Introduction
Lung cancer remains the leading cause of
malignancy-related mortality worldwide, in both men and women, with
over a million cases diagnosed annually; non-small cell lung cancer
(NSCLC) accounts for ~85% of all lung cancer cases (1). Few patients are diagnosed at an early
stage of NSCLC when curative resection is possible, and the
objective response rate of advanced disease to chemotherapy is very
low (2). Therefore, more effective
and less toxic therapeutic agents are being sought for the
treatment of NSCLC. The current state of knowledge has revealed
that cancer cells have aberrant signaling pathways in cell cycle
control, proliferation, invasion, and angiogenesis (3). Treatments targeted against these
abnormal processes in cancer cells have shown promise in the
management of lung cancer.
The phosphatidylinositol 3 kinase
(PI3K)/serine-threonine protein kinase AKT pathway is a prototypic
survival pathway that is constitutively activated in many types of
cancer. This pathway is an attractive therapeutic target in cancer
because: i) it is constitutively activated in many types of cancer;
ii) it serves as a convergence point for many growth stimuli; and
iii) it controls, through its downstream substrates, cellular
processes that contribute to the initiation and maintenance of
cancer cell proliferation. Moreover, the activation of the PI3K/AKT
pathway confers resistance to many types of cancer therapy and is a
negative prognostic factor for different tumor types (4–6).
mTOR, the mammalian target of rapamycin, is a
well-preserved serine/threonine kinase (MW, 289 kDa), with 95% of
its amino acid identity conserved from yeast to human and mouse. It
acts as a key kinase downstream of PI3K/AKT activation (7,8). mTOR
activation regulates downstream processes that ultimately result in
increased cell size and proliferation through translational
regulation by the downstream effectors: 4E-binding protein 1 and
phosphorylated S6 kinase (p-S6k) (4). Several lines of evidence have
suggested a critical role for mTOR in the development of lung
cancer. For example, positive staining for phosphorylated mTOR was
observed in 74% of human NSCLC tissue (9). Immunohistochemical staining for p-S6K,
a marker for mTOR activation, has revealed that malignant
progression from the normal lung, atypical alveolar hyperplasia
(AAH), bronchoalveolar carcinoma, to adenocarcinoma was accompanied
by progressively increasing levels of p-S6K (10). Therefore, mTOR is a promising
molecular target for lung cancer.
The mTOR inhibitor rapamycin is a natural macrolide
antibiotic produced by Streptomyces hygroscopicus, which
binds to FKBP-12 (FK506-binding protein) and the resultant complex
inhibits the protein kinase activity of mTOR. Rapamycin was
originally discovered as a potent antifungal agent, but rapamycin
and its derivatives have been explicitly designed and utilized for
their effectiveness as anticancer agents, stemming from their
superior solubility and stability properties (CCI-779, RAD001, and
AP23573). Rapamycin and its derivatives CCI-779, RAD001, and
AP23573 have been tested both as single agents and in combination
with EGFR inhibitors in phase I and II clinical trials against
several types of cancer, including NSCLC (6,11,12).
The safety and efficacy of rapamycin and its derivatives in these
clinical trials are indicative of the promising antitumor activity
of these agents over a broad range of dosage levels.
Rapamycin and its derivatives have antitumor
activities functioning through several mechanisms. As mTOR
regulates the translation of mRNAs that encode protein required for
G1 cell cycle progression and S-phase initiation, mTOR inhibition
of mTOR by rapamycin fundamentally results in cell cycle arrest.
Although the effect of rapamycin for tumor cells in the clinical
trials was thereby expected to be cytostatic, tumor regression was
observed, indicating that rapamycin may also induce apoptosis. To
date, reports on the ability of rapamycin to actually induce
apoptosis vary from one type of cancer to another, and the
mechanism by which rapamycin induces apoptosis in cancer cells is
poorly understood. Rapamycin has been shown to induce apoptosis in
BKS-2 immature B cell lymphoma (13), JN-desmoplastic small round cell
tumors-1 cells (14),
hepatocellular carcinoma cells (15), anaplastic large cell lymphoma
(16), IGROV1 ovarian carcinoma
cells (17), and rhabdomyosarcoma
cells (18,19). Conversely, rapamycin does not induce
apoptosis in SU-DHL-4 B lymphoma cells (20). Regarding lung cancer cell lines,
limited data are available, and they are inconsistent. Apoptosis
was not induced in KLN-205 and A549 NSCLC cell lines by rapamycin
alone (21), whereas apoptosis was
induced in Calu6 NSCLC cells when treated with the combination
rapamycin plus erlotinib (22).
The ability to induce apoptosis is an important
component of an antitumor profile when further development of
rapamycin and its derivatives are considered as anticancer agents,
and when combined with other cytotoxic anticancer agents.
Therefore, we examined the apoptotic ability of mTOR inhibitor
rapamycin and its mechanism of action in NSCLC cell lines.
