeting both
EGFRwt and EGFRvIII such as D2C7-IT, are under investigation, with
certain drugs showing potential in early studies (85,89).
The Ras/Raf/MEK/ERK pathway
Among the pathways that regulate cell division,
proliferation and survival is the Ras/Raf/MEK/ERK pathway. Its
dysregulation has been associated with the development of several
types of cancer, including GBM. Aberrant activation of this pathway
in GBM has been associated with poor prognosis and resistance to
therapy. Several bioinformatic analyses have been performed to
investigate the prevalence and functional significance of mutations
in the genes of this pathway. One study examined GBM patient
samples using bioinformatics to identify mutations and changes in
the copy number of genes encoding components of this pathway. The
most commonly mutated genes in GBM are EGFR, KRAS, NRAS and BRAF
(Figs. 1A and B and 2B), and these mutations are associated
with an increased activity of the Ras/Raf/MEK/ERK pathway (90,91).
Inhibitors that block this pathway, such as sorafenib or U0126,
reduced GBM cell proliferation while increasing apoptosis in
vitro. Brennan et al (26) used a combination of bioinformatic
and experimental approaches to investigate the functional
significance of Ras/Raf/MEK/ERK pathway mutations in GBM. They
discovered that mutations in the BRAF and KRAS genes were linked to
pathway activation and increased cell proliferation. They also
demonstrated that small-molecular-weight inhibitors selectively
reduced the viability of GBM cells with BRAF or KRAS mutations
(26). An additional study examined
the role of this pathway in the mesenchymal subtype of GBM, which
is associated with increased pathway activity. Inhibition of the
pathway using the MEK1/2 inhibitor trametinib led to a decrease in
the in vitro migration and invasion of mesenchymal GBM
cells. Along with genetic alterations, epigenetic modifications,
including DNA methylation, have been associated with the
dysregulation of the Ras/Raf/MEK/ERK pathway in GBM. Specific DNA
methylation changes in GBM regulate gene expression, influencing
tumor progression and prognosis. Changes like EZH2 activation in
the mesenchymal subtype, Wnt signaling demethylation and promoter
methylation of genes like FNDC3B and SOX10 are linked to poor
survival and treatment resistance. Methylation-regulated biomarkers
offer therapeutic potential (92–94).
One study found that increased pathway activity in GBM is linked to
hypermethylation of the RASSF1A promoter region, causing lower
expression of the RASSF1A tumor suppressor gene (95).
These results suggest that targeting the
Ras/Raf/MEK/ERK pathway may be a potential strategy for treating
GBM and that dysregulation of this pathway plays a significant role
in the development of this type of cancer. Studies suggested that
BRAF-targeted therapy is an effective treatment for patients with
BRAF-mutated GBM. BRAF inhibitors such as vemurafenib, dabrafenib
and encorafenib are generally well-tolerated and lead to rapid
clinical improvement. These inhibitors inactivate Raf kinase
through competitive occupation of the ATP binding pocket, disrupt
the MAPK signaling cascade, cause G1 cell-cycle arrest and induce
apoptosis. However, no BRAF inhibitor is currently Food and Drug
Administration (FDA)-approved for use in patients with GBM
(96). However, dabrafenib and
encorafenib are tested in phase II clinical trials alongside the
MEK inhibitors trametinib and binimetinib, respectively (97).
In addition, MEK1/2 inhibitors show promise as
targeted treatments for GBM. Blocking MEK signaling in GBM results
in antiproliferative effects, reducing cell division and
Ki67-positive cells. Trametinib, a MEK1/2 inhibitor, inhibits the
Ras-Raf-MEK-ERK pathway, limiting GBM proliferation, migration and
invasion (96). Other MEK
inhibitors, such as cobimetinib, are also considered potential
treatments, demonstrating good tolerance and effectiveness, as
evidenced by tumor size reduction (98).
On the other hand, atorvastatin, a Ras/MAPK
inhibitor, enhances the efficacy of TMZ both in vitro and in
GBM xenografts by inhibiting Ras signaling through a
prenylation-dependent mechanism (99). However, when evaluated in
combination with radiotherapy and TMZ in patients with GBM (phase
II, NCT02029573), atorvastatin does not improve PFS (100).
The PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway plays a pivotal role in
several biological processes, such as cell survival, growth and
proliferation. Dysregulation of this pathway has been associated
with the onset and proliferation of various malignant tumors,
including GBM. In addition, aberrant activation of this pathway has
also been associated with poor prognosis and resistance to
radiation and chemotherapy (101).
Several studies examined GBM patient samples using
bioinformatics to identify mutations and changes in copy number
changes of genes implicated in the PI3K/AKT/mTOR cascade (Figs. 1A and B and 2C-F). Changes in genes such as PIK3CA,
PTEN and AKT1 have been commonly found in GBM, and these changes
have been associated with an elevated activity of the PI3K/AKT/mTOR
pathway. A cBioPortal analysis of 592 GBM samples (TCGA, PanCancer
Atlas-GBM) suggested that PIK3CA exhibits oncogenic E545K/A
mutations, which are known to most frequently occur in the helical
domain of the protein (102).
Parsons et al (101)
examined the genomic alterations in a large cohort of patients with
GBM and discovered that PIK3CA is mutated in ~15% of cases. Further
investigation revealed that these mutations are mostly missense and
clustered in the kinase domain of its p110 subunit. In addition,
the researchers discovered that PIK3CA mutations are associated
with increased cell proliferation, PI3K/AKT/mTOR pathway activation
and reduced responsiveness to chemotherapy (101). In another study, Liang and
Slingerland (103) aimed to
explore the functional significance of PIK3CA mutations in GBM
using a combination of bioinformatics and experimental approaches.
As a result, they were able to identify an association between
PIK3CA mutations and an elevated activation of the PI3K/AKT/mTOR
signaling cascade along with increased cell proliferation.
Furthermore, they showed that inhibiting the PI3K/AKT/mTOR pathway
using a small molecule inhibitor selectively reduced the viability
of GBM cells with PIK3CA mutations (103). Dysregulated Wnt signaling
interacts with the PI3K/Akt pathway to enhance GBM invasiveness and
therapy resistance. Aberrant Wnt/β-catenin activation promotes
epithelial-to-mesenchymal transition, increasing cell migration and
survival. Cross-talk between the Wnt and PI3K/Akt pathways
amplifies invasive behavior and supports tumor progression by
creating a tumor-promoting microenvironment and enhancing
angiogenesis (104,105).
Apart from genetic alterations, GBM has been
associated with dysregulation of the PI3K/AKT/mTOR pathway due to
epigenetic modifications, including DNA methylation. According to
one study, increased activity of the PI3K/AKT/mTOR pathway in GBM
is associated with hypermethylation of the PTEN promoter region,
which results in decreased expression of the PTEN tumor suppressor
gene (106). Based on the lollipop
model produced by cBioPortal, five of the 132 PTEN mutations were
R173H/C alterations, while six of the mutations were R130*/Q. Among
the 6 R130 mutations, three were missense mutations (R130Q),
involving the conversion of Arginine to Glutamine, while the
remaining three were nonsense mutations (R130*). On the contrary,
each of the 5 R173H/C mutations was of the missense type.
Furthermore, a truncation occurring at residue 223 results in an
alteration in the amino acid sequence, leading to a nonsense
mutation (R223*). The pair of missense mutations, R130Q and R173H,
are identified within the phosphatase domain of PTEN, impacting its
phosphatase activity. On the other hand, R233 is located in the C2
domain, a crucial region responsible for PTEN's binding to the
lipid plasma membrane (107).
Earlier research indicated that mutations at R130Q and R173C/H
result in the loss of PTEN function. Likewise, the truncations at
R130* and R233* lead to functional loss (108).
These results imply that the dysregulation of the
PI3K/AKT/mTOR pathway is crucial for the onset and advancement of
GBM. Hence, targeting this pathway may be a promising approach for
treating the disease. PI3K inhibitors are classified into pan-PI3K,
isoform-selective and dual PI3K/mTOR inhibitors. Although >50
PI3K inhibitors have been developed for cancer treatment, only a
few, such as BKM120, XL147 and XL765, have progressed to clinical
trials for GBM (109).
