Introduction
Skin cutaneous melanoma (SKCM) is a highly
aggressive malignancy with strong invasive potential and a markedly
high mortality rate (0.6% of all patients with stage-4 melanoma)
(1). It tends to progress rapidly,
frequently metastasizing to regional lymph nodes and distant
organs. The risk of recurrence is positively associated with the
clinical stage at diagnosis (1,2).
Research indicates that early-stage melanoma is often treatable
with radical surgical excision alone, without the need for adjuvant
therapy (3). However, in patients
with lymph node involvement, the recurrence rate remains high even
after complete resection. Notably, the 5-year survival rate for
patients with metastatic SKCM is only ~27% (2). Therefore, elucidating the mechanisms
underlying melanoma metastasis is essential for improving early
diagnosis, prognostic evaluation and the development of novel
targeted therapies. A recent comprehensive review highlighted the
evolving molecular subclasses of melanoma and emerging roles of
vitamin D signaling in tumor-immune crosstalk (4).
The skin functions as an integrated
neuro-immuno-endocrine organ that senses environmental cues and
coordinates systemic homeostasis (5). Ultraviolet radiation, beyond its
carcinogenic potential, modulates cutaneous vitamin D synthesis,
neuropeptide release and local immunoregulation, a concept termed
‘photo-neuro-immuno-endocrinology’ (6). These layers of regulation provide
additional context for understanding melanoma biology.
N6-methyladenosine (m6A), the most abundant internal modification
of eukaryotic mRNA, serves a pivotal role in regulating mRNA
stability, protein translation, viral infection and embryonic
development (7). This modification
is dynamically reversible and is orchestrated by three main classes
of regulators: ‘Writers’ [methyltransferases such as Vir like m6A
methyltransferase associated (VIRMA), methyltransferase m6A complex
catalytic subunit (METTL)3/14, RNA binding motif protein
(RBM)15/15B and Wilms tumor 1 associated protein (WTAP)], ‘erasers’
[demethylases including fat mass and obesity-associated protein
(FTO) and AlkB homolog 5, RNA demethylase (ALKBH5)] and ‘readers’
[m6A-binding proteins such as heterogeneous nuclear
ribonucleoprotein (HNRNP)C, HNRNPA2B1, YTH m6A RNA binding protein
(YTHD)F1/2/3 and YTHDC1/2] (8).
Extensive research has reported that m6A modifications are involved
in a wide range of biological processes, including the heat shock
response, tissue development, DNA damage repair, and stem cell
self-renewal and differentiation (9–12).
Moreover, dysregulation of m6A regulators has been increasingly
implicated in tumor initiation, progression, metastasis and
resistance to therapy (13).
Aberrant m6A modification has been increasingly
recognized as a critical contributor to cancer initiation and
progression. In acute myeloid leukemia (AML), for example, the m6A
methyltransferases METTL14 and METTL3 are markedly upregulated and
notably influence patient prognosis (14,15).
Similarly, the m6A demethylases FTO and ALKBH5 are overexpressed in
several tumor types, where they enhance cancer stemness and promote
oncogenic processes (16–18). In uveal melanoma and conjunctival
melanoma, m6A-mediated methylation of β-secretase 2 has been
reported to trigger intracellular calcium release, thereby
accelerating tumor progression (19). Moreover, Dahal et al
(20) reported that METTL3 is
markedly upregulated in melanoma and promotes cellular invasiveness
by upregulating MMP2 and N-cadherin expression. Despite growing
evidence supporting the role of m6A modifications in tumor
development and prognosis, the specific regulatory mechanisms by
which m6A influences metastasis and invasion in SKCM remain poorly
understood.
Therefore, the aim of the present study was to
uncover the molecular mechanisms by which m6A modification
contributes to SKCM metastasis and to provide a theoretical
foundation for the development of novel therapeutic targets and
prognostic biomarkers. To elucidate the potential role of m6A
modification in SKCM metastasis, the present study integrated
expression profiles from primary and metastatic SKCM samples in the
Gene Expression Omnibus (GEO) database to systematically identify
metastasis-related differentially expressed genes (DEGs) and
associated signaling pathways. A prognostic prediction model was
subsequently constructed using survival data from The Cancer Genome
Atlas (TCGA)-SKCM cohort. Key mRNAs strongly associated with both
m6A regulation and patient prognosis were further screened. In
addition, the present study explored their interactions within a
long noncoding (lnc)RNA-micro (mi)RNA-mRNA competing endogenous
(ce)RNA network and analyzed their correlation with immune cell
infiltration.
Materials and methods
Patients and data collection
Gene expression data for 46 metastatic SKCM samples,
77 primary SKCM samples and 21 normal skin samples were obtained
from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), specifically from
the GSE8401 (21), GSE15605
(22), GSE46517 (23) and GSE65904 (24) datasets. Raw data were
log-transformed and normalized prior to downstream analyses
(Fig. S1). A summary of patient
characteristics is presented in Tables
I and SI. Moreover, RNA
sequencing (RNA-seq) data from the TCGA-SKCM dataset were retrieved
from the University of California, Santa Cruz Xena platform
(https://xena.ucsc.edu/) and converted to
transcripts per million (TPM) using the ‘count2tpm’ R package
(version 4.3.0; http://www.r-project.org/). Following quality control
and filtering for complete survival data, 461/472 patients in the
TCGA-SKCM dataset with matched RNA-seq and survival information
were included in the subsequent analyses.
 | Table I.Sample information. |
Table I.
Sample information.
| Dataset | Total number of
samples | Sample type | Number of
samples |
|---|
| GSE8401 | 83 | Primary
melanoma | 31 |
|
|
| Metastatic
melanoma | 52 |
| GSE15605 | 74 | Primary
melanoma | 46 |
|
|
| Metastatic
melanoma | 12 |
|
|
| Normal skin
samples | 16 |
| GSE46517 | 104 | Primary
melanoma | 31 |
|
|
| Metastatic
melanoma | 73 |
| GSE65904 | 210 | Melanoma | 210 |
In addition, seven pairs of SKCM and adjacent normal
tissue samples were obtained from patients who underwent surgical
treatment at the Institute of Dermatology, Peking Union Medical
College and Chinese Academy of Medical Sciences (Nanjing, China)
between 2018 and 2021. A total of 7 patients, aged 26–92 years (4
males and 3 females) were included. All paraffin-embedded tissue
sections were confirmed as cutaneous melanoma by two or more
dermatopathologists, with cases declining to participate excluded.
The fresh tumor and adjacent tissues were promptly frozen and
stored at −80°C for subsequent analyses. The present study was
approved by the Ethics Committee of the Institute of Dermatology,
Peking Union Medical College and Chinese Academy of Medical
Sciences (approval no. 2017-KY-044), and all procedures were
performed in accordance with approved institutional guidelines.
Written informed consent was obtained from all participants.
Construction of a metastatic SKCM risk
prediction model and survival analysis
TCGA-SKCM cases were classified into primary and
metastatic groups based on patient clinical information. Least
absolute shrinkage and selection operator (LASSO) Cox regression
analysis was performed using the ‘glmnet’ package in R (version
4.3.0; http://www.r-project.org/), with the
optimal λ value determined using 10-fold cross-validation (25). This approach identified the most
predictive gene set, which, combined with primary-metastatic
status, was used to construct a risk score model. Individual risk
scores were calculated for each patient, who were then stratified
into high- and low-risk groups using the median risk score as the
cutoff. Survival differences between the two groups were assessed
using Kaplan-Meier analysis and compared using the log-rank test,
with significance defined as P<0.05. The predictive accuracy of
the risk model was evaluated by receiver operating characteristic
(ROC) curve analysis, and model performance was quantified using
the area under the curve. Internal robustness was assessed using
10-fold cross-validation of the LASSO-Cox model. The resulting risk
scores for each fold and the coefficients selected at the λ_min and
λ_1se penalty parameters are summarized in Table SII. To address potential violations
of the proportional hazard rate assumption due to survival curve
crossover for certain genes, the analyzed time range was restricted
to periods preceding the crossover points. Survival analyses were
re-performed within these truncated time intervals to ensure
robustness of the log-rank test results (26).
Transcriptome sequencing data
bioinformatics analysis
Based on the risk scores generated by the machine
learning model, the TCGA-SKCM cohort was stratified into high- and
low-risk groups. Differential expression analyses were performed to
compare gene expression profiles among primary tumors, metastatic
tumors and normal tissues from the GEO datasets, as well as between
the high- and low-risk groups in the TCGA cohort. For the TCGA-SKCM
RNA-seq data, raw read-count matrices were analyzed using the
‘DESeq2’ package (R version 4.3.0; http://www.r-project.org/) and the median-of-ratios
procedure was used to normalize library size and estimate
dispersion, followed by Wald tests to identify DEGs. Genes with
adjusted P<0.05 and |log2FoldChange (FC)|>1 were
deemed significant. TPM values, calculated using the count2tpm tool
(R version 4.3.0; http://www.r-project.org/), were used only for
downstream visualization (27). For
the GEO datasets, raw or processed expression matrices were
analyzed using the ‘limma’ package (R version 4.3.0; http://www.r-project.org/) with empirical Bayes
moderation to identify DEGs, applying the same adjusted P-value and
fold-change thresholds. DEGs identified from multiple GEO datasets
were then integrated using the ‘RobustRankAggreg’ (RRA) package (R
version 4.3.0; http://www.r–project.org/) (28) to obtain a robust DEG list.
For single-gene differential expression analyses,
thresholds were set at |log2FC|>1 and adjusted P<0.05 to
identify differentially expressed lncRNAs and mRNAs associated with
each of the four candidate genes. Overlapping lncRNAs and mRNAs
were determined using the ‘VennDiagram’ package in R (version
4.3.0; http://www.r-project.org/).
Visualization of DEGs was achieved using volcano plots and heatmaps
generated using the ‘ggplot2’ package (R version 4.3.0; http://www.r-project.org/).
To assess the biological processes and signaling
pathways underlying SKCM metastasis, enrichment analyses were
performed on DEGs identified from GEO datasets comparing primary
and metastatic melanoma, and co-expressed DEGs associated with the
risk-related genes [keratin (KRT)17, plakophilin 1 (PKP1), cadherin
3 (CDH3) and cellular retinoic acid binding protein 2 (CRABP2)].
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analyses were performed using the
Metascape database (http://metascape.org/) and the ‘clusterProfiler’ R
package (version 4.3.0; http://www.r–project.org/) (29). The top 20 enriched GO and KEGG terms
were visualized using network analysis in Cytoscape 3.6.1 (30). Additionally, gene set enrichment
analysis (GSEA) was performed using clusterProfiler, with
significance thresholds set at false discovery rate (FDR) <0.25
and adjusted P<0.05 (31).
Correlation analysis of m6A-regulators
with metastatic SKCM-associated genes
Based on previous studies (32), 21 key m6A regulatory genes [FTO,
ALKBH5, RBM15, RBM15B, METTL3, METTL14, METTL16, WTAP, VIRMA, zinc
finger CCCH-type containing 13 (ZC3H13), HNRNPC, HNRNPA2B1, insulin
like growth factor 2 mRNA binding protein (IGF2BP)1, IGF2BP2,
IGF2BP3, RBMX, YTHDC1, YTHDC2, YTHDF1, YTHDF2 and YTHDF3] were
selected to assess the potential association between m6A
methylation regulation and metastatic SKCM. Pearson correlation
analysis was performed to assess the relationships between the
expression levels of these m6A regulators and key genes, with
|R|>0.2 and P<0.05 considered statistically significant.
Establishment of an m6A-regulated
metastatic SKCM-associated ceRNA network
Potential miRNAs targeting key mRNAs were predicted
using the miRWalk database (http://mirwalk.umm.uni-heidelberg.de/). Concurrently,
lncRNA-miRNA interactions were identified using the TarBase v8
database (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex).
Common miRNAs predicted by both approaches were determined using
Venn diagram analysis using the VennDiagram R package. A
lncRNA-miRNA-mRNA ceRNA network was subsequently constructed and
visualized using Cytoscape (v3.6.1; http://cytoscape.org/release_notes_3_6_1.html). Key
regulatory nodes within the network were identified using the
‘cytoHubba’ plugin. To further characterize the lncRNAs, sequences
were retrieved from LNCipedia (https://lncipedia.org/), and their subcellular
localizations were predicted using lncLocator (http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/).
The subcellular localization of mRNAs was determined using the
DM3Loc web tool (http://dm3loc.lin-group.cn/). Potential miRNA binding
sites within genomic sequences were identified using miRanda
(https://www.bioinformatics.com.cn/local_miranda_miRNA_target_prediction_120)
(33). Only lncRNAs and mRNAs
predicted to localize mainly in the cytoplasm (lncLocator or DM3Loc
probability ≥0.6) were retained, ensuring structural plausibility
for ceRNA crosstalk.
Relationships between the level of
immune cell infiltration and the expression of KRT17, PKP1, CDH3
and CRABP2
Relative enrichment scores for 24 immune subsets
were calculated from log2(TPM+1) matrices with the GSVA
package (v1.34.0; method=‘ssgsea’, kcdf=‘Gaussian’) (34,35).
Absolute cell fractions were inferred using CIBERSORTx (https://cibersortx.stanford.edu/index.php) in
‘absolute mode’ with the LM22 signature and 1,000 permutations;
samples with P≥0.05 were discarded. Deconvolution was repeated
using the EPIC algorithm implemented in TIMER2.0 (http://timer.cistrome.org/TIMER2.0) to provide an
additional independent estimate. To evaluate the immune and stromal
components of the tumor microenvironment, the Estimation of STromal
and Immune cells in MAlignant Tumor tissues using Expression data
(ESTIMATE) algorithm was applied using the ‘estimate’ R package
(version 4.3.0; http://www.r-project.org/). Immune scores, stromal
scores and tumor purity were calculated for each sample according
to the original algorithm. These packages were used to assess the
correlation between the expression of m6A-associated genes (KRT17,
PKP1, CDH3 and CRABP2) and the infiltration levels of 24 immune
cell types. Pearson correlation analysis was applied, with
|R|>0.2 and P<0.05 considered statistically significant.
Immunohistochemical staining of
patient tumor tissues
Tissues were fixed in 10% neutral buffered formalin
at 4°C overnight, followed by paraffin embedding and sectioning at
a 4–5 µm thickness. Sections were sequentially deparaffinized in
xylene, dehydrated and rehydrated through a graded ethanol series,
and finally immersed in distilled water. Subsequently, 10% goat
serum (Gibco; Thermo Fisher Scientific, Inc.) was applied to the
sections and incubated at room temperature for 1 h to complete
blocking. Sections were then incubated overnight at 4°C with
primary antibodies against PKP1 (1:500; cat. no. ab154622; Abcam),
KRT17 (1:200; cat. no. ab109725; Abcam), CRABP2 (1:1,000; cat. no.
ab211927; Abcam) and CDH3 (1:250; cat. no. ab242060; Abcam). After
washing, the slides were incubated with HRP-conjugated secondary
antibody (1:2,000; cat. no. ab6759; Abcam) in a humidified chamber
for 30 min, followed by color development using a DAB detection kit
(Beyotime Institute of Biotechnology). A total of two independent
pathologists evaluated the stained sections under an optical
microscope (Olympus Corporation). The proportion of positively
stained cells was scored as follows: 0, 0–5%; 1, 6–25%; 2, 26–50%;
3, 51–75%; and 4, >75%. Staining intensity was graded as
follows: 0, negative; 1, light yellow; 2, yellow-to-brown; and 3,
brown. The final immunohistochemical score was calculated as the
product of the proportion and intensity scores, and classified into
four categories: Negative (−; 0), weakly positive (+; 1–4),
moderately positive (++; 5–8) and strongly positive (+++;
9–12).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from SKCM and matched
adjacent normal tissues using TRIzol™ reagent (Invitrogen™; Thermo
Fisher Scientific, Inc.). cDNA synthesis was performed using the
EVO M-MLV RT Mix Kit [cat. no. AG11728; Accurate Biotechnology
(Hunan) Co., Ltd.] according to the manufacturer's instructions.
qPCR was then performed using the SYBR Green Premix Pro Taq HS qPCR
Kit [cat. no. AG11701; Accurate Biotechnology (Hunan) Co., Ltd.] on
a LightCycler® 480 Real-Time PCR System (Roche
Diagnostics). Each 10 µl reaction mixture contained 1 µl cDNA, 5 µl
2X SYBR Green Pro Taq HS Premix, 0.5 µM of each primer and
nuclease-free water. The qPCR cycling conditions were as follows:
Initial denaturation at 94°C for 30 sec, followed by 40 cycles at
94°C for 5 sec, 60°C for 10 sec and 50°C for 30 sec. Relative gene
expression levels were calculated using the 2−ΔΔCq
method (36). The primer sequences
used in the present study are listed in Table II.
| Table II.Primers used in the reverse
transcription-quantitative PCR assay. |
Table II.
Primers used in the reverse
transcription-quantitative PCR assay.
|
|
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|
|
|---|
| Gene | Species | Amplicon length,
bp | Forward | Reverse |
|---|
| KRT17 | Human | 119 |
GGTGGGTGGTGAGATCAATGT |
CGGCATCCTTGCGGTTCTT |
| PKP1 | Human | 120 |
TCAGCAACAAGAGCGACAAG |
TCAGGTAGGTGCGGATGG |
| CDH3 | Human | 121 |
TGGAGATCCTTGATGCCAATGA |
GCGTCCAGATCAGTGACCG |
| CRABP2 | Human | 99 |
TGAATGTGATGCTGAGGAAGAT |
TGGTGGAGGTTTTGATGTAGAA |
| β-actin | Human | 73 |
GTGGCCGAGGACTTTGATTG |
CCTGTAACAACGCATCTCATATT |
Statistical analysis
All statistical analyses, except those involving
transcriptomic sequencing data, were performed using GraphPad Prism
(version 8.0; Dotmatics) and SPSS software (version 23.0; IBM
Corp.). For comparisons of immunohistochemistry scores between
tumor and adjacent normal tissues, non-parametric tests were
applied: The Mann-Whitney U test for two-group comparisons or the
Kruskal-Wallis test for multiple groups, followed by Dunn's post
hoc test with Benjamini-Hochberg correction for multiple
comparisons. For comparisons among more than two groups with
normally distributed data, one-way ANOVA was used, followed by
Tukey's post hoc test for multiple pairwise comparisons. RT-qPCR
data were analyzed using the Wilcoxon signed-rank test to compare
expression levels between tumor and matched normal tissues. Unless
otherwise specified, P-values were adjusted for multiple
comparisons using the Benjamini-Hochberg false-discovery-rate (FDR)
procedure. P<0.05 was considered to indicate a statistically
significant difference.
Results
Comprehensive bioinformatics
identification of SKCM during metastasis
To identify key genes associated with SKCM
metastasis, a comprehensive analysis of multiple GEO datasets was
performed. Prior to analysis, gene expression data were normalized
(Fig. S1), and the ‘limma’ package
in R was used to identify DEGs within each microarray dataset,
using adjusted P<0.05 and |log2FC|>1 as cutoff
criteria (Fig. S2). Subsequently,
DEGs from all datasets were integrated using the RRA package,
resulting in the identification of 94 consistently differentially
expressed mRNAs, 84 upregulated and 10 downregulated, in metastatic
SKCM samples compared with in primary SKCM samples across the four
datasets (Fig. 1A).
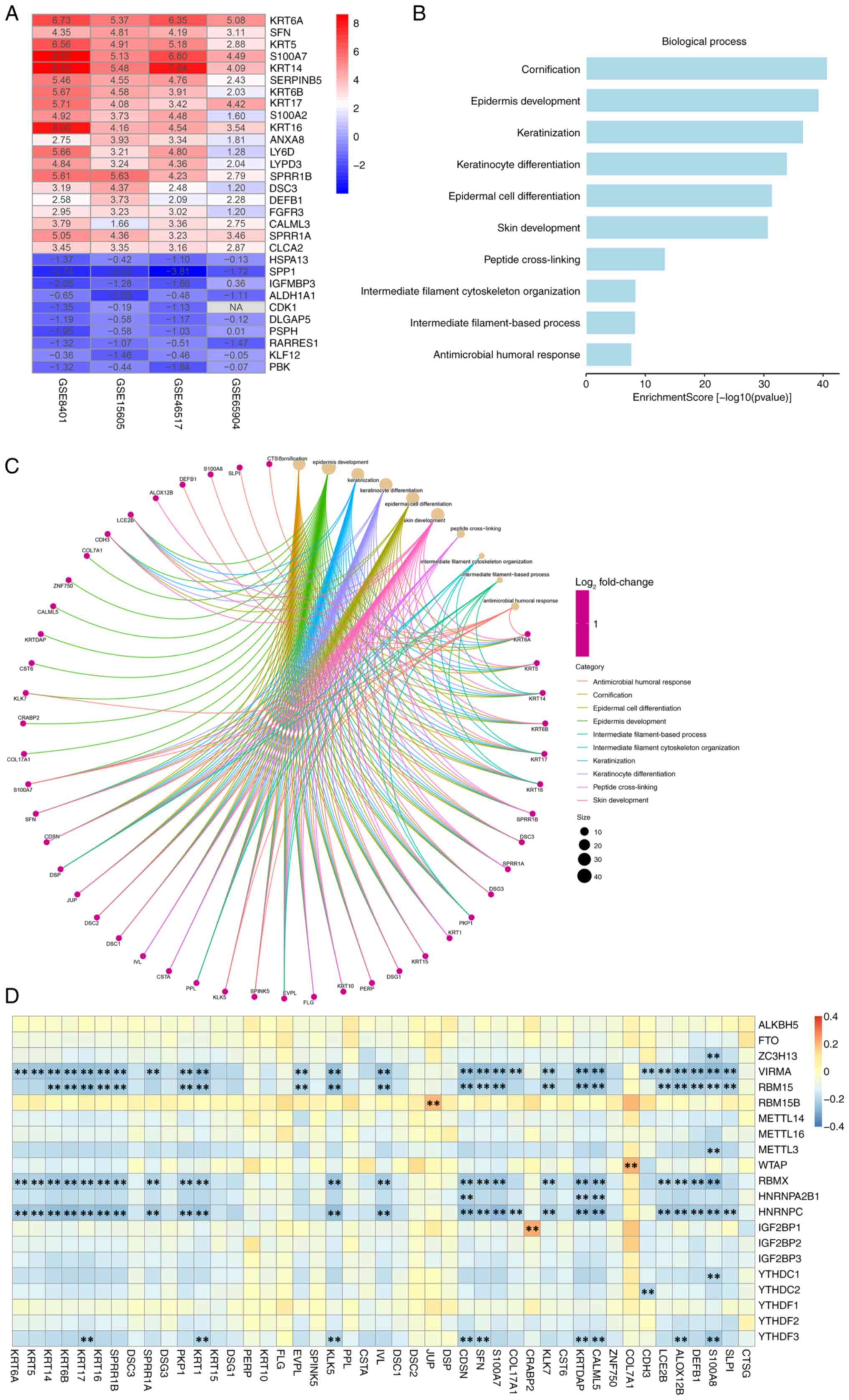 | Figure 1.Critical SKCM metastasis-related
genes and signaling pathways in the GEO SKCM cohort. (A)
Identification of DEGs in 4 GEO datasets using the RobustRankAggreg
package. A heatmap of the top 20 upregulated and 10 downregulated
DEGs in the integrated GEO datasets analysis is presented: The rows
represent the genes, and the columns represent the GEO dataset.