First-generation pan-PI3K inhibitors, like
wortmannin and LY294002, had limited clinical utility due to poor
solubility, short half-life, off-target effects and high toxicity.
In response, second-generation pan-PI3K inhibitors, including
BKM120, XL147, PX-866, GDC-0941 and GDC-0032, were developed with
improved safety, efficacy and pharmacokinetics, and are now
undergoing clinical trials (109).
Among these, Buparlisib (BKM120) is the most
frequently used PI3K inhibitor in GBM clinical trials, as it is
well-tolerated and able to cross the blood-brain barrier (BBB)
(109). Buparlisib induces G2/M
cell cycle arrest and apoptosis in GBM cells through p53-dependent
microtubule misalignment and mitotic dysfunction. It also enhances
apoptosis induced by TNF-related apoptosis-inducing ligand (TRAIL)
and B-cell lymphoma 2 (Bcl-2) inhibitors by increasing Noxa
expression, sequestering Mcl-1 and releasing pro-apoptotic proteins
Bim and Bak. However, a phase II study of BKM120 in recurrent GBM
showed mild toxicities, including elevated liver enzymes, rash and
hyperglycemia, but limited efficacy in patients with PTEN loss or
PIK3CA mutations, despite its ability to inhibit Akt
phosphorylation (NCT01339052) (110).
Similarly, Pilaralisib (XL147), an oral, reversible
pan-PI3K inhibitor, has shown promise. A phase I study combining
XL147 and XL765 (a dual PI3K/mTOR inhibitor) in recurrent GBM
demonstrated moderate BBB penetration, with tumor-to-plasma ratios
of 0.27–0.40. Both drugs reduced S6K1 phosphorylation and Ki67
expression, suggesting their potential for GBM growth inhibition
(111).
On the mTOR inhibitor front, early agents like
rapamycin faced challenges as cancer treatments due to
immunosuppressive effects. Improved analogs, such as everolimus and
temsirolimus, are FDA-approved for specific cancers but still
encounter issues like persistent mTOR signaling and Akt activation
through an mTORC1-mediated feedback loop. To address these
limitations, second-generation mTOR inhibitors that target both
mTORC1 and mTORC2 by binding the ATP-binding pocket have been
developed. This category includes dual PI3K/mTOR inhibitors (e.g.,
NVP-BEZ235, XL765) and mTORC1/2 inhibitors (e.g., Torin1, AZD8055),
which are currently being tested in GBM clinical trials (109,112).
However, resistance to first- and second-generation
inhibitors has spurred the development of third-generation mTOR
inhibitors. RapaLink-1, a BBB-penetrating compound, shows greater
potency in GBM models compared to its predecessors. Another
promising strategy involves selective mTORC2 inhibitors, such as
CID613034 and JR-AB2-011, which avoid mTORC1-related feedback loops
and exhibit strong anti-tumor effects in GBM studies. These
advancements renew optimism for the development of effective
mTOR-targeted therapies for GBM (112).
Analysis of genes involved in cell cycle and
apoptosis pathway
The cell cycle is a complex sequence of processes
that govern cellular growth and division. Cancer is characterized
by the unregulated growth and invasion of cancerous cells that
result from dysregulations in cell cycle control. Over the years,
several studies have looked into the cell cycle in GBM and
discovered several key molecular pathways that are dysregulated
(25).
TP53
GBM commonly exhibits mutations in the tumor
suppressor gene TP53 (Figs. 1A and
B and 2G). TP53 regulates DNA
repair, cell cycle progression and apoptosis, and loss of TP53
function is linked to increased genomic instability and oncogenesis
(113). Numerous studies have
revealed a high prevalence of TP53 mutations in GBM, with mutation
rates ranging from 30–60% in different cohorts (114). Furthermore, mutations in TP53 have
been associated with tumor cell survival and invasion, as well as
resistance to therapy and poor prognosis. For instance, Kim et
al (115) utilized TCGA data
to identify gene signatures associated with GBM prognosis. Their
findings revealed a correlation between TP53 mutations and an
unfavorable prognosis in this type of cancer (115). Data from the Ensmbl genome
database for the p53 rs121912651 variant reveal that these
mutations are considered pathogenic and are referred to as contact
mutations. These mutations disrupt the normal binding of p53 to
DNA, thereby contributing to the invasiveness of GBM cells
(108). Familial GBM is linked to
germline mutations in tumor suppressor and DNA repair genes. TP53
mutations in Li-Fraumeni syndrome, NF1/NF2 in neurofibromatosis,
APC and mismatch repair genes in Turcot syndrome, and BRCA1/BRCA2
mutations are associated with a higher risk of glioblastoma. These
mutations predispose individuals by disrupting cell-cycle
regulation and DNA repair, causing the genetic basis of
susceptibility in some families (116,117).
Research has been carried out to examine the
potential of the TP53 pathway as a therapeutic avenue for treating
GBM. One approach is to use gene therapy to restore TP53 function,
either by delivering wt-TP53 or by using small molecules that
activate TP53 signaling (118).
Another strategy is to target TP53 downstream effectors, such as
the p53-regulated gene mouse double minute 2/homolog (MDM2), which
is commonly overexpressed in GBM (119,120). Research has also discovered
possible interactions between TP53 and other signaling pathways in
GBM. According to one study, co-deletion of TP53 and PTEN tumor
suppressor gene were frequently encountered in GBM, increased AKT
signaling and decreased apoptosis in GBM cells (121).
The high prevalence of p53 mutations in GBM
highlights their potential as key targets for precision medicine.
Strategies to restore wt-p53 function in mutant p53 (mut-p53)
tumors, inhibit Gain-of-function (GOF) mut-p53 and inhibit the
MDM2/p53 complex to prevent wt-p53 degradation offer promising
therapeutic avenues for GBM and other cancers (122,123).
GOF p53 mutations in GBM can be corrected by
restoring wt-p53 function through additional point mutations that
stabilize the p53 protein (124).
Various compounds aimed at reactivating wt-p53 by altering the
conformation of mut-p53 have been developed (123). Among these, PRIMA-1 {2,2-bis
(hydroxymethyl)-1-azabicyclo[2.2.2]octan-3-one} stands out as a
highly effective molecule identified for its ability to restore
wt-p53 properties in select missense mutants (125). PRIMA-1 promotes p21 expression,
cell cycle arrest and apoptosis by refolding mut-p53 to a wt-p53
conformation (125,126). Its analog, PRIMA-1MET (APR-246),
has demonstrated efficacy in inhibiting cell growth, reducing
stemness and inducing apoptosis in GBM, as well as suppressing
tumor growth in mouse models (127–129). While PRIMA-1 has not been tested
in patients with GBM, PRIMA-1MET has undergone phase I/IIa trials
in hematological malignancies and prostate cancer (130).
Enhancing protein turnover using histone deacetylase
(HDAC) inhibitors is another approach for p53-based therapies.
Mut-p53 relies on the chaperone complex of Hsp70 and Hsp90, which
requires HDAC6 for stability. HDAC6 inhibition disrupts this
complex, promoting mut-p53 degradation (131). Several HDAC inhibitors, including
vorinostat, SAHA, CUDC-907, CCNU and CUDC-101, have shown promise
in GBM models (132–135). Vorinostat, combined with
tranylcypromine, reduces GBM stem cell viability and alters
apoptosis-regulatory genes (132).
SAHA destabilizes mut-p53 by disrupting HDAC6-Hsp90 interactions,
but it also downregulates wt-p53, limiting its use to homozygous
mut-p53 tumors (131,136–139). CUDC-907, a dual HDAC and PI3K
inhibitor, degrades mut-p53, radiosensitizing pediatric high-grade
GBMs (135). CCNU sensitizes adult
GBM to chemotherapy by degrading mut-p53 (134), while CUDC-101 enhances mut-p53
degradation, improving responses to EGFR inhibitors (133). These findings highlight HDAC
inhibitors' potential in mut-p53-targeted GBM therapies.
Mouse double minute 2/4 homolog
(MDM2/4)
MDM2 and MDM4 are essential genes responsible for
regulating p53, a commonly mutated or dysregulated tumor suppressor
protein in GBM (Figs. 1A and B and
2H-I). In that context, MDM2 and
MDM4 act as negative regulators and their overexpression has been
associated with the progression of GBM (140,141). MDM2 functions as an E3 ubiquitin
ligase that degrades p53, while MDM4 (also recognized as MDMX)
serves as a suppressor of p53, inhibiting its transcriptional
activity. In GBM, both genes are typically overexpressed, and this
overexpression is associated with both therapeutic resistance and
an unfavorable prognosis.
Biernat et al (142) illustrated that the MDM2 gene,
which is implicated in PI3K/AKT/mTOR pathway regulation, is
upregulated in nearly 10% of primary cases of GBM. Several studies
have looked into MDM2 and MDM4′s roles in GBM and identified
potential therapeutic targets for their inhibition. For instance,
researchers have examined the effect of small-molecule inhibitors
of MDM2 and MDM4 in preclinical models of GBM. The results revealed
a reduction in tumor growth and enhanced survival. Several other
strategies, such as using RNA interference to target MDM2 and MDM4
expression, and developing immunotherapies targeting MDM2 and MDM4,
have also been investigated (143). Overall, TP53 and cell cycle
regulators are critical targets for GBM research, and further
research is needed to unravel the impact of their mutations on the
development of GBM and to develop effective therapeutic strategies
targeting these proteins and their downstream effectors.
MDM2 inhibitors have gained attention as potential
therapies for GBM, as blocking the MDM2/p53 interaction to
reactivate p53 function represents a promising approach for cancer
and GBM treatment (123). Nutlins,
identified through chemical library screenings, are MDM2
inhibitors, with RG7112 being the first in class (144). RG7112 restored p53 activity in
MDM2-amplified, TP53 wt-GBM cell lines, crossed the BBB and
blood-tumor barrier, reduced tumor growth and improved survival in
xenograft models. Being the first MDM2 inhibitor to enter clinical
trials, it demonstrated p53 activation and pro-apoptotic effects
but was limited by significant toxicities at high doses. Its
second-generation analogue, RG7388 (idasanutlin), addressed these
limitations with improved potency, selectivity and pharmacokinetics
and has been tested in the NOA-20 trial alongside radiotherapy for
patients with GBM with unmethylated MGMT promoters (73,145).
Other MDM2 inhibitors, including MI77301, CGM097,
MK8242 and AMG232, have been developed (146), with AMG232 showing high potency
and selectivity for wt-p53 GBM stem cells. AMG232 effectively
inhibited tumor spheroid growth and stemness-related factors
(147). Nutlin3a demonstrated the
ability to impair DNA repair and enhance temozolomide-induced cell
death in GBM models (148). In
addition, novel indolylglyoxylyldipeptides targeting both MDM2 and
the translocator protein (TSPO) reactivated p53, disrupted the
mitochondrial membrane potential and inhibited GBM-cell viability,
suggesting dual TSPO/MDM2 targeting as a potential anti-GBM therapy
(149).
Analysis of genes involved telomerase
activity
The key gene responsible for telomerase activity in
GBM is TERT, which encodes the enzyme's catalytic subunit. This
enzyme is involved in the maintenance of telomere length and
prevention of cellular senescence. Various malignant cancers,
including GBM, have been shown to overexpress TERT (Fig. 1A). This overexpression has been
implicated in the development of GBM and associated with lower
overall survival and poor prognosis (150–152). By analyzing the genomic landscape
of gliomas, Killela et al (153) showed that TERT promoter mutations
are present in 83% of GBM cases. They also discovered that these
mutations are linked to increased telomerase activity and decreased
patient survival (153).
Telomerase, as a target for drug development,
appears to be an attractive candidate due to its pivotal role in
cancer progression. Various anti-telomerase strategies are under
development, including small-molecule enzymatic inhibitors and
indirect approaches. One such inhibitor, BIBR1532, a selective
noncompetitive inhibitor targeting the telomerase thumb domain
(154,155), has shown effectiveness in
vitro by halting cancer cell proliferation and inducing
telomere shortening at high doses (156). However, its poor pharmacokinetic
properties, such as low cellular permeability, have hindered its
clinical advancement (157).
In parallel, eribulin, a mitotic inhibitor, is
undergoing clinical investigation for its potential against GBM. It
has demonstrated potent activity, particularly in TERT
promoter-mutant GBM cells, both in vitro and in vivo,
with effective tumor tissue penetration (158). These promising findings have led
to the initiation of clinical trials exploring its
telomerase-inhibiting potential in GBM (159).
Telomerase-based vaccines, which aim to trigger
anti-tumoral immune responses, have also been in clinical
development for decades (160).
These vaccines utilize telomerase-derived peptides to generate
immune responses in CD8+ cytotoxic lymphocytes, with trials across
various cancers. However, clinical efficacy has been limited,
particularly for GBM, which is resistant to immune checkpoint
blockade (161). For instance, the
GV1001 peptide vaccine trial in advanced pancreatic cancer showed
no survival benefit over chemotherapy alone, underscoring the
challenges in achieving clinical success (162).
To address these challenges, newer generations of
anti-telomerase vaccines, such as INO5401, have been developed.
INO5401 incorporates a modified full-length TERT gene to reduce
immune tolerance and enhance CD8+ T-cell responses (163). In a phase I/II clinical trial,
this vaccine, combined with standard care, showed promising early
results. Specifically, patients withO-6-methylguanine-DNA
methyltransferase (MGMT)-unmethylated tumors had a 12-month overall
survival rate of 84.4%, while those with MGMT-methylated tumors had
a rate of 85%, suggesting potential clinical benefits (164).
In addition, the use of telomerase-specific
oncolytic viruses, which selectively replicate in cancer cells and
express therapeutic agents like TERT-specific siRNAs or
immunostimulatory cytokines, is emerging as a novel approach.
Preclinical studies have shown promising results for these viruses,
and they are currently being assessed in clinical trials
(NCT03491683 and NCT03548571) (73). This combination of strategies
underscores the ongoing efforts to harness telomerase for cancer
therapy, offering new hope for GBM and other cancers.
Conclusion
Over the years, numerous significant molecular
mechanisms underlying the development of gliomas have been
unraveled. These discoveries changed the classification of gliomas
and offered insight into the initiation and progression of these
tumors. GBM stands out as the predominant and highly aggressive
primary brain tumor in adults with a median survival of merely
12–15 months despite treatment efforts. Progress in bioinformatics
has enabled a more thorough comprehension of the genetic
alterations underlying GBM, offering new perspectives of
understanding the development of the disease and novel potential
targets for therapeutic interventions. The analysis of large-scale
genomic datasets, such as TCGA, has been used in several studies to
identify recurrently altered genes in GBM, including EGFR, TP53,
PTEN and IDH1, among others. Further bioinformatics analysis of
these genes revealed information regarding the pathways and
processes disrupted in GBM, including cell cycle control and the
PI3K/AKT/mTOR pathway. Furthermore, the utilization of whole-genome
sequencing has been instrumental in pinpointing recurring mutations
in GBM, such as those found in TP53 and PTEN, alongside less
frequent mutations in other genes like RB1 and NF1. Validating
bioinformatics algorithms in GBM research requires benchmarking
against established datasets like TCGA, cross-validation to
minimize overfitting and external cohort testing to ensure
generalizability. Biological validation through wet-lab experiments
confirms prediction relevance, while reproducibility is supported
by open-source code and transparent workflows. Metrics such as
accuracy, sensitivity, specificity and Area Under Curve are
critical for evaluation, alongside testing on synthetic data and
alignment with known biological pathways. Addressing scalability
and computational efficiency ensures the practical application of
these algorithms in large-scale GBM studies (165–167).