Each cell contains the log2FoldChange value. Red
represents upregulated genes and blue represents downregulated
genes. GO analysis based on DEGs between metastatic and primary
SKCM. (B) GO enrichment analysis was performed using the
clusterProfiler package to evaluate the related biological
processes. (C) The line color of the ring graph represents the GO
terms, the points represent the genes and the size of the points
represents the count number. (D) Heatmap illustrating Pearson
correlation coefficients between metastasis-associated genes and
N6-methyladenosine regulators in SKCM, with color intensity
indicating the correlation coefficient (blue, negative; yellow,
neutral; and orange/red, positive). **P<0.01. SKCM, skin
cutaneous melanoma; GEO, Gene Expression Omnibus; DEGs,
differentially expressed genes; GO, Gene Ontology. |
Subsequently, GO biological process enrichment
analysis was performed using the ‘clusterProfiler’ package in R.
The results indicated that pathways related to cornification,
epidermis development, and keratinization were significantly
enriched, suggesting their critical involvement in SKCM metastasis.
Notably, genes such as cathepsin G (CTSG), serine peptidase
inhibitor kazal type 1 and S100 calcium binding protein A8 (S100A8)
were identified as key contributors within these pathways (Fig. 1B and C).
In addition, the correlations between the 94
metastasis-related DEGs and 21 m6A regulatory genes were evaluated
using Pearson correlation analysis, with thresholds set at
|R|>0.5 and P<0.05. DEGs whose expression levels were
significantly correlated with ≥1 m6A regulator were defined as
m6A-related mRNAs. Based on this criterion, a total of 45 mRNAs
were identified that showed significant correlations with m6A
(Fig. 1D).
m6A regulatory-related genes in SKCM
in the TCGA dataset
A LASSO-Cox analysis of the upregulated pathways and
related genes involved in SKCM metastasis was subsequently
performed. The partial likelihood deviance in relation to log(λ)
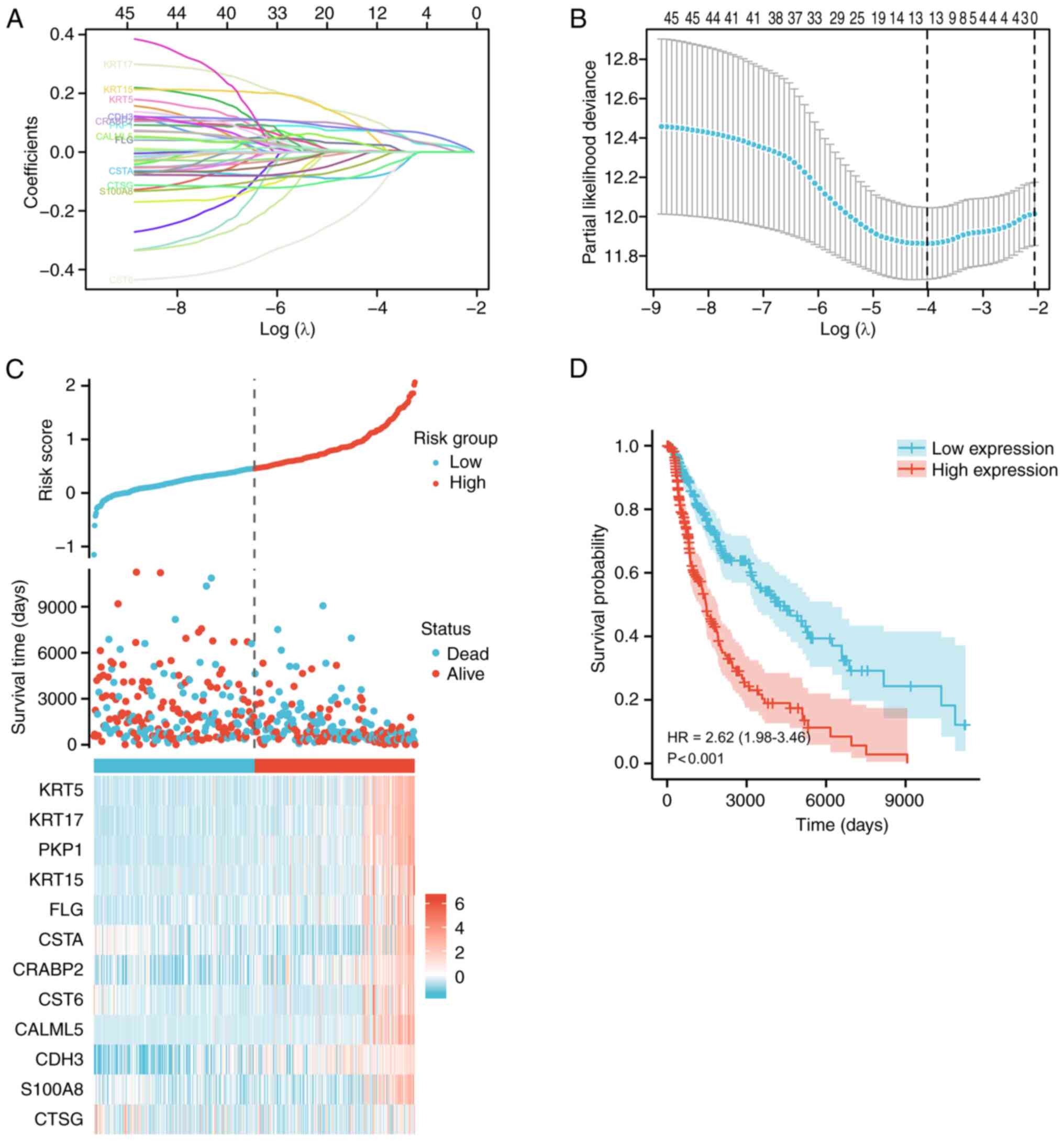
was derived from the LASSO-Cox regression model (Fig. 2A and B). Using the TCGA
transcriptomic data and survival information, 12 SKCM metastasis
regulators associated with overall survival (OS) were identified:
KRT5, KRT17, PKP1, KRT15, filaggrin (FLG), cystatin A (CSTA),
CRABP2, cystatin E/M (CST6), calmodulin like (CALML)5, CDH3, S100A8
and CTSG. The risk score and survival status distribution indicated
that patient survival time decreased with an elevated risk score.
Moreover, combined with the gene expression distribution, higher
expression of these genes correlated with a higher risk score
(Fig. 2C). Furthermore, survival
analysis revealed that patients in the high-risk group exhibited
significantly worse long-term survival than those in the low-risk
group [hazard ratio (HR), 2.62; P<0.001; Fig. 2D]. Ultimately, the present study
focused on four mRNAs strongly related to m6A regulation (CDH3,
KRT17, PKP1 and CRABP2). These genes appear to serve crucial roles
in SKCM metastasis.
Construction and prognostic analysis
of the ceRNA network
According to previous studies, mRNA expression is
modulated by miRNAs, whilst lncRNAs can competitively bind miRNAs
and thus indirectly regulate mRNA expression. This
lncRNA-miRNA-mRNA triple regulatory network is known as a ceRNA
network (37). To clarify the
regulation of metastasis-related mRNAs in SKCM by lncRNAs via
miRNAs, the present study constructed a ceRNA network centered on
CDH3, KRT17, PKP1 and CRABP2. First, single-gene differential
expression analysis was performed for each of the four genes in the
TCGA-SKCM cohort, identifying significantly co-expressed or
differentially up or downregulated transcripts, and for common
differentially expressed mRNAs and lncRNAs (Fig. S3). Ultimately, 494 differentially
expressed mRNAs and 91 differentially expressed lncRNAs were
identified (Fig. 3A).
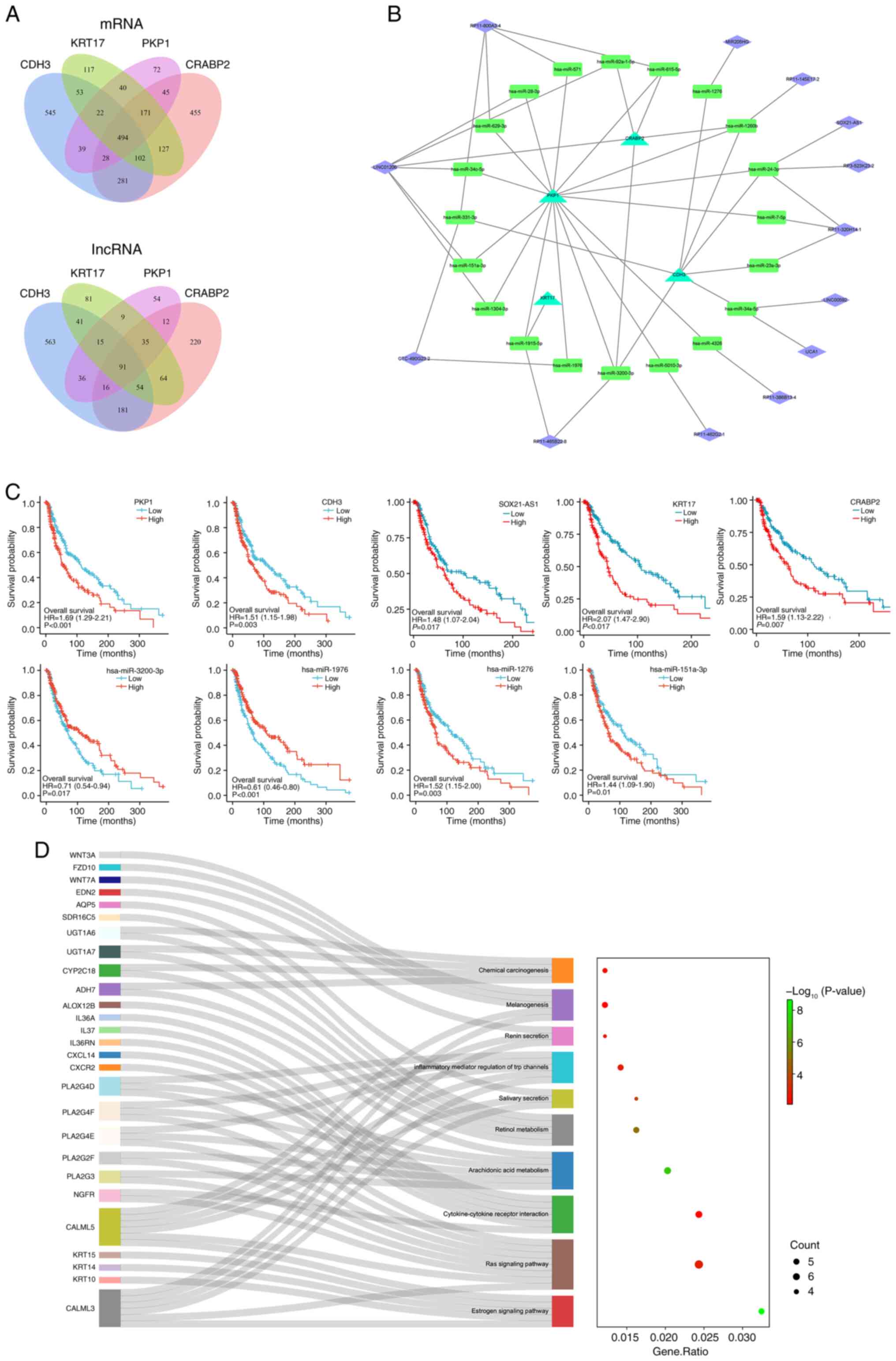 | Figure 3.Construction, prognosis analysis and
enrichment analysis of the ceRNA network. (A) Venn diagram of the
co-expressed differential mRNAs and lncRNAs of CDH3, KRT17, PKP1
and CRABP2 in SKCM. (B) Global view of the ceRNA network, which
consists of 13 lncRNAs, 20 miRNAs and 4 mRNAs. Triangles,
rectangles and diamonds represent mRNAs, miRNAs and lncRNAs,
respectively. (C) Prognostic analysis of the ceRNA network
expression level in SKCM assessed using the Kaplan-Meier plotter.