Additional research will continue to use
bioinformatic tools to uncover new insights into GBM and improve
patient outcomes. Bioinformatics is critical to further the
understanding of GBM by allowing the analysis of large amounts of
genomic data and to identify genetic changes that contribute to
tumor development and progression. These discoveries have not only
improved our understanding of GBM biology, but have also led to the
recognition of novel therapeutic targets for treating this cancer.
In conclusion, bioinformatics analysis is a critical tool for
furthering the understanding of GBM and identifying new treatment
strategies for this devastating disease.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
MAG designed the review article and wrote the
majority of the article. NEJ, TM and MM researched references and
contributed to the writing. RAH and MES wrote the final draft and
edited the manuscript. Data authentication is not applicable. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Bauchet L, Davis FG, Deltour I,
Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh
KM, et al: The epidemiology of glioma in adults: A ‘state of the
science’ review. Neuro Oncol. 16:896–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bailey P and Cushing H: Microchemical
color reactions as an aid to the identification and classification
of brain tumors. Proc Natl Acad Sci USA. 11:82–84. 1925. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Masui K, Cloughesy TF and Mischel PS:
Review: Molecular pathology in adult high-grade gliomas: From
molecular diagnostics to target therapies. Neuropathol Appl
Neurobiol. 38:271–291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van den Bent MJ: Interobserver variation
of the histopathological diagnosis in clinical trials on glioma: A
clinician's perspective. Acta Neuropathol. 120:297–304. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen R, Smith-Cohn M, Cohen AL and Colman
H: Glioma subclassifications and their clinical significance.
Neurotherapeutics. 14:284–297. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry A and Wesseling P: Histologic
classification of gliomas. Handb Clin Neurol. 134:71–95. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cancer Genome Atlas Research Network, .
Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA,
Rheinbay E, Miller CR, Vitucci M, et al: Comprehensive, integrative
genomic analysis of diffuse lower-grade gliomas. N Engl J Med.
372:2481–2498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kleihues P, Burger PC and Scheithauer BW:
The new WHO classification of brain tumours. Brain Pathol.
3:255–268. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Neftel C, Laffy J, Filbin MG, Hara T,
Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM,
et al: An integrative model of cellular states, plasticity, and
genetics for glioblastoma. Cell. 178:835–849.e21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Couturier CP, Ayyadhury S, Le PU, Nadaf J,
Monlong J, Riva G, Allache R, Baig S, Yan X, Bourgey M, et al:
Single-cell RNA-seq reveals that glioblastoma recapitulates a
normal neurodevelopmental hierarchy. Nat Commun. 11:34062020.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martínez AH, Madurga R, García-Romero N
and Ayuso-Sacido Á: Unraveling glioblastoma heterogeneity by means
of single-cell RNA sequencing. Cancer Lett. 527:66–79. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martinez R, Schackert HK, Plaschke J,
Baretton G, Appelt H and Schackert G: Molecular mechanisms
associated with chromosomal and microsatellite instability in
sporadic glioblastoma multiforme. Oncology. 66:395–403. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tepeoglu M, Borcek P, Ozen O and Altinors
N: Microsatellite instability in glioblastoma: Is it really
relevant in tumor prognosis? Turk Neurosurg. 29:778–784.
2019.PubMed/NCBI
|
|
17
|
Agnihotri S and Zadeh G: Metabolic
reprogramming in glioblastoma: The influence of cancer metabolism
on epigenetics and unanswered questions. Neuro Oncol. 18:160–172.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deshmukh R, Allega MF and Tardito S: A map
of the altered glioma metabolism. Trends Mol Med. 27:1045–1059.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Virtuoso A, Giovannoni R, De Luca C,
Gargano F, Cerasuolo M, Maggio N, Lavitrano M and Papa M: The
glioblastoma microenvironment: Morphology, metabolism, and
molecular signature of glial dynamics to discover metabolic
rewiring sequence. Int J Mol Sci. 22:33012021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee H, Kim D and Youn B: Targeting
oncogenic rewiring of lipid metabolism for glioblastoma treatment.
Int J Mol Sci. 23:138182022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Andersen RS, Anand A, Harwood DSL and
Kristensen BW: Tumor-associated microglia and macrophages in the
glioblastoma microenvironment and their implications for therapy.
Cancers (Basel). 13:42552021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park JH and Lee HK: Current understanding
of hypoxia in glioblastoma multiforme and its response to
immunotherapy. Cancers (Basel). 14:11762022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lamborn KR, Chang SM and Prados MD:
Prognostic factors for survival of patients with glioblastoma:
Recursive partitioning analysis. Neuro Oncol. 6:227–235. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stoyanov GS, Dzhenkov D, Ghenev P, Iliev
B, Enchev Y and Tonchev AB: Cell biology of glioblastoma
multiforme: From basic science to diagnosis and treatment. Med
Oncol. 35:272018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1608. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Godek KM, Venere M, Wu Q, Mills KD, Hickey
WF, Rich JN and Compton DA: Chromosomal instability affects the
tumorigenicity of glioblastoma tumor-initiating cells. Cancer
Discov. 6:532–545. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Buruiană A, Florian ȘI, Florian AI, Timiș
TL, Mihu CM, Miclăuș M, Oșan S, Hrapșa I, Cataniciu RC, Farcaș M,
et al: The roles of miRNA in glioblastoma tumor cell communication:
Diplomatic and aggressive negotiations. Int J Mol Sci. 21:19502020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mafi A, Rahmati A, Aghdam ZB, Salami R,
Salami M, Vakili O and Aghadavod E: Recent insights into the
microRNA-dependent modulation of gliomas from pathogenesis to
diagnosis and treatment. Cell Mol Biol Lett. 27:652022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tluli O, Al-Maadhadi M, Al-Khulaifi AA,
Akomolafe AF, Al-Kuwari SY, Al-Khayarin R, Maccalli C and Pedersen
S: Exploring the role of microRNAs in glioma progression,
prognosis, and therapeutic strategies. Cancers (Basel).
15:42132023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stackhouse CT, Gillespie GY and Willey CD:
Exploring the roles of lncRNAs in GBM pathophysiology and their
therapeutic potential. Cells. 9:23692020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bagheri-Mohammadi S, Karamivandishi A,
Mahdavi SA and Siahposht-Khachaki A: New sights on long non-coding
RNAs in glioblastoma: A review of molecular mechanism. Heliyon.
10:e397442024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hashemi M, Roshanzamir SM, Orouei S,
Daneii P, Raesi R, Zokaee H, Bikarannejad P, Salmani K, Khorrami R,
Paskeh MDA, et al: Shedding light on function of long non-coding
RNAs (lncRNAs) in glioblastoma. Noncoding RNA Res. 9:508–522. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brat DJ, Aldape K, Colman H, Holland EC,
Louis DN, Jenkins RB, Kleinschmidt-DeMasters BK, Perry A,
Reifenberger G, Stupp R, et al: cIMPACT-NOW update 3: Recommended
diagnostic criteria for ‘Diffuse astrocytic glioma, IDH-wildtype,
with molecular features of glioblastoma, WHO grade IV’. Acta
Neuropathol. 136:805–810. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tesileanu CMS, Dirven L, Wijnenga MMJ,
Koekkoek JAF, Vincent AJPE, Dubbink HJ, Atmodimedjo PN, Kros JM,
van Duinen SG, Smits M, et al: Survival of diffuse astrocytic
glioma, IDH1/2 wildtype, with molecular features of glioblastoma,
WHO grade IV: A confirmation of the cIMPACT-NOW criteria. Neuro
Oncol. 4:515–523. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang B, Gu X, Han X, Gao Q, Liu J, Guo T
and Gao D: Crosstalk between DNA methylation and histone
acetylation triggers GDNF high transcription in glioblastoma cells.