KRT17, PKP1, CDH3, CRABP2, SOX21-AS1, hsa-miR-1276 and
hsa-miR-151a-3p were demonstrated to be risk factors for SKCM,
whilst hsa-miR-3200-3p, hsa-miR-1976 were protective factors for
SKCM. (D) Functional enrichment analyses of co-expressed
differential mRNAs of CDH3, KRT17, PKP1 and CRABP2 in SKCM. The
Sankey diagram shows the pathways in which the co-expressed
differential mRNAs were mainly enriched. The bubble pattern
demonstrates the top 10 enrichment pathways with Entities ratio and
Entities count. The y-axis represents the pathway and the x-axis
represents the rich factor. The size and color of each bubble
represent the number of differentially expressed genes enriched in
the pathway and the -log10(P-value), respectively.
ceRNA, competing endogenous RNA; lncRNA, long noncoding RNA; KRT17,
keratin 17; PKP1, plakophilin 1; CDH3, cadherin 3; CRABP2, cellular
retinoic acid binding protein 2; SKCM, skin cutaneous melanoma;
miRNA/miR, microRNA. |
Subsequently, miRNAs predicted to target the 91
lncRNAs (TarBase) and the 4 mRNAs (miRWalk) were screened,
identifying the shared miRNA interactions between lncRNAs and
mRNAs. Consequently, 13 lncRNAs, 20 miRNAs and 4 mRNAs were
integrated into the ceRNA network (Fig.
3B). Kaplan-Meier survival analysis revealed that KRT17, PKP1,
CDH3, CRABP2, SOX21-AS1, hsa-miR-3200-3p, hsa-miR-1976,
hsa-miR-1276 and hsa-miR-151a-3p were all significantly associated
with OS in SKCM. HR analysis also demonstrated that KRT17, PKP1,
CDH3, CRABP2, SOX21-AS1, hsa-miR-1276 and hsa-miR-151a-3p are risk
factors for SKCM, whereas hsa-miR-3200-3p and hsa-miR-1976 serve as
protective factors (Fig. 3C).
Additionally, co-expressed differentially expressed
mRNAs associated with KRT17, PKP1, CDH3, and CRABP2 were subjected
to functional enrichment analysis using the Metascape online tool.
The results revealed that these genes are closely associated with
the estrogen signaling pathway, Ras signaling pathway,
cytokine-cytokine receptor interaction, melanogenesis and chemical
carcinogenesis, with the CALML3/5 and phospholipase A2 group IVA
families serving important roles in these pathways (Fig. 3D).
Association between m6A regulators and
the development and progression of SKCM
To further elucidate the abnormal m6A regulatory
mechanisms in SKCM, the expression of 21 m6A regulators in SKCM and
normal tissues from TCGA were analyzed. A total of four
downregulated regulators (METTL3, WTAP, YTHDC1 and YTHDC2) and 13
upregulated regulators (ALKBH5, VIRMA, RBM15, ZC3H13, IGF2BP1,
IGF2BP2, IGF2BP3, YTHDF1, YTHDF2, YTHDF3, HNRNPC, HNRNPA2B1 and
RBMX) were identified in SKCM (Fig.
4A). The present study also evaluated how these 21 m6A
regulators vary across different clinical stages (I–IV), and the
ANOVA results revealed that ALKBH5, METTL3, WTAP, RBM15, YTHDF2,
HNRNPC, HNRNPA2B1, RBMX, YTHDC1 and IGF2BP3 displayed continuous
patterns of increased or decreased mRNA expression with advancing
SKCM stage (Fig. 4B). This suggests
that certain m6A regulators may be closely associated with SKCM
tumorigenesis and progression.
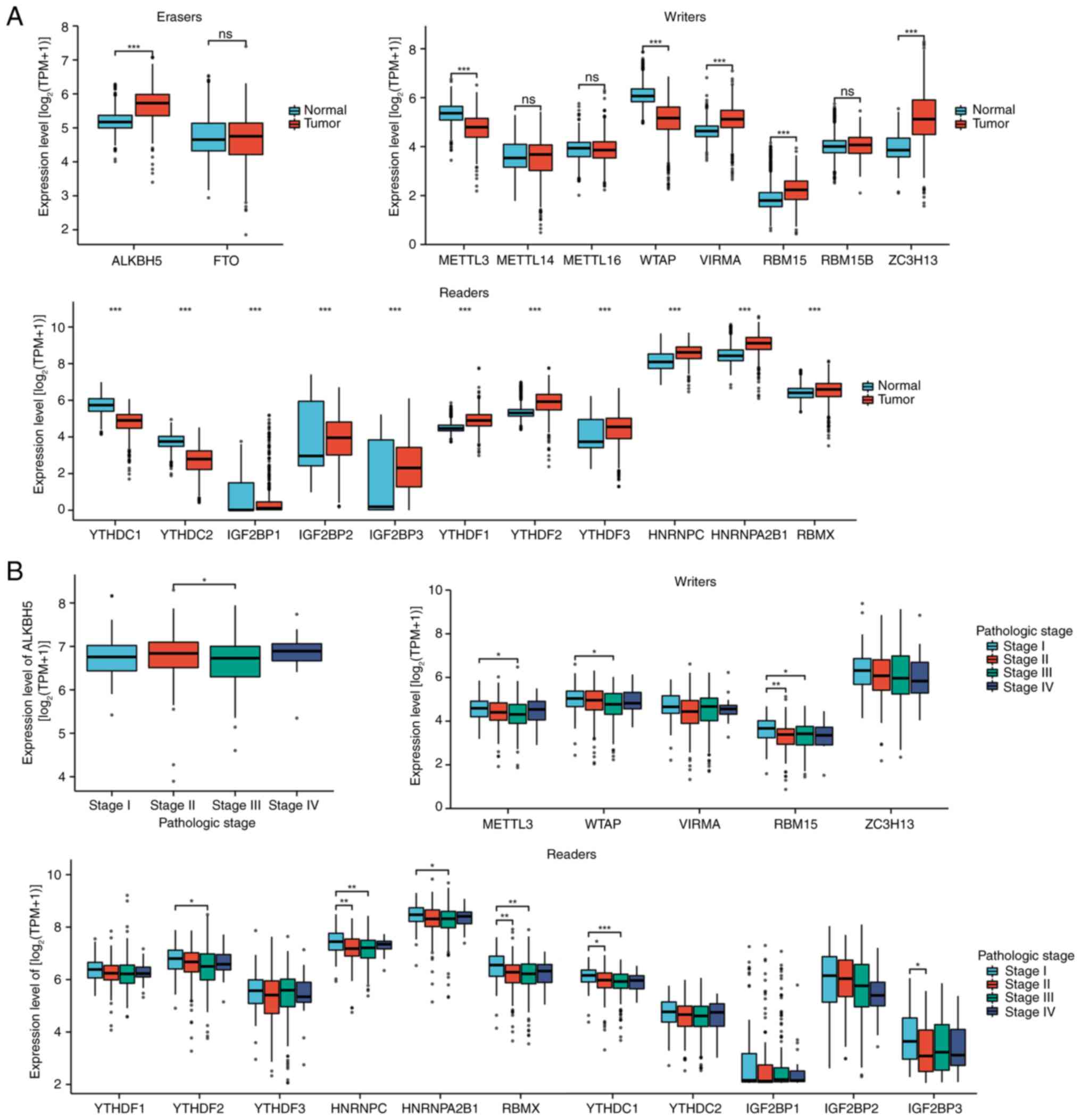 | Figure 4.Association of m6A regulators with
the development and progression of SKCM. (A) Compared with normal
samples, four downregulated (METTL3, WTAP, YTHDC1 and YTHDC2) and
13 upregulated regulators (ALKBH5, VIRMA, RBM15, ZC3H13, IGF2BP1,
IGF2BP2, IGF2BP3, YTHDF1, YTHDF2, YTHDF3, HNRNPC, HNRNPA2B1 and
RBMX) were identified in tumor samples. (B) ALKBH5, METTL3, WTAP,
RBM15, YTHDF2, HNRNPC, HNRNPA2B1, RBMX, YTHDC1 and IGF2BP3
exhibited consistently higher or lower mRNA levels as SKCM clinical
staging progressed. *P<0.05; **P<0.01; ***P<0.001. m6A,
N6-methyladenosine; SKCM, skin cutaneous melanoma; METTL,
methyltransferase, m6A complex catalytic subunit; WTAP, Wilms'
tumor associated protein; YTHD, YTH m6A RNA binding protein;
ALKBH5, AlkB homolog 5, RNA demethylase; VIRMA, Vir Like m6A
methyltransferase associated; RBM, RNA binding motif protein;
ZC3H13, zinc finger CCCH-type containing 13; IGF2BP, insulin like
growth factor 2 mRNA binding protein; HNRNP heterogeneous nuclear
ribonucleoprotein; ns, not significant; TPM, transcripts per
million. |
Association between m6A regulators and
the expression levels of KRT17, PKP1, CDH3 and CRABP2 in SKCM
The associations between the four key metastatic
prognostic mRNAs (KRT17, PKP1, CDH3 and CRABP2) and the
aforementioned 21 m6A regulators were subsequently assessed. The
results showed that the expression level of KRT17 was significantly
associated with METTL3, RBM15, ALKBH5, IGF2BP3 and HNRNPC. The
expression level of PKP1 was significantly associated with METTL16,
ALKBH5, IGF2BP3, YTHDF1 and YTHDF2. The expression level of CDH3
was significantly associated with VIRMA, RBM15B, YTHDC2, IGF2BP1
and RBMX, whereas the expression level of CRABP2 was significantly
associated with METTL3, RBM15B, FTO, ALKBH5, IGF2BP1 and YTHDF1
(Fig. 5).
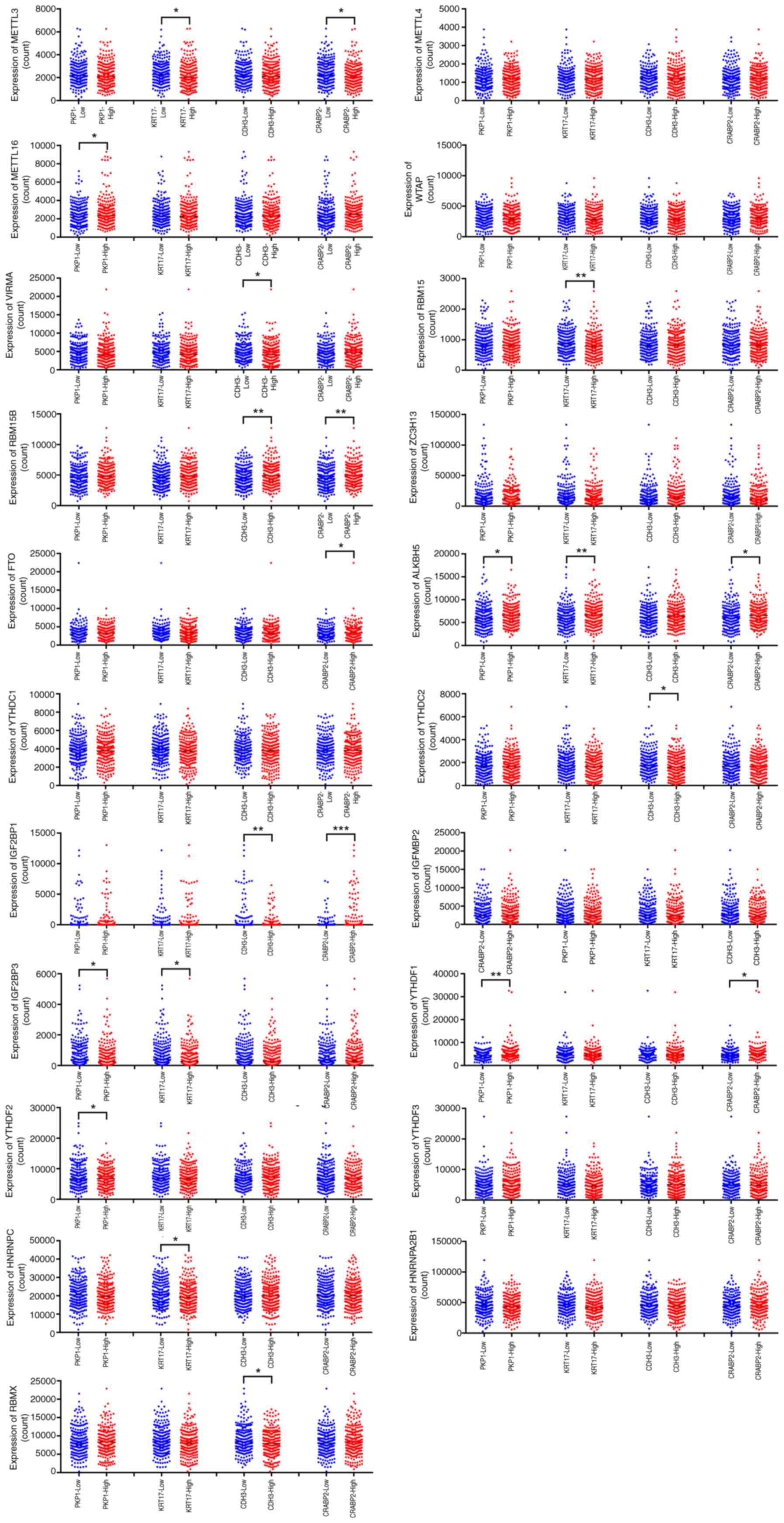 | Figure 5.Association between m6A regulators
with the expression levels of KRT17, PKP1, CDH3 and CRABP2 in SKCM.