Clin Epigenetics. 12:472020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Azab MA: The potential role of histone
modifications in glioblastoma therapy: Review article. J Mol
Pathol. 4:Article 4. 2023. View Article : Google Scholar
|
|
39
|
McCornack C, Woodiwiss T, Hardi A, Yano H
and Kim AH: The function of histone methylation and acetylation
regulators in GBM pathophysiology. Front Oncol. 13:11441842023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rama AR, Alvarez PJ, Madeddu R and Aranega
A: ABC transporters as differentiation markers in glioblastoma
cells. Mol Biol Rep. 41:4847–4851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ahmed M, Verreault M, Declèves X and
Idbaih A: Role of multidrug resistance in glioblastoma
chemoresistance: Focus on ABC transporters. Cancer sensitizing
agents for chemotherapy, glioblastoma resistance to chemotherapy:
Molecular mechanisms and innovative reversal strategies.
Paulmurugan R and Massoud TF: Academic Press; 15. pp. 243–261.
2021
|
|
42
|
Canzoneri R, Lacunza E and Abba MC:
Genomics and bioinformatics as pillars of precision medicine in
oncology. Medicina (B Aires). 79:587–592. 2019.PubMed/NCBI
|
|
43
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu P, Heins ZJ, Muller JT, Katsnelson L,
de Bruijn I, Abeshouse AA, Schultz N, Fenyö D and Gao J:
Integration and analysis of CPTAC proteomics data in the context of
cancer genomics in the cBioPortal. Mol Cell Proteomics.
18:1893–1898. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brlek P, Kafka A, Bukovac A and
Pećina-Šlaus N: Integrative cBioPortal analysis revealed molecular
mechanisms that regulate EGFR-PI3K-AKT-mTOR pathway in diffuse
gliomas of the brain. Cancers (Basel). 13:32472021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ahsan H, Asghar M and Malik SI: Potential
diagnostic and drug target markers in glioblastoma. Sci Rep.
14:72922024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dhar C: Utilizing publicly available
cancer clinicogenomic data on cBioPortal to compare epidermal
growth factor receptor mutant and wildtype non-small cell lung
cancer. Cureus. 13:e146832021.PubMed/NCBI
|
|
48
|
Reimer N, Unberath P, Busch H, Börries M,
Metzger P, Ustjanzew A, Renner C, Prokosch HU and Christoph J:
Challenges and experiences extending the cBioPortal for cancer
genomics to a molecular tumor board platform. Stud Health Technol
Inform. 18:139–143. 2021.PubMed/NCBI
|
|
49
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Methods Mol Biol. 696:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Donaldson IM: Protein interaction data
resources. Handbook of cell signaling. 2nd Edition. Academic Press;
San Diego, CA: pp. 1375–1385. 2010, View Article : Google Scholar
|
|
52
|
Jean-Quartier C, Jeanquartier F and
Holzinger A: Open data for differential network analysis in glioma.
Int J Mol Sci. 21:5472020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ma T and Zhang A: Integrate multi-omics
data with biological interaction networks using multi-view
factorization AutoEncoder (MAE). BMC Genomics. 20 (Suppl
11):S9442019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Picard M, Scott-Boyer MP, Bodein A, Périn
O and Droit A: Integration strategies of multi-omics data for
machine learning analysis. Comput Struct Biotechnol J.
19:3735–3746. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sanches PHG, de Melo NC, Porcari AM and de
Carvalho LM: Integrating molecular perspectives: Strategies for
comprehensive multi-omics integrative data analysis and machine
learning applications in transcriptomics, proteomics, and
metabolomics. Biology (Basel). 13:8482024.PubMed/NCBI
|
|
56
|
Zhou Y, Yang L, Zhang X, Chen R, Chen X,
Tang W and Zhang M: Identification of potential biomarkers in
glioblastoma through bioinformatic analysis and evaluating their
prognostic value. Biomed Res Int. 2019:65815762019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Erasimus H, Gobin M, Niclou S and Van Dyck
E: DNA repair mechanisms and their clinical impact in glioblastoma.
Mutat Res Rev Mutat Res. 769:19–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu Y, Song Y, Wang R and Wang T: Molecular
mechanisms of tumor resistance to radiotherapy. Mol Cancer.
22:962023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stockhausen MT, Kristoffersen K and
Poulsen HS: The functional role of notch signaling in human
gliomas. Neuro Oncol. 12:199–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bazzoni R and Bentivegna A: Role of notch
signaling pathway in glioblastoma pathogenesis. Cancers (Basel).
11:2922019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stenson PD, Mort M, Ball EV, Shaw K,
Phillips A and Cooper DN: The human gene mutation database:
Building a comprehensive mutation repository for clinical and
molecular genetics, diagnostic testing and personalized genomic
medicine. Hum Genet. 133:1–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
1,000 Genomes Project Consortium, .
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker
RE, Kang HM, Marth GT and McVean GA: An integrated map of genetic
variation from 1,092 human genomes. Nature. 491:56–65. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Garcia FAO, de Andrade ES and Palmero EI:
Insights on variant analysis in silico tools for pathogenicity
prediction. Front Genet. 13:10103272022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Steinhaus R, Proft S, Schuelke M, Cooper
DN, Schwarz JM and Seelow D: MutationTaster2021. Nucleic Acids Res.
49:W446–W451. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Crespo I, Vital AL, Gonzalez-Tablas M,
Patino MDC, Otero A, Lopes MC, de Oliveira C, Domingues P, Orfao A
and Tabernero MD: Molecular and genomic alterations in glioblastoma
multiforme. Am J Pathol. 185:1820–1833. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Huang PH, Xu AM and White FM: Oncogenic
EGFR signaling networks in glioma. Sci Signal. 2:re62009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Binder ZA, Thorne AH, Bakas S, Wileyto EP,
Bilello M, Akbari H, Rathore S, Ha SM, Zhang L, Ferguson CJ, et al:
Epidermal growth factor receptor extracellular domain mutations in
glioblastoma present opportunities for clinical imaging and
therapeutic development. Cancer Cell. 34:163–177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lu KV, Zhu S, Cvrljevic A, Huang TT,
Sarkaria S, Ahkavan D, Dang J, Dinca EB, Plaisier SB, Oderberg I,
et al: Fyn and SRC are effectors of oncogenic epidermal growth
factor receptor signaling in glioblastoma patients. Cancer Res.
69:6889–6898. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nagane M, Coufal F, Lin H, Bögler O,
Cavenee WK and Huang HJ: A common mutant epidermal growth factor
receptor confers enhanced tumorigenicity on human glioblastoma
cells by increasing proliferation and reducing apoptosis. Cancer
Res. 56:5079–5086. 1996.PubMed/NCBI
|
|
70
|
Jain A, Penuel E, Mink S, Schmidt J, Hodge
A, Favero K, Tindell C and Agus DB: HER kinase axis receptor dimer
partner switching occurs in response to EGFR tyrosine kinase
inhibition despite failure to block cellular proliferation. Cancer
Res. 70:1989–1999. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sathornsumetee S and Rich JN: Designer
therapies for glioblastoma multiforme. Ann N Y Acad Sci.
1142:108–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang TT, Sarkaria SM, Cloughesy TF and
Mischel PS: Targeted therapy for malignant glioma patients: Lessons
learned and the road ahead. Neurotherapeutics. 6:500–512. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
El Atat O, Naser R, Abdelkhalek M, Habib
RA and El Sibai M: Molecular targeted therapy: A new avenue in
glioblastoma treatment. Oncol Lett. 25:462022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ezzati S, Salib S, Balasubramaniam M and
Aboud O: Epidermal growth factor receptor inhibitors in
glioblastoma: Current status and future possibilities. Int J Mol
Sci. 25:23162024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vivanco I, Robins HI, Rohle D, Campos C,
Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N,
et al: Differential sensitivity of glioma-versus lung
cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer
Discov. 2:458–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hegi ME, Diserens AC, Bady P, Kamoshima Y,
Kouwenhoven MCM, Delorenzi M, Lambiv WL, Hamou MF, Matter MS, Koch
A, et al: Pathway analysis of glioblastoma tissue after
preoperative treatment with the EGFR tyrosine kinase inhibitor
gefitinib-A phase II trial. Mol Cancer Ther. 10:1102–1112. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Neyns B, Sadones J, Joosens E, Bouttens F,
Verbeke L, Baurain JF, D'Hondt L, Strauven T, Chaskis C, Veld PI,
et al: Stratified phase II trial of cetuximab in patients with
recurrent high-grade glioma. Ann Oncol. 20:1596–1603. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Westphal M, Heese O, Steinbach JP, Schnell
O, Schackert G, Mehdorn M, Schulz D, Simon M, Schlegel U, Senft C,
et al: A randomized, open-label phase III trial with nimotuzumab,
an anti-epidermal growth factor receptor monoclonal antibody in the
treatment of newly diagnosed adult glioblastoma. Eur J Cancer.