The expression level of KRT17 was associated with METTL3, RBM15,
ALKBH5, IGF2BP3 and HNRNPC; the expression level of PKP1 was
associated with METTL16, ALKBH5, IGF2BP3, YTHDF1 and YTHDF2; the
expression level of CDH3 was associated with VIRMA, RBM15B, YTHDC2,
IGF2BP1 and RBMX; the expression level of CRABP2 was associated
with METTL3, RBM15B, FTO, ALKBH5, IGF2BP1 and YTHDF1. *P<0.05;
**P<0.01; ***P<0.001. m6A, N6-methyladenosine; KRT17, keratin
17; PKP1, plakophilin 1; CDH3, cadherin 3; CRABP2, cellular
retinoic acid binding protein 2; SKCM, skin cutaneous melanoma;
METTL, methyltransferase, m6A complex catalytic subunit; WTAP,
Wilms' tumor associated protein; VIRMA, Vir Like m6A
methyltransferase associated; RBM, RNA binding motif protein;
ZC3H13, zinc finger CCCH-type containing 13; FTO, fat mass and
obesity-associated protein; ALKBH5, AlkB homolog 5, RNA
demethylase; YTHD, YTH m6A RNA binding protein; IGF2BP, insulin
like growth factor 2 mRNA binding protein; HNRNP heterogeneous
nuclear ribonucleoprotein. |
Immune landscape associated with
KRT17, PKP1, CDH3 and CRABP2 expression in SKCM
To thoroughly assess the influence of KRT17, PKP1,
CDH3 and CRABP2 expression on immune cell infiltration, we used
single-sample (ss)GSEA to assess correlations between these genes
and 24 tumor-infiltrating immune cell types in 472 SKCM samples.
The results indicated that PKP1 is positively correlated with mast
cells and Th17 cells (Pearson-value >0.2; P<0.05; (Fig. 6A and B); KRT17 is negatively
correlated with Tγδ (Tgd) cells, T helper cells (Th cells), and Th1
cells (Pearson-value <-0.2; P<0.05); CDH3 is positively
correlated with mast cells and Th17 cells but negatively correlated
with B cells and T helper cells; and CRABP2 is positively
correlated with mast cells, natural killer (NK) cells, immature
dendritic cells and neutrophils, and negatively correlated with B
cells (Fig. 6A and B). To
corroborate the ssGSEA-derived landscape, the immune infiltration
data was re-evaluated using ESTIMATE and CIBERSORTx. The results
revealed that tumors with a high expression of PKP1, KRT17, CRABP2
or CDH3 displayed significantly lower StromalScore, ImmuneScore and
composite ESTIMATEScore than their low-expression counterparts (all
FDR <0.01; Fig. S4), mirroring
the global immune-suppressed phenotype observed with ssGSEA.
Consistent with the ssGSEA results, CIBERSORTx deconvolution
analysis further characterized the tumor immune microenvironment.
Specifically, high CRABP2 expression was associated with
significantly reduced levels of CD8+ T cells (Figs. 6B and S4). By contrast, both high-PKP1 and
high-CRABP2 tumors showed increased levels of NK cells (Fig. 6B). Furthermore, both tumor subtypes
shared a common feature of an increased M0-to-M2 macrophage
fraction (all FDR <0.05; Fig.
S4). The concordant results from the three independent
algorithms strengthen the conclusion that elevated expression of
these keratin-related genes is associated with an immunologically
‘cold’ microenvironment in melanoma.
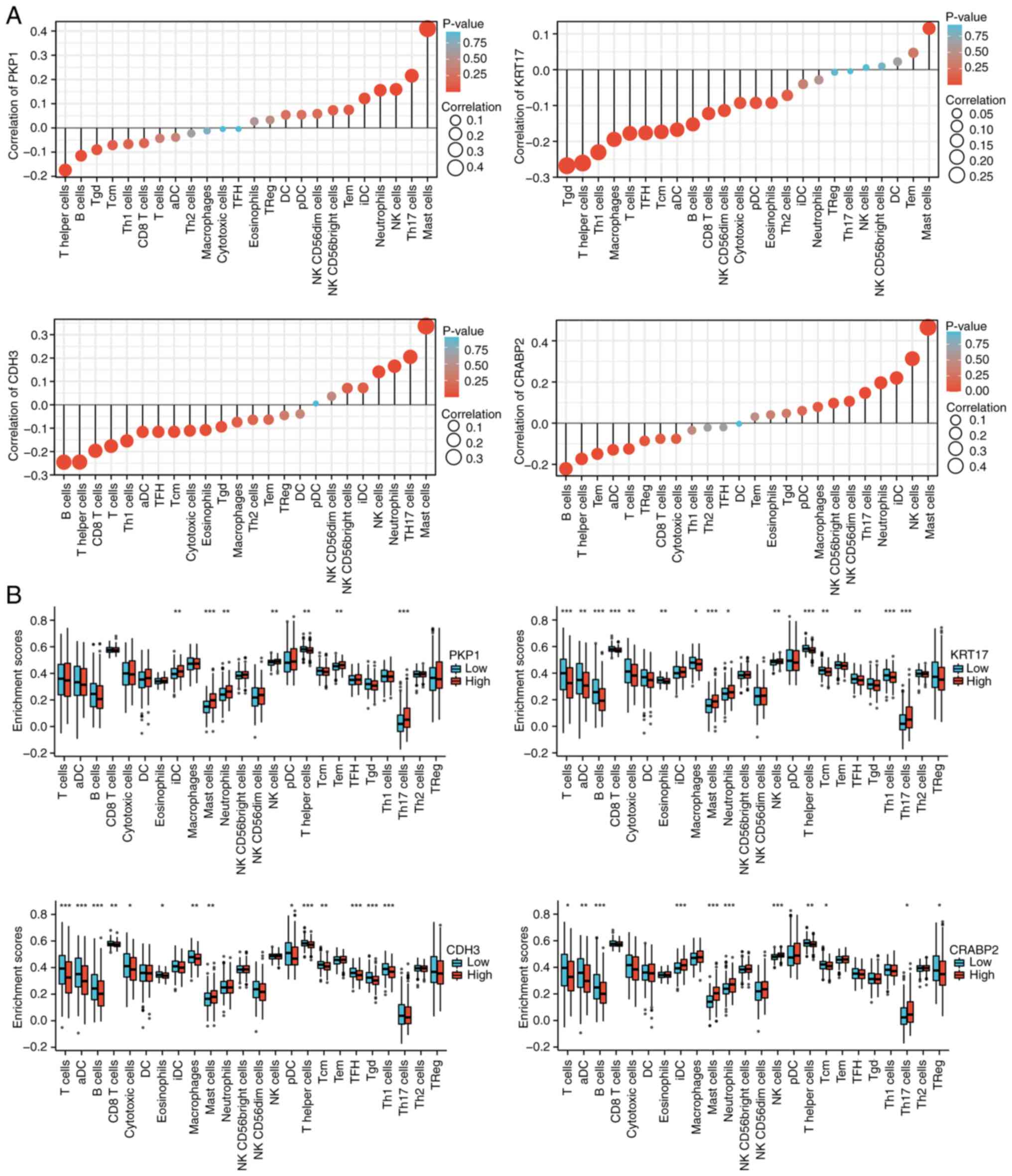 | Figure 6.Immune landscape of KRT17, PKP1, CDH3
and CRABP2 expression levels in SKCM. (A) Correlations of 24 immune
cells with KRT17, PKP1, CDH3 and CRABP2 expression levels,
respectively. The size of the dots represents the absolute
Pearson's correlation coefficient values. (B). Enrichment
differences of 24 immune cell types between high- and
low-expression groups of KRT17, PKP1, CDH3 and CRABP2. *P<0.05;
**P<0.01; ***P<0.001. KRT17, keratin 17; PKP1, plakophilin 1;
CDH3, cadherin 3; CRABP2, cellular retinoic acid binding protein 2;
SKCM, skin cutaneous melanoma; NK, natural killer; DC, dendritic
cell; aDC, activated DC; TFH, follicular helper T cells; Tcm,
central memory T cells; Tgd, γδ T cells; Tem, effector memory T
cells; pDC, plasmacytoid DC; iDC, immature DC. |
Clinical significance of KRT17, PKP1,
CDH3 and CRABP2 in patients with SKCM
ROC curves revealed that KRT17, PKP1, CDH3 and
CRABP2 demonstrated predictive power for OS in the TCGA cohort of
patients with SKCM (Fig. 7A). The
associations of these genes with clinical factors was then further
assessed. KRT17, PKP1, CDH3, and CRABP2 demonstrated a positive
association with pathological stage (Fig. 7B). For tumor (T)-node (N)-metastasis
(M) staging, their expression levels were significantly associated
with the T stage, but had no discernable association with N or M
stages (Fig. 7C). Similarly, no
significant association was revealed between their expression and
patient age (Fig. 7D).