51:522–532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nitta Y, Shimizu S, Shishido-Hara Y,
Suzuki K, Shiokawa Y and Nagane M: Nimotuzumab enhances
temozolomide-induced growth suppression of glioma cells expressing
mutant EGFR in vivo. Cancer Med. 5:486–499. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hasselbalch B, Lassen U, Hansen S,
Holmberg M, Sorensen M, Kosteljanetz M, Broholm H, Stockhausen MT
and Poulsen HS: Cetuximab, bevacizumab, and irinotecan for patients
with primary glioblastoma and progression after radiation therapy
and temozolomide: A phase II trial. Neuro Oncol. 12:508–516.
2010.PubMed/NCBI
|
|
81
|
McCrea HJ, Ivanidze J, O'Connor A, Hersh
EH, Boockvar JA, Gobin YP, Knopman J and Greenfield JP:
Intraarterial delivery of bevacizumab and cetuximab utilizing
blood-brain barrier disruption in children with high-grade glioma
and diffuse intrinsic pontine glioma: Results of a phase I trial. J
Neurosurg Pediatr. 28:371–379. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pérez-Soler R, Delord JP, Halpern A, Kelly
K, Krueger J, Sureda BM, von Pawel J, Temel J, Siena S, Soulières
D, et al: HER1/EGFR inhibitor-associated rash: Future directions
for management and investigation outcomes from the HER1/EGFR
inhibitor rash management forum. Oncologist. 10:345–356. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Thomas M: Cetuximab: Adverse event profile
and recommendations for toxicity management. Clin J Oncol Nurs.
9:332–338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Keir ST, Chandramohan V, Hemphill CD,
Grandal MM, Melander MC, Pedersen MW, Horak ID, Kragh M, Desjardins
A, Friedman HS and Bigner DD: Sym004-induced EGFR elimination is
associated with profound anti-tumor activity in EGFRvIII
patient-derived glioblastoma models. J Neurooncol. 138:489–498.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Shikalov A, Koman I and Kogan NM: Targeted
glioma therapy-clinical trials and future directions.
Pharmaceutics. 16:1002024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lim Y, Yoo J, Kim MS, Hur M, Lee EH, Hur
HS, Lee JC, Lee SN, Park TW, Lee K, et al: GC1118, an anti-EGFR
antibody with a distinct binding epitope and superior inhibitory
activity against high-affinity EGFR ligands. Mol Cancer Ther.
15:251–263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Choi SW, Jung HA, Cho H, Kim TM, Park CK,
Nam DH and Lee SH: A multicenter, phase II trial of GC1118, a novel
anti-EGFR antibody, for recurrent glioblastoma patients with EGFR
amplification. Cancer Med. 12:15788–15796. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lassman AB, van den Bent MJ, Gan HK,
Reardon DA, Kumthekar P, Butowski N, Lwin Z, Mikkelsen T, Nabors
LB, Papadopoulos KP, et al: Safety and efficacy of depatuxizumab
mafodotin + temozolomide in patients with EGFR-amplified, recurrent
glioblastoma: Results from an international Phase I multicenter
trial. Neuro Oncol. 21:106–114. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Desjardins A, Chandramohan V, Landi DB,
Johnson MO, Khasraw M, Peters KB, Low J, Herndon JE, Threatt S,
Bullock CA, et al: A phase I trial of D2C7-IT in combination with
an Fc-engineered anti-CD40 monoclonal antibody (2141-V11)
administered intratumorally via convection-enhanced delivery for
adult patients with recurrent malignant glioma (MG). J Clin Oncol.
40 (Suppl):e140152022. View Article : Google Scholar
|
|
90
|
Kowalewski A, Durślewicz J, Zdrenka M,
Grzanka D and Szylberg Ł: Clinical relevance of BRAF V600E mutation
status in brain tumors with a focus on a novel management
algorithm. Target Oncol. 15:531–540. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Etcheverry A, Aubry M, de Tayrac M,
Vauleon E, Boniface R, Guenot F, Saikali S, Hamlat A, Riffaud L,
Menei P, et al: DNA methylation in glioblastoma: Impact on gene
expression and clinical outcome. BMC Genomics. 11:7012010.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Klughammer J, Kiesel B, Roetzer T,
Fortelny N, Kuchler A, Nenning KH, Furtner J, Sheffield NC,
Datlinger P, Peter N, et al: The DNA methylation landscape of
glioblastoma disease progression shows extensive heterogeneity in
time and space. Nat Med. 24:1611–1624. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Mao YK, Liu ZB and Cai L: Identification
of glioblastoma-specific prognostic biomarkers via an integrative
analysis of DNA methylation and gene expression. Oncol Lett.
20:1619–1628. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sun X, Yuan W, Hao F and Zhuang W:
Promoter methylation of RASSF1A indicates prognosis for patients
with stage II and III colorectal cancer treated with
oxaliplatin-based chemotherapy. Med Sci Monit. 23:5389–5395. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Szklener K, Mazurek M, Wieteska M,
Wacławska M, Bilski M and Mańdziuk S: New directions in the therapy
of glioblastoma. Cancers (Basel). 14:53772022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bouchè V, Aldegheri G, Donofrio CA,
Fioravanti A, Roberts-Thomson S, Fox SB, Schettini F and Generali
D: BRAF signaling inhibition in glioblastoma: Which clinical
perspectives? Front Oncol. 11:7720522021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Shannon S, Jia D, Entersz I, Beelen P, Yu
M, Carcione C, Carcione J, Mahtabfar A, Vaca C, Weaver M, et al:
Inhibition of glioblastoma dispersal by the MEK inhibitor
PD0325901. BMC Cancer. 17:1212017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Peng P, Wei W, Long C and Li J:
Atorvastatin augments temozolomide's efficacy in glioblastoma via
prenylation-dependent inhibition of ras signaling. Biochem Biophys
Res Commun. 489:293–298. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Altwairgi AK, Alghareeb WA, AlNajjar FH,
Alhussain H, Alsaeed E, Balbaid AAO, Aldanan S, Orz Y and Alsharm
AA: Atorvastatin in combination with radiotherapy and temozolomide
for glioblastoma: A prospective phase II study. Invest New Drugs.
39:226–231. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ranjbar R, Mohammadpour S, Esfahani AT,
Namazian S, Yaghob-Taleghani M, Baghaei K, Tabatabaei SA,
Pasharavesh L and Nazemalhosseini-Mojarad E: Prevalence and
prognostic role of PIK3CA E545K mutation in iranian colorectal
cancer patients. Gastroenterol Hepatol Bed Bench. 12:S22–S29.
2019.PubMed/NCBI
|
|
103
|
Liang J and Slingerland JM: Multiple roles
of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell
Cycle. 2:339–345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zuccarini M, Giuliani P, Ziberi S,
Carluccio M, Iorio PD, Caciagli F and Ciccarelli R: The role of Wnt
signal in glioblastoma development and progression: A possible new
pharmacological target for the therapy of this tumor. Genes
(Basel). 9:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dreyer CA, VanderVorst K, Natwick D, Bell
G, Sood P, Hernandez M, Angelastro JM, Collins SR and Carraway KL:
A complex of Wnt/planar cell polarity signaling components Vangl1
and Fzd7 drives glioblastoma multiforme malignant properties.