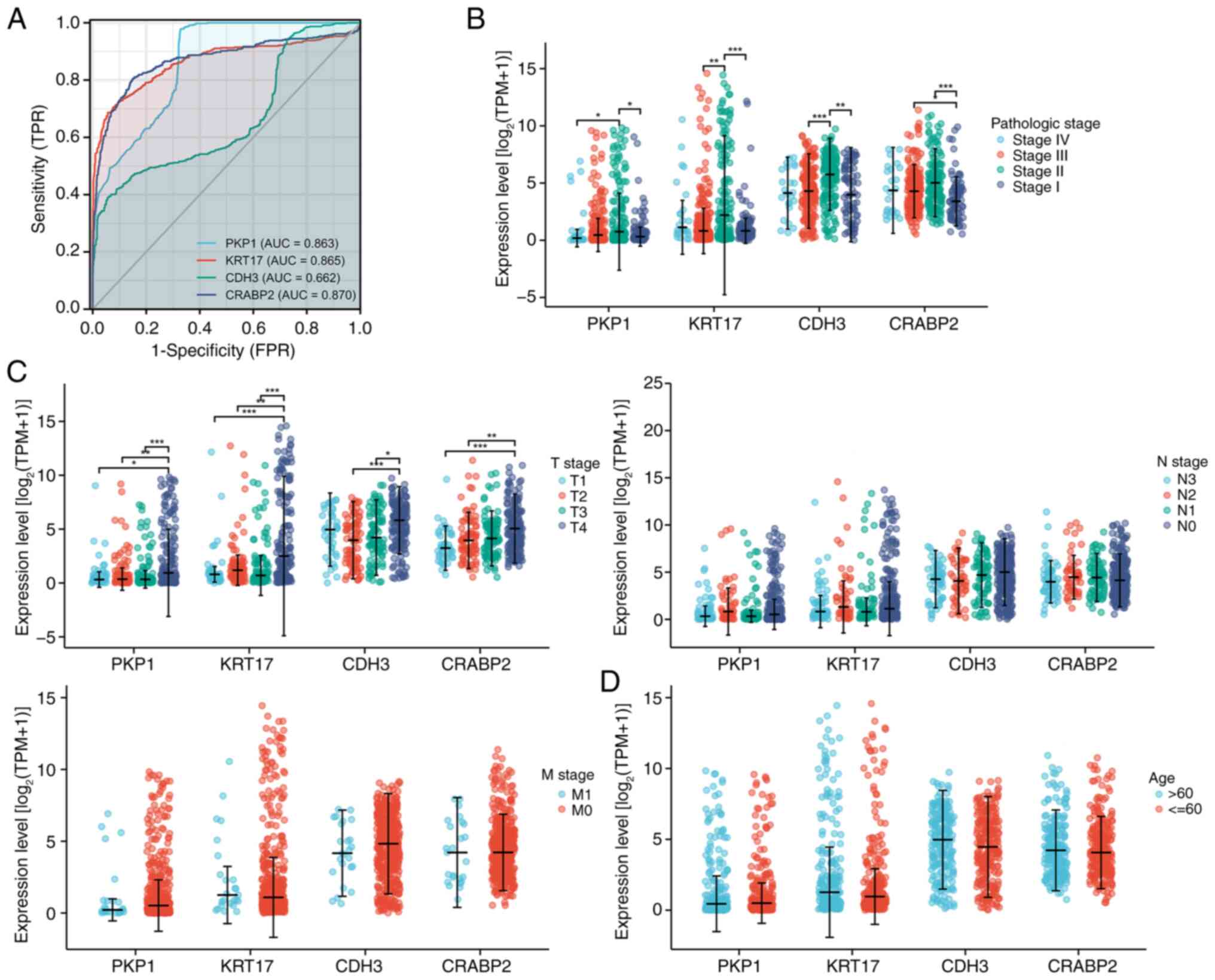 | Figure 7.Clinical significance of KRT17, PKP1,
CDH3 and CRABP2 expression in patients with SKCM. (A) Receiver
operating characteristic curves of KRT17, PKP1, CDH3 and CRABP2
expression of patients with SKCM in The Cancer Genome Atlas cohort.
(B) Expression levels of KRT17, PKP1, CDH3 and CRABP2 were
positively associated with the pathological stage. (C) In TNM
staging, the expression of KRT17, PKP1, CDH3 and CRABP2 were only
significantly associated with T stage, but not with N and M stage.
(D) No association was demonstrated between KRT17, PKP1, CDH3 and
CRABP2 expression and age. *P<0.05; **P<0.01; ***P<0.001.
KRT17, keratin 17; PKP1, plakophilin 1; CDH3, cadherin 3; CRABP2,
cellular retinoic acid binding protein 2; SKCM, skin cutaneous
melanoma; TPR, true positive rate; FPR, false positive rate; AUC,
area under the curve; TPM, transcripts per million; T, tumor; N,
node; M, metastasis. |
Expression of KRT17, PKP1, CDH3 and
CRABP2 in SKCM tissues
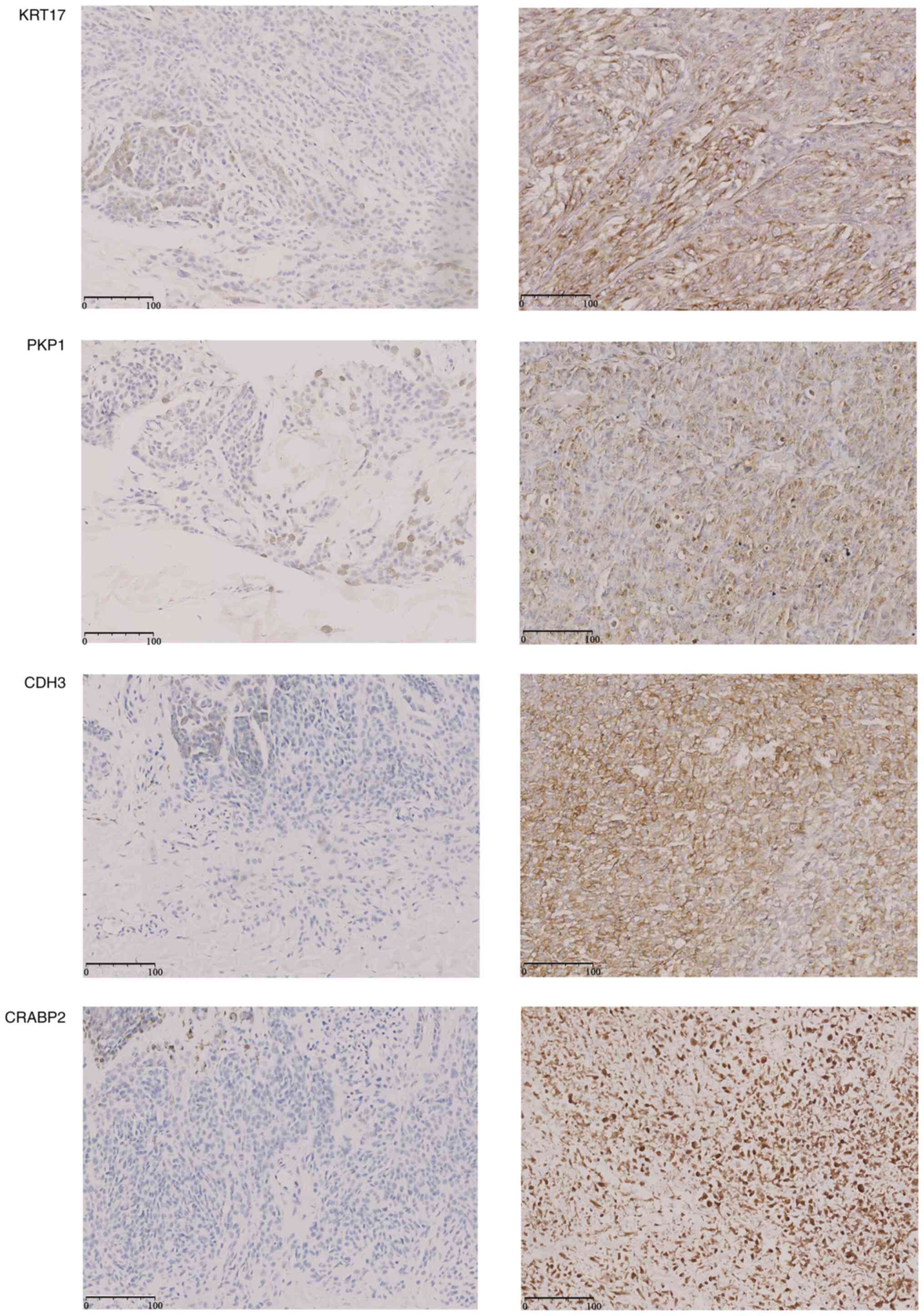
Representative immunohistochemistry images
demonstrated markedly higher cytoplasmic staining for all four
genes in tumor tissues compared with in adjacent skin (Fig. 8). Consistently, RT-qPCR analysis on
the same sample set showed 2.3- to 4.5-fold upregulation at the
mRNA level (Fig. S5). These
concordant protein and transcript results corroborate the
bioinformatic prediction of the present study that the four hub
genes are overexpressed in metastatic SKCM.
Discussion
SKCM is a highly malignant tumor derived from
melanocytes, known for its poor prognosis and strong invasiveness.
Risk factors such as ultraviolet exposure, excessive pigmented
nevi, a family history of melanoma and a higher number of nevi lead
to DNA damage that can initiate carcinogenesis (38,39).
Despite emerging diagnostic and therapeutic approaches, clinical
options remain limited as SKCM often progresses rapidly and is
frequently asymptomatic in the early stages (1). Hence, elucidating the molecular
mechanisms underlying SKCM is paramount for improving treatment
outcomes and patient prognosis.
Research has reported that abnormal m6A modification
is closely associated with tumorigenesis and progression, as m6A
modulates numerous processes (such as mRNA splicing, 3′-end
processing, nuclear export, translational regulation, mRNA decay
and miRNA biogenesis) and that its dynamic reversibility influences
tumor cell growth and differentiation (40). This role has been reported in
several cancers, including cervical cancer (41), acute myeloid leukemia (AML)
(42), pancreatic cancer (43) and hepatocellular carcinoma (44). However, the specific molecular
mechanisms mediated by m6A in SKCM are not yet fully
understood.
The present study comprehensively analyzed four
metastatic SKCM cohorts from the GEO database (GSE8401, GSE15605,
GSE46517 and GSE65904), identifying a total of 823 DEGs, comprising
283 upregulated and 540 downregulated genes. GO enrichment analysis
indicated that the DEGs are predominantly involved in biological
pathways such as cornification, epidermis development and
keratinization, with genes including CTSG, sphingosine-1-phosphate
lyase 1 and S100A8 serving pivotal roles. These findings underscore
that SKCM metastasis is accompanied by proliferation and
keratinization in epidermal/epithelial cells. Similarly,
keratinization has been associated with a poor prognosis in human
papillomavirus-negative oropharyngeal carcinoma (45).
Subsequently, the present study constructed a
survival prediction model in the TCGA database using LASSO-Cox
analysis. The results revealed that KRT5, KRT17, PKP1, KRT15, FLG,
CSTA, CRABP2, CST6, CALML5, CDH3, S100A8 and CTSG were
significantly associated with OS. Elevated expression of these
genes was closely associated with poor outcomes. Multiple studies
have reported that m6A RNA modification, such as DNA methylation
and histone modifications, exerts regulatory control over
tumorigenesis via methyltransferases and demethylases (46). However, the precise mechanism by
which m6A may affect melanoma progression in an mRNA-dependent
manner remains elusive. Therefore, the present study employed
Pearson correlation analysis to identify m6A-related metastasis
genes in SKCM, and the findings revealed that KRT17, PKP1, CDH3 and
CRABP2 exhibit strong associations with m6A regulation and OS in
patients with melanoma.
Further single-gene differential expression analysis
of KRT17, PKP1, CDH3 and CRABP2 revealed 91 commonly differentially
expressed lncRNAs. A lncRNA-miRNA-mRNA triple regulatory network
comprising 20 miRNAs and 13 lncRNAs was then constructed. In
addition, enrichment analysis demonstrated that 494
co-differentially expressed mRNAs associated with KRT17, PKP1, CDH3
and CRABP2 are primarily enriched in the estrogen signaling
pathway, Ras signaling pathway, cytokine-cytokine receptor
interaction, melanogenesis and chemical carcinogenesis. Notably,
melanin and its synthesis process itself can affect melanoma
behavior by regulating oxidative stress, drug sensitivity and
immune escape, thus having a profound impact on prognosis (47).
Previous studies indicate that KRTs are crucial in
epidermal development, intermediate filament organization,
cytoskeletal reorganization, keratinization and keratinocyte
migration in patients with melanoma (48–50).
In particular, KRT17 has been identified as a diagnostic and
prognostic marker in breast cancer (51), epithelial ovarian cancer (52), cervical squamous cell carcinoma
(53) and gastric cancer (54). Moreover, PKP1, a key component of
desmosomes, has been reported to be abnormally expressed in
multiple cancers, including prostate cancer (54), oral squamous cell carcinoma
(55) and esophageal adenocarcinoma
(56), regulating tumor cell
proliferation, colony formation, migration/invasion and apoptosis.
In lung cancer, high PKP1 expression has been markedly associated
with favorable clinical prognosis, whereas its downregulation is
associated with hypermethylation of its promoter region (57). CDH3, which encodes P-cadherin, is a
core component of adherens junctions and contributes to the
development and progression of several malignancies (58). In patients with adenomyosis,
aberrant expression of m6A regulators has been associated with
dysregulated CDH3, sodium voltage-gated channel β subunit 4 and
placenta associated 8 (59). CRABP2
also serves important roles in multiple tumors. In hepatocellular
carcinoma, downregulation of CRABP2 has been reported to inhibit
tumor formation in vivo (59). Additionally, Yan et al
(60) reported that mutations in
ciliary neurotrophic factor receptor, CRABP2, galanin And GMAP
prepropeptide, and progestagen associated endometrial protein
markedly affected immune infiltration in melanoma.
Furthermore, the investigation of 21 m6A regulators
revealed significant differences in the expression of several
factors (METTL3, WTAP, YTHDC1, YTHDF2, ALKBH5, VIRMA, RBM15,
ZC3H13, IGF2BP1, IGF2BP2, IGF2BP3, YTHDF1, YTHDF2, YTHDF3, HNRNPC,
HNRNPA2B1 and RBMX) between normal and tumor samples. Among these,
ALKBH5, an important ‘eraser’ in the m6A regulatory system,
influences tumor growth and metastasis by demethylating specific
transcripts. In breast cancer, ALKBH5-mediated demethylation
upregulates NANOG expression, promoting cancer stem cell
specification and metastatic capacity (61). ALKBH5 is also highly expressed in
lung adenocarcinoma cells, and its knockout can suppress cancer
cell proliferation and invasion by increasing m6A modification of
forkhead box M1 (62).
Additionally, the m6A methyltransferase WTAP forms a
complex with METTL3 and METTL14 and co-localizes in the nucleus to
participate in RNA methylation. Research has reported that, under
the influence of the hepatocellular carcinoma suppressor ETS1, WTAP
regulates the cell cycle in liver cancer via a p21/p27-dependent
mechanism (63,64). METTL3 and METTL14 are likewise
implicated in oncogenic proliferation and metastasis (65,66).
In the cohort in the present study, all three genes were
significantly upregulated in metastatic SKCM relative to normal
skin, suggesting a potential but as-yet unproven role in melanoma
progression. To move beyond bioinformatic association, follow-up
experiments, such as chromatin immunoprecipitation-qPCR to confirm
direct binding of the complex to target promoters and RNA
immunoprecipitation-qPCR to assess m6A modification on specific
transcripts, will be necessary to clarify causality in future
studies.
However, the ceRNA network presented in the present
study is exploratory. Although most miRNA-RNA edges were retrieved
from the TarBase and miRWalk records supported by cross-linking
immunoprecipitation (CLIP)-sequencing, immunoprecipitation or
reporter assays, in-situ validation (such as argonaute-CLIP,
biotinylated RNA pull-down or dual-luciferase assays) was not
performed in the present study. Furthermore, the localization
filter relied on computational predictors rather than fractionation
experiments. Future work will need to verify key triplets,
particularly the SOX21-AS1/hsa-miR-1276/KRT17 and
hsa-miR-151a-3p/PKP1 axes, in melanoma cell lines and
patient-derived samples.
Subsequently, the association between KRT17, PKP1,
CDH3 and CRABP2 expression and immune infiltration in SKCM was
assessed, and the results indicated that these four genes are
positively correlated with mast cells and Th17 cells and negatively
correlated with B cells and Th1 cells. Immunomodulatory shifts in
the tumor microenvironment serve vital roles in tumor metastasis.
In a murine melanoma endothelial model, upregulated indoleamine
2,3-dioxygenase 1 was reported to respond to CD40 agonist
immunotherapy-induced IFN-γ production, suggesting a novel
immunosuppressive feedback mechanism (67). Similarly, Singh et al
(23) reported that focal
ultrasound heating combined with anti-CD40 agonist antibodies
markedly improved T-cell and macrophage function in melanoma. These
findings initially suggest that m6A regulators may alter SKCM
progression and prognosis by influencing KRT17, PKP1, CDH3 and
CRABP2 expression and remodeling the tumor immune microenvironment.
Notably, in the present study, KRT17, PKP1, CDH3 and CRABP2
demonstrated significant differential expression across T stages
but not in N and M stages, implying that these genes may have a
greater role in local tumor growth and tissue infiltration rather
than lymph node or distal metastasis. However, further large-scale
multicenter studies are needed to validate these findings and
explore the underlying molecular mechanisms. Furthermore, there is
growing evidence that cancer cells can ‘rewire’ the neuro-endocrine
network of the host by secreting hormone-like factors and
neurotransmitters to shape a supportive microenvironment (68). This autonomous regulation capability
suggests that when targeting m6A-related pathways, its potential
interaction with the whole-body steady-state axis should also be
considered. If future experiments confirm that genes such as KRT17
or PKP1 critically regulate m6A modifications and the immune
microenvironment, they may hold promise for early diagnosis, risk
stratification and novel immunotherapeutic approaches in SKCM.
Lastly, RT-qPCR experiments demonstrated higher
expression levels of KRT17, PKP1, CDH3 and CRABP2 in SKCM tumor
tissues compared with in adjacent normal tissues. Thus, we
hypothesize that m6A modification may influence SKCM metastasis by
modulating the expression of these four genes. Nevertheless,
certain limitations should be noted. First, although the in
silico analyses in the present study are generally in-line with
previous findings, larger, multicenter clinical cohorts are
required to enhance reliability. Moreover, further in vitro
and in vivo functional experiments are needed to clarify the
mechanisms by which m6A modification and KRT17, PKP1, CDH3 and
CRABP2 operate in SKCM. Furthermore, although analyzing the
correlation between these four genes and clinicopathological
variables has important research value for the prognosis of SKCM
patients, disease-free survival (DFS) and tumor-stage-stratified
correlations were not incorporated as in the TCGA-SKCM cohort. This
is since, in the dataset used for this study, DFS data are missing
for 65% of patients and the recorded follow-up periods are highly
heterogeneous (median, 6.2 months; interquartile range, 1.8–11.5
months). Preliminary modelling under these conditions yielded
unstable HR estimates that risk over-interpretation. Consequently,
survival analyses were restricted to OS, for which follow-up is
more complete, and immune-infiltration profiling plus experimental
validation was focused on. Future studies in prospective cohorts
with comprehensive DFS information will be required to extend and
confirm the prognostic relevance of the selected markers across
multiple clinical endpoints.
In summary, the results of the present study
demonstrate that KRT17, PKP1, CDH3 and CRABP2 expression are
regulated by m6A modification and may influence SKCM metastasis,
ultimately altering patient survival. Moreover, immune
microenvironment analyses suggested that mast cells, Th17 cells, B
cells and Th1 cells serve critical roles in this process. These
findings provide new insights into the mechanisms of SKCM
metastasis and potential therapeutic targets. Nonetheless, further
research and larger clinical samples are required for more in-depth
validation and exploration.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81772916 and 82103470) and the
Natural Science Foundation of Jiangsu Province (grant no.
BK20171132).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YG was responsible for study concept design, data
organization, formal analysis and drafting the initial manuscript.
SZ contributed to data visualization and analysis, manuscript
drafting, revision, and final approval. TG was responsible for data
validation, study concept design, data review and editing. XLX
contributed to study concept design, provided and obtained research
resources, and participated in manuscript review and revision. XZ
contributed to study concept design, data organization, manuscript
writing and final manuscript approval. YG and XZ confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Institute of Dermatology, Peking Union Medical
College and Chinese Academy of Medical Sciences (approval no.
2017-KY-044). Written informed consent was obtained from all
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schadendorf D, van Akkooi ACJ, Berking C,
Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A and Ugurel
S: Melanoma. Lancet. 392:971–984. 2018. View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gershenwald JE, Scolyer RA, Hess KR,
Sondak VK, Long GV, Ross MI, Lazar AJ, Faries MB, Kirkwood JM,
McArthur GA, et al: Melanoma staging: Evidence-Based changes in the
American joint committee on cancer eighth edition cancer staging
manual. CA Cancer J Clin. 67:472–492. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Slominski RM, Kim TK, Janjetovic Z,
Brożyna AA, Podgorska E, Dixon KM, Mason RS, Tuckey RC, Sharma R,
Crossman DK, et al: Malignant melanoma: An overview, new
perspectives, and vitamin D signaling. Cancers (Basel).
16:22622024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Slominski RM, Raman C, Jetten AM and
Slominski AT: Neuro-immuno-endocrinology of the skin: How
environment regulates body homeostasis. Nat Rev Endocrinol.
21:495–509. 2025. View Article : Google Scholar
|
|
6
|
Slominski RM, Chen JY, Raman C and
Slominski AT: Photo-neuro-immuno-endocrinology: How the ultraviolet
radiation regulates the body, brain, and immune system. Proc Natl
Acad Sci USA. 121:e23083741212024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan B, Liu H, Zhang S, da Silva SR, Zhang
L, Meng J, Cui X, Yuan H, Sorel O, Zhang SW, et al: Viral and
cellular N6-Methyladenosine and
N6,2′-O-Dimethyladenosine epitranscriptomes in the KSHV
life cycle. Nat Microbiol. 3:108–120. 2018. View Article : Google Scholar
|
|
8
|
Pfeifer GP: Defining driver DNA
methylation changes in human cancer. Int J Mol Sci. 19:11662018.
View Article : Google Scholar
|
|
9
|
Wang Y, Li Y, Toth JI, Petroski MD, Zhang
Z and Zhao JC: N6-Methyladenosine modification destabilizes
developmental regulators in embryonic stem cells. Nat Cell Biol.
16:191–198. 2014. View
Article : Google Scholar
|
|
10
|
Alarcón CR, Lee H, Goodarzi H, Halberg N
and Tavazoie SF: N6-Methyladenosine marks primary microRNAs for
processing. Nature. 519:482–485. 2015. View Article : Google Scholar
|
|
11
|
Huang H, Weng H and Chen J: m6A
modification in coding and non-coding RNAs: Roles and therapeutic
implications in cancer. Cancer Cell. 37:270–288. 2020. View Article : Google Scholar
|
|
12
|
Huang H, Weng H and Chen J: The biogenesis
and precise control of RNA m6A methylation. Trends
Genet. 36:44–52. 2020. View Article : Google Scholar
|
|
13
|
Huang H, Weng H, Deng X and Chen J: RNA
modifications in cancer: Functions, mechanisms, and therapeutic
implications. Annu Rev Cancer Biol. 4:221–240. 2020. View Article : Google Scholar
|
|
14
|
Weng H, Huang H, Wu H, Qin X, Zhao BS,
Dong L, Shi H, Skibbe J, Shen C, Hu C, et al: METTL14 inhibits
hematopoietic stem/progenitor differentiation and promotes
leukemogenesis via mRNA m6A modification. Cell Stem
Cell. 22:191–205. 2018. View Article : Google Scholar
|
|
15
|
Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma
D, Lv J, Heng J, Ding Y, Xue Y, et al: m6A modulates
haematopoietic stem and progenitor cell specification. Nature.
549:273–276. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C,
Huang H, Nachtergaele S, Dong L, Hu C, et al: FTO plays an
oncogenic role in acute myeloid leukemia as a
N6-methyladenosine RNA demethylase. Cancer Cell.
31:127–141. 2017. View Article : Google Scholar
|
|
17
|
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S,
Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al: m6A demethylase
ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by
Sustaining FOXM1 expression and cell proliferation program. Cancer
Cell. 31:591–606.e6. 2017. View Article : Google Scholar
|
|
18
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA.
113:E2047–E2056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He F, Yu J, Yang J, Wang S, Zhuang A, Shi
H, Gu X, Xu X, Chai P and Jia R: m6A RNA
hypermethylation-induced BACE2 boosts intracellular calcium release
and accelerates tumorigenesis of ocular melanoma. Mol Ther.