Cancer Lett. 567:2162802023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Mueller S, Phillips J, Onar-Thomas A,
Romero E, Zheng S, Wiencke JK, McBride SM, Cowdrey C, Prados MD,
Weiss WA, et al: PTEN promoter methylation and activation of the
PI3K/Akt/mTOR pathway in pediatric gliomas and influence on
clinical outcome. Neuro Oncol. 14:1146–1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chang H, Cai Z and Roberts TM: The
Mechanisms underlying PTEN loss in human tumors suggest potential
therapeutic opportunities. Biomolecules. 9:7132019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Carico C, Nuño M, Mukherjee D, Elramsisy
A, Dantis J, Hu J, Rudnick J, Yu JS, Black KL, Bannykh SI and Patil
CG: Loss of PTEN is not associated with poor survival in newly
diagnosed glioblastoma patients of the temozolomide era. PLoS One.
7:e336842012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhao HF, Wang J, Shao W, Wu CP, Chen ZP,
To ST and Li WP: Recent advances in the use of PI3K inhibitors for
glioblastoma multiforme: Current preclinical and clinical
development. Mol Cancer. 16:1002017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wen PY, Wen PY, Yung WKA, Mellinghoff IK,
Ramkissoon S, Alexander B, Rinne M, Colman H, Omuro AM, DeAngelis
LM, et al: Rockich Xu Lager Mellinghoff Phase II trial of the
phosphatidyinositol-3 kinase (PI3K) inhibitor buparlisib (bkm120)
in recurrent glioblastoma conducted by the ivy foundation early
phase clinical trials consortium. Neuro Oncol. 16 (Suppl
3):iii472014. View Article : Google Scholar
|
|
111
|
Cloughesy TF, Mischel PS, Omuro AMP,
Prados M, Wen PY and Wu B K Y JJ IK: Tumor pharmacokinetics (PK)
and pharmacodynamics (PD) of SAR245409 (XL765) and SAR245408
(XL147) administered as single agents to patients with recurrent
glioblastoma (GBM): An Ivy Foundation early-phase clinical trials
consortium study. J Clin Oncol. 31:2012. 2013. View Article : Google Scholar
|
|
112
|
Papavassiliou KA and Papavassiliou AG: The
bumpy road towards mTOR inhibition in glioblastoma: Quo vadis?
Biomedicines. 9:18092021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
England B, Huang T and Karsy M: Current
understanding of the role and targeting of tumor suppressor p53 in
glioblastoma multiforme. Tumour Biol. 34:2063–2074. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ohgaki H: Genetic pathways to
glioblastomas. Neuropathology. 25:1–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kim YW, Koul D, Kim SH, Lucio-Eterovic AK,
Freire PR, Yao J, Wang J, Almeida JS, Aldape K and Yung WK:
Identification of prognostic gene signatures of glioblastoma: A
study based on TCGA data analysis. Neuro Oncol. 15:829–839. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Kyritsis AP, Bondy ML, Rao JS and Sioka C:
Inherited predisposition to glioma. Neuro Oncol. 12:104–113. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Rice T, Lachance DH, Molinaro AM,
Eckel-Passow JE, Walsh KM, Barnholtz-Sloan J, Ostrom QT, Francis
SS, Wiemels J, Jenkins RB, et al: Understanding inherited genetic
risk of adult glioma: A review. Neurooncol Pract. 3:10–16.
2016.PubMed/NCBI
|
|
118
|
Mohyeldin A and Chiocca EA: Gene and viral
therapy for glioblastoma: A review of clinical trials and future
directions. Cancer J. 18:82–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Olafson LR, Gunawardena M, Nixdorf S,
McDonald KL and Rapkins RW: The role of TP53 gain-of-function
mutation in multifocal glioblastoma. J Neurooncol. 147:37–47. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chow LM, Endersby R, Zhu X, Rankin S, Qu
C, Zhang J, Broniscer A, Ellison DW and Baker SJ: Cooperativity
within and among Pten, p53, and Rb pathways induces high-grade
astrocytoma in adult brain. Cancer Cell. 19:305–316. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhang Y, Dube C, Gibert M Jr, Cruickshanks
N, Wang B, Coughlan M, Yang Y, Setiady I, Deveau C, Saoud K, et al:
The p53 pathway in glioblastoma. Cancers (Basel). 10:2972018.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53. Annu Rev Biochem. 77:557–582.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Bykov VJ, Issaeva N, Shilov A, Hultcrantz
M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G:
Restoration of the tumor suppressor function to mutant p53 by a
low-molecular-weight compound. Nat Med. 8:282–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Lambert JM, Gorzov P, Veprintsev DB,
Soderqvist M, Segerback D, Bergman J, Fersht AR, Hainaut P, Wiman
KG and Bykov VJ: PRIMA-1 reactivates mutant p53 by covalent binding
to the core domain. Cancer Cell. 15:376–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bykov VJ, Zache N, Stridh H, Westman J,
Bergman J, Selivanova G and Wiman KG: PRIMA-1MET synergizes with
cisplatin to induce tumor cell apoptosis. Oncogene. 24:3484–3491.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zache N, Lambert JM, Wiman KG and Bykov
VJ: PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53.
Cell Oncol. 30:411–418. 2008.PubMed/NCBI
|
|
129
|
Patyka M, Sharifi Z, Petrecca K, Mansure
J, Jean-Claude B and Sabri S: Sensitivity to PRIMA-1MET is
associated with decreased MGMT in human glioblastoma cells and
glioblastoma stem cells irrespective of p53 status. Oncotarget.
7:60245–60269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lehmann S, Bykov VJ, Ali D, Andren O,
Cherif H, Tidefelt U, Uggla B, Yachnin J, Juliusson G, Moshfegh A,
et al: Targeting p53 in vivo: A first-in-human study with
p53-targeting compound APR-246 in refractory hematologic
malignancies and prostate cancer. J Clin Oncol. 30:3633–3639. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Li D, Marchenko ND, Schulz R, Fischer V,
Velasco-Hernandez T, Talos F and Moll UM: Functional inactivation
of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization
of mutant p53 in human cancer cells. Mol Cancer Res. 9:577–588.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Singh MM, Johnson B, Venkatarayan A,
Flores ER, Zhang J, Su X, Barton M, Lang F and Chandra J:
Preclinical activity of combined HDAC and KDM1A inhibition in
glioblastoma. Neuro Oncol. 17:1463–1473. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Liffers K, Kolbe K, Westphal M, Lamszus K
and Schulte A: Histone deacetylase inhibitors resensitize
EGFR/EGFRvIII-overexpressing, erlotinib-resistant glioblastoma
cells to tyrosine kinase inhibition. Target Oncol. 11:29–40. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Staberg M, Michaelsen SR, Rasmussen RD,
Villingshoj M, Poulsen HS and Hamerlik P: Inhibition of histone
deacetylases sensitizes glioblastoma cells to lomustine. Cell Oncol
(Dordr). 40:21–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Pal S, Kozono D, Yang X, Fendler W, Fitts
W, Ni J, Alberta JA, Zhao J, Liu KX, Bian J, et al: Dual HDAC and
PI3K inhibition abrogates NFκB- and FOXM1-mediated DNA damage
response to radiosensitize pediatric high-grade gliomas. Cancer
Res. 78:4007–4021. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Kitange GJ, Mladek AC, Carlson BL,
Schroeder MA, Pokorny JL, Cen L, Decker PA, Wu W, Lomberk GA, Gupta
SK, et al: Inhibition of histone deacetylation potentiates the
evolution of acquired temozolomide resistance linked to MGMT
upregulation in glioblastoma xenografts. Clin Cancer Res.
18:4070–4079. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Rasmussen RD, Gajjar MK, Jensen KE and
Hamerlik P: Enhanced efficacy of combined HDAC and PARP targeting
in glioblastoma. Mol Oncol. 10:751–763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Choi SA, Kwak PA, Park CK, Wang KC, Phi
JH, Lee JY, Lee CS, Lee JH and Kim SK: A novel histone deacetylase
inhibitor, CKD5, has potent anti-cancer effects in glioblastoma.