29:2121–2133. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dahal U, Le K and Gupta M: RNA m6A
methyltransferase METTL3 regulates invasiveness of melanoma cells
by matrix metallopeptidase 2. Melanoma Res. 29:382–389. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu L, Shen SS, Hoshida Y, Subramanian A,
Ross K, Brunet JP, Wagner SN, Ramaswamy S, Mesirov JP and Hynes RO:
Gene expression changes in an animal melanoma model correlate with
aggressiveness of human melanoma metastases. Mol Cancer Res.
6:760–769. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cabrita R, Lauss M, Sanna A, Donia M,
Larsen MS, Mitra S, Johansson I, Phung B, Harbst K,
Vallon-Christersson J, et al: Tertiary lymphoid structures improve
immunotherapy and survival in melanoma. Nature. 577:561–565. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Singh MP, Sethuraman SN, Ritchey J,
Fiering S, Guha C, Malayer J and Ranjan A: In-situ vaccination
using focused ultrasound heating and anti-CD-40 agonistic antibody
enhances T-cell mediated local and abscopal effects in murine
melanoma. Int J Hyperthermia. 36:64–73. 2019. View Article : Google Scholar
|
|
24
|
Raskin L, Fullen DR, Giordano TJ, Thomas
DG, Frohm ML, Cha KB, Ahn J, Mukherjee B, Johnson TM and Gruber SB:
Transcriptome profiling identifies HMGA2 as a biomarker of melanoma
progression and prognosis. J Invest Dermatol. 133:2585–2592. 2013.
View Article : Google Scholar
|
|
25
|
Xu F, Lin H, He P, He L, Chen J, Lin L and
Chen Y: A TP53-associated gene signature for prediction of
prognosis and therapeutic responses in lung squamous cell
carcinoma. Oncoimmunology. 9:17319432020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Han D, Hou Y, Chen H and Chen Z:
Statistical inference methods for two crossing survival curves: A
comparison of methods. PLoS One. 10:e01167742015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-Seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar
|
|
28
|
Kolde R, Laur S, Adler P and Vilo J:
Robust rank aggregation for gene list integration and
meta-analysis. Bioinformatics. 28:573–580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Y, Zhou B, Pache L, Chang M,
Khodabakhshi A.H, Tanaseichuk O, Benner C and Chanda SK: Metascape
provides a biologist-oriented resource for the analysis of
systems-level datasets. Nat Commun. 10:15232019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zaccara S, Ries RJ and Jaffrey SR:
Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell
Biol. 20:608–624. 2019. View Article : Google Scholar
|
|
33
|
Enright AJ, John B, Gaul U, Tuschl T,
Sander C and Marks DS: MicroRNA targets in drosophila. Genome Biol.
5:R12003. View Article : Google Scholar
|
|
34
|
Hänzelmann S, Castelo R and Guinney
J.GSVA: Gene set variation analysis for microarray and RNA-Seq
data. BMC Bioinformatics. 14:72013.
|
|
35
|
Bindea G, Mlecnik B, Tosolini M,
Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T,
Lafontaine L, Berger A, et al: Spatiotemporal dynamics of
intratumoral immune cells reveal the immune landscape in human
cancer. Immunity. 39:782–795. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar
|
|
38
|
Markovic SN, Erickson LA, Rao RD, Weenig
RH, Pockaj BA, Bardia A, Vachon CM, Schild SE, McWilliams RR, Hand
JL, et al: Malignant melanoma in the 21st century, part 1:
Epidemiology, risk factors, screening, prevention, and diagnosis.
Mayo Clin Proc. 82:364–380. 2007. View Article : Google Scholar
|
|
39
|
Osipov M and Sokolnikov M: Previous
malignancy as a risk factor for the second solid cancer in a cohort
of nuclear workers. SciMedicine J. 3:8–15. 2021. View Article : Google Scholar
|
|
40
|
Jiang X, Liu B, Nie Z, Duan L, Xiong Q,
Jin Z, Yang C and Chen Y: The role of m6A modification in the
biological functions and diseases. Signal Transduct Target Ther.
6:742021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Li Z, Kong B, Song C, Cong J, Hou
J and Wang S: Reduced m6A mRNA methylation is correlated
with the progression of human cervical cancer. Oncotarget.
8:98918–98930. 2017. View Article : Google Scholar
|
|
42
|
Kwok CT, Marshall AD, Rasko JEJ and Wong
JJL: Genetic alterations of m6A regulators predict
poorer survival in acute myeloid leukemia. J Hematol Oncol.
10:392017. View Article : Google Scholar
|
|
43
|
Cho SH, Ha M, Cho YH, Ryu JH, Yang K, Lee
KH, Han ME, Oh SO and Kim YH: ALKBH5 gene is a novel biomarker that
predicts the prognosis of pancreatic cancer: A retrospective
multicohort study. Ann Hepatobiliary Pancreat Surg. 22:305–309.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao X, Chen Y, Mao Q, Jiang X, Jiang W,
Chen J, Xu W, Zhong L and Sun X: Overexpression of YTHDF1 is
associated with poor prognosis in patients with hepatocellular
carcinoma. Cancer Biomarkers. 21:859–868. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Slebos RJC, Jehmlich N, Brown B, Yin Z,
Chung CH, Yarbrough WG and Liebler DC: Proteomic analysis of
oropharyngeal carcinomas reveals novel HPV-associated biological
pathways. Int J Cancer. 132:568–579. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Deng X, Su R, Feng X, Wei M and Chen J:
Role of N6-methyladenosine modification in cancer. Curr
Opin Genet Dev. 48:1–7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Slominski RM, Sarna T, Płonka PM, Raman C,
Brożyna AA and Slominski AT: Melanoma, melanin, and melanogenesis:
The yin and yang relationship. Front Oncol. 12:8424962022.
View Article : Google Scholar
|
|
48
|
Han W, Hu C, Fan ZJ and Shen GL:
Transcript levels of keratin 1/5/6/14/15/16/17 as potential
prognostic indicators in melanoma patients. Sci Rep. 11:10232021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Han Y, Li X, Yan J, Ma C, Wang X, Pan H,
Zheng X, Zhang Z, Gao B and Ji XY: Bioinformatic analysis
identifies potential key genes in the pathogenesis of melanoma.
Front Oncol. 10:5819852020. View Article : Google Scholar
|
|
50
|
Kodet O, Lacina L, Krejčí E, Dvořánková B,
Grim M, Štork J, Kodetová D, Vlček Č, Šáchová J, Kolář M, et al:
Melanoma cells influence the differentiation pattern of human
epidermal keratinocytes. Mol Cancer. 14:12015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
van de Rijn M, Perou CM, Tibshirani R,
Haas P, Kallioniemi O, Kononen J, Torhorst J, Sauter G, Zuber M,
Köchli OR, et al: Expression of cytokeratins 17 and 5 identifies a
group of breast carcinomas with poor clinical outcome. Am J Pathol.
161:1991–1996. 2002. View Article : Google Scholar
|
|
52
|
Wang YF, Lang HY, Yuan J, Wang J, Wang R,
Zhang XH, Zhang J, Zhao T, Li YR, Liu JY, et al: Overexpression of
keratin 17 is associated with poor prognosis in epithelial ovarian
cancer. Tumour Biol. 34:1685–1689. 2013. View Article : Google Scholar
|
|
53
|
Escobar-Hoyos LF, Yang J, Zhu J, Cavallo
JA, Zhai H, Burke S, Koller A, Chen EI and Shroyer KR: Keratin 17
in premalignant and malignant squamous lesions of the cervix:
Proteomic discovery and immunohistochemical validation as a
diagnostic and prognostic biomarker. Mod Pathol. 27:621–630. 2014.
View Article : Google Scholar
|
|
54
|
Chivu-Economescu M, Dragu DL, Necula LG,
Matei L, Enciu AM, Bleotu C and Diaconu CC: Knockdown of KRT17 by
siRNA induces antitumoral effects on gastric cancer cells. Gastric
Cancer. 20:948–959. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Harris TM, Du P, Kawachi N, Belbin TJ,
Wang Y, Schlecht NF, Ow TJ, Keller CE, Childs GJ, Smith RV, et al:
Proteomic analysis of oral cavity squamous cell carcinoma specimens
identifies patient outcome-associated proteins. Arch Pathol Lab
Med. 139:494–507. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kaz AM, Luo Y, Dzieciatkowski S, Chak A,
Willis JE, Upton MP, Leidner RS and Grady WM: Aberrantly methylated
PKP1 in the progression of Barrett's esophagus to esophageal
adenocarcinoma. Genes Chromosomes Cancer. 51:384–393. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Haase D, Cui T, Yang L, Ma Y, Liu H, Theis
B, Petersen I and Chen Y: Plakophilin 1 is methylated and has a
tumor suppressive activity in human lung cancer. Exp Mol Pathol.
108:73–79. 2019. View Article : Google Scholar
|
|
58
|
Xu Y, Zhao J, Dai X, Xie Y and Dong M:
High expression of CDH3 predicts a good prognosis for colon
adenocarcinoma patients. Exp Ther Med. 18:841–847. 2019.PubMed/NCBI
|
|
59
|
Zhai J, Li S, Sen S, Opoku-Anane J, Du Y,
Chen ZJ and Giudice LC: m6A RNA methylation regulators
contribute to eutopic endometrium and myometrium dysfunction in
adenomyosis. Front Genet. 11:71620202020. View Article : Google Scholar
|
|
60
|
Yan J, Wu X, Yu J, Zhu Y and Cang S:
Prognostic role of tumor mutation burden combined with immune
infiltrates in skin cutaneous melanoma based on multi-omics
analysis. Front Oncol. 10:5706542020. View Article : Google Scholar
|
|
61
|
Zhang C, Zhi WI, Lu H, Samanta D, Chen I,
Gabrielson E and Semenza GL: Hypoxia-Inducible factors regulate
pluripotency factor expression by ZNF217- and ALKBH5-mediated
modulation of RNA methylation in breast cancer cells. Oncotarget.
7:64527–64542. 2016. View Article : Google Scholar
|
|
62
|
Chao Y, Shang J and Ji W:
ALKBH5-m6A-FOXM1 signaling axis promotes proliferation
and invasion of lung adenocarcinoma cells under intermittent
hypoxia. Biochem Biophys Res Commun. 521:499–506. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Schöller E, Weichmann F, Treiber T, Ringle
S, Treiber N, Flatley A, Feederle R, Bruckmann A and Meister G:
Interactions, localization, and phosphorylation of the
m6A generating METTL3-METTL14-WTAP complex. RNA.
24:499–512. 2018. View Article : Google Scholar
|
|
64
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH,
Wang F, Wang TT, Xu QG, Zhou WP and Sun SH: METTL14 suppresses the
metastatic potential of hepatocellular carcinoma by modulating
N6-methyladenosine-dependent primary MicroRNA
processing. Hepatology. 65:529–543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen M, Wei L, Law CT, Tsang FHC, Shen J,
Cheng CLH, Tsang LH, Ho DWH, Chiu DKC, Lee JMF, et al: RNA
N6-methyladenosine methyltransferase-like 3 promotes liver cancer
progression through YTHDF2-dependent posttranscriptional silencing
of SOCS2. Hepatology. 67:2254–2270. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Georganaki M, Ramachandran M, Tuit S,
Núñez NG, Karampatzakis A, Fotaki G, van Hooren L, Huang H, Lugano
R, Ulas T, et al: Tumor endothelial cell up-regulation of IDO1 is
an immunosuppressive feed-back mechanism that reduces the response
to CD40-stimulating immunotherapy. Oncoimmunology. 9:17305382020.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Slominski RM, Raman C, Chen JY and
Slominski AT: How cancer hijacks the body's homeostasis through the
neuroendocrine system. Trends Neurosci. 46:263–275. 2023.
View Article : Google Scholar
|