Oncotarget. 8:9123–9133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Yang XF, Zhao ZJ, Liu JJ, Yang XH, Gao Y,
Zhao S, Shi S, Huang KQ and Zheng HC: SAHA and/or MG132 reverse the
aggressive phenotypes of glioma cells: An in vitro and vivo study.
Oncotarget. 8:3156–3169. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Reifenberger G, Liu L, Ichimura K, Schmidt
EE and Collins VP: Amplification and overexpression of the MDM2
gene in a subset of human malignant gliomas without p53 mutations.
Cancer Res. 53:2736–2739. 1993.PubMed/NCBI
|
|
141
|
Riemenschneider MJ, Büschges R, Wolter M,
Reifenberger J, Boström J, Kraus JA, Schlegel U and Reifenberger G:
Amplification and overexpression of the MDM4 (MDMX) gene from 1q32
in a subset of malignant gliomas without TP53 mutation or MDM2
amplification. Cancer Res. 59:6091–6096. 1999.PubMed/NCBI
|
|
142
|
Biernat W, Kleihues P, Yonekawa Y and
Ohgaki H: Amplification and overexpression of MDM2 in primary (de
novo) glioblastomas. J Neuropathol Exp Neurol. 56:180–185. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wade M, Li YC and Wahl GM: MDM2, MDMX and
p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 13:83–96.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Vu B, Wovkulich P, Pizzolato G, Lovey A,
Ding Q, Jiang N, Liu JJ, Zhao C, Glenn K, Wen Y, et al: Discovery
of RG7112: A small-molecule MDM2 inhibitor in clinical development.
ACS Med Chem Lett. 4:466–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Miles X, Vandevoorde C, Hunter A and
Bolcaen J: MDM2/X inhibitors as radiosensitizers for glioblastoma
targeted therapy. Front Oncol. 11:7034422021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Canon J, Osgood T, Olson SH, Saiki AY,
Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, et al: The
MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and
potentiates the activity of p53-inducing cytotoxic agents. Mol
Cancer Ther. 14:649–658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Her NG, Oh JW, Oh YJ, Han S, Cho HJ, Lee
Y, Ryu GH and Nam DH: Potent effect of the MDM2 inhibitor AMG232 on
suppression of glioblastoma stem cells. Cell Death Dis. 9:7922018.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Wang H, Cai S, Bailey BJ, Saadatzadeh MR,
Ding J, Tonsing-Carter E, Georgiadis TM, Gunter TZ, Long EC, Minto
RE, et al: Combination therapy in a xenograft model of
glioblastoma: Enhancement of the antitumor activity of temozolomide
by an MDM2 antagonist. J Neurosurg. 126:446–459. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Daniele S, Taliani S, Da Pozzo E,
Giacomelli C, Costa B, Trincavelli ML, Rossi L, La Pietra V,
Barresi E, Carotenuto A, et al: Apoptosis therapy in cancer: The
first single-molecule co-activating p53 and the translocator
protein in glioblastoma. Sci Rep. 4:47492014. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Mosrati MA, Malmström A, Lysiak M,
Krysztofiak A, Hallbeck M, Milos P, Hallbeck AL, Bratthäll C,
Strandéus M, Stenmark-Askmalm M and Söderkvist P: TERT promoter
mutations and polymorphisms as prognostic factors in primary
glioblastoma. Oncotarget. 6:16663–16673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Xu J, Xu FP, Liu ZH, Cui Q, Zhang KP and
Li Z: The correlation analysis of TERT promoter mutations with
IDH1/2 mutations and 1p/19q detected in human gliomas. Medicine
(Baltimore). 101:e296682022. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Yaltirik CK, Yilmaz SG, Ozdogan S, Bilgin
EY, Barut Z, Ture U and Isbir T: Determination of IDH1, IDH2, MGMT,
TERT and ATRX gene mutations in glial tumors. In Vivo.
36:1694–1702. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda
C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL,
Giovanella BC, et al: TERT promoter mutations occur frequently in
gliomas and a subset of tumors derived from cells with low rates of
self-renewal. Proc Natl Acad Sci USA. 110:6021–6026. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Pascolo E, Wenz C, Lingner J, Hauel N,
Priepke H, Kauffmann I, Garin-Chesa P, Rettig WJ, Damm K and
Schnapp A: Mechanism of human telomerase inhibition by BIBR1532, a
synthetic, non-nucleosidic drug candidate. J Biol Chem.
277:15566–15572. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Bryan C, Rice C, Hoffman H, Harkisheimer
M, Sweeney M and Skordalakes E: Structural basis of telomerase
inhibition by the highly specific BIBR1532. Structure.
23:1934–1942. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Nakashima M, Nandakumar J, Sullivan KD,
Espinosa JM and Cech TR: Inhibition of telomerase recruitment and
cancer cell death. J Biol Chem. 288:33171–33180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Aquilanti E, Kageler L, Wen PY and
Meyerson M: Telomerase as a therapeutic target in glioblastoma.
Neuro Oncol. 23:2004–2013. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Takahashi M, Miki S, Fujimoto K, Fukuoka
K, Matsushita Y, Maida Y, Yasukawa M, Hayashi M, Shinkyo R, Kikuchi
K, et al: Eribulin penetrates brain tumor tissue and prolongs
survival of mice harboring intracerebral glioblastoma xenografts.
Cancer Sci. 110:2247–2257. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Takahashi M, Miki S, Fukuoka K, Maida Y,
Hayashi M, Hamada A, Nishikawa R, Nagane M, Maruyama T, Mukasa A,
et al: EXTH-50. Development of investigator initiated clinical
trial of TERT-targeting therapy using eribulin mesylate in patients
with recurrent glioblastoma. Neuro Oncol. 19 (Suppl 6):vi832017.
View Article : Google Scholar
|
|
160
|
Zanetti M: A second chance for telomerase
reverse transcriptase in anticancer immunotherapy. Nat Rev Clin
Oncol. 14:115–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Reardon DA, Brandes AA, Omuro A,
Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr
O, et al: Effect of nivolumab vs bevacizumab in patients with
recurrent glioblastoma: The CheckMate 143 phase 3 randomized
clinical trial. JAMA Oncol. 6:1003–1010. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Middleton G, Silcocks P, Cox T, Valle J,
Wadsley J, Propper D, Coxon F, Ross P, Madhusudan S, Roques T, et
al: Gemcitabine and capecitabine with or without telomerase peptide
vaccine GV1001 in patients with locally advanced or metastatic
pancreatic cancer (TeloVac): An open-label, randomised, phase 3
trial. Lancet Oncol. 15:829–840. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Yan J, Pankhong P, Shin TH, Obeng-Adjei N,
Morrow MP, Walters JN, Khan AS, Sardesai NY and Weiner DB: Highly
optimized DNA vaccine targeting human telomerase reverse
transcriptase stimulates potent antitumor immunity. Cancer Immunol
Res. 1:179–189. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Reardon DA, Brem S, Desai AS, Bagley SJ,
Kurz SC, De La Fuente MI, Nagpal S, Welch MR, Hormigo A, Forsyth P,
et al: LTBK-01. INO-5401 AND INO-9012 delivered intramuscularly
(IM) with electroporation (EP) in combination with cemiplimab
(REGN2810) in newly diagnosed glioblastoma. Neuro Oncol.
22:ii2372020. View Article : Google Scholar
|
|
165
|
Richters MM, Xia H, Campbell KM,
Gillanders WE, Griffith OL and Griffith M: Best practices for
bioinformatic characterization of neoantigens for clinical utility.
Genome Med. 11:562019. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
You Y, Ru X, Lei W, Li T, Xiao M, Zheng H,
Chen Y and Zhang L: Developing the novel bioinformatics algorithms
to systematically investigate the connections among survival time,
key genes and proteins for Glioblastoma multiforme. BMC
Bioinformatics. 21 (Suppl 13):S3832020. View Article : Google Scholar
|
|
167
|
Shi J: Machine learning and bioinformatics
approaches for classification and clinical detection of bevacizumab
responsive glioblastoma subtypes based on miRNA expression. Sci
Rep. 12:86852022. View Article : Google Scholar : PubMed/NCBI
|