Materials and methods
Cell lines and culture conditions
The NSCLC cell line Lu99 was provided by the Cell
Resource Center for Biomedical Research (Institute of Development,
Aging and Cancer, Tohoku University, Sendai, Japan). The 86-2 cell
line was provided by Dr S. Kobayashi (Miyagi Prefectural Semine
Hospital, Miyagi, Japan) through the Cell Resource Center for
Biomedical Research. The RERF-LC-AI lung cancer cell line was
obtained from RIKEN cell bank (Tsukuba, Japan). The Ma10 and Ma25
cell lines were provided by Dr T. Hirashima (Osaka Prefectural
Habikino Hospital, Osaka, Japan). Cells were cultured in DMEM
(Wako, Osaka, Japan) or RPMI 1640 medium (Wako) containing 10%
(v/v) fetal bovine serum, penicillin (100 IU/ml), and streptomycin
(100 μg/ml). Cells were grown at 37°C in humidified atmosphere with
5% CO2. Cells from exponentially growing cultures were
used in all experiments.
Preparation of mitochondrial and
cytosolic extracts
Cells were collected by centrifugation at 300 × g at
4°C for 5 min and then washed with ice-cold PBS. The cell pellets
were resuspended in MT buffer (250 mM sucrose, 20 mM HEPES, pH 7.5,
10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT,
and 0.1 mM PMAF), and homogenized. The homogenates were centrifuged
at 10,000 × g at 4°C for 30 min. The supernatant was used as the
cytosol fraction, and the pellet was resuspended in MT buffer
containing 0.1% Triton X-100 and 0.1% SDS as the mitochondrial
fraction. The protein concentration of each fraction was determined
by Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA, USA).
Western blot analysis
Cells were treated with various concentrations of
drugs. Cell lysates were denatured in sample buffer containing SDS,
and equal amount of total protein were separated on 15% SDS-PAGE
and transferred to Immobilon-P (Millipore, Bedford, MA, USA)
membranes. After blocking, the membranes were incubated with the
following primary antibodies: anti-Bcl-2 (Cell Signaling
Technology, Beverly, MA, USA), anti-cytochrome c (BD Biosciences
Pharmingen, San Diego, CA, USA), and anti-β-actin (Sigma-Aldrich,
St. Louis, MO, USA). The membranes were incubated with appropriate
horseradish peroxidase-conjugated secondary antibodies, and
detection was performed using ECL reagent (Amersham Biosciences,
Amersham, UK).
Cell proliferation assay
Cellular proliferation assay was performed by
modified MTT assay. Briefly, 1×104 cells were treated
with rapamycin in flat bottom 96-well plates at different
concentrations as indicated for 12, 24, 48, 60, and 72 h. WST-8
(2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt) solution (Dojindo Laboratories, Co., Kumamoto,
Japan) was added to each well and incubated for 2 h at 37°C. The
absorbance was measured at 450 nm using a Model 680 microplate
reader (Bio-Rad). Cell viability was calculated as the mean
absorbance of the wells containing treated cells divided by the
mean for the untreated control wells. All experiments were
performed at least in triplicate and were repeated 3 times.
Caspase activity assay
The activity of caspase-3 was determined by a
caspase colorimetric assay kit (Medical & Biological
Laboratories, Co., Nagoya, Japan), according to the manufacturer’s
protocol. Briefly, rapamycin-treated cells were washed with
ice-cold PBS and lysed in a lysis buffer. The cell lysates were
tested for caspase-3 activities by incubating with a
caspase-specific peptide conjugated to the molecule
p-nitroaniline. The chromophore p-nitroaniline
cleaved by caspase-3 was quantitated by measuring the absorbance at
a wavelength of 405 nm with a microplate reader (Bio-Rad).
Apoptosis assay
Cells were harvested after incubation with or
without 1 μM rapamycin for up to 5 days and treated with 5 μl of PE
Annexin V solution and 7-amino-actinomycin D (7-AAD) (PE Annexin V
Apoptosis Detection kit I, BD Biosciences Pharmingen). After
washing, dual parameter flow cytometric analysis was performed to
determine the percentage of apoptosis cells using a flow cytometer
(FACSCalibur, BD Biosciences Pharmingen).
Statistical analysis
The statistical calculations were performed with
SPSS 16.0 software (SPSS, Chicago, IL, USA). Student’s t-test was
used for a comparison between 2 groups, and P<0.05 was
considered statistically significant.
Results
Growth inhibitory effect of rapamycin on
NSCLC cell lines with p53 mutation
To evaluate the ability of rapamycin to induce
apoptosis without its confounding effects on cell cycle
progression, we used 5 NSCLC cell lines with p53 mutation (Table I). The histological subtypes of lung
cancer cell lines examined were adenocarcinoma (Ma10), giant cell
carcinoma (Lu99), large cell carcinoma (86-2 and Ma25), and
squamous cell carcinoma (RERF-LC-AI). Mutational status in
p53 of these cell lines have been described (23–27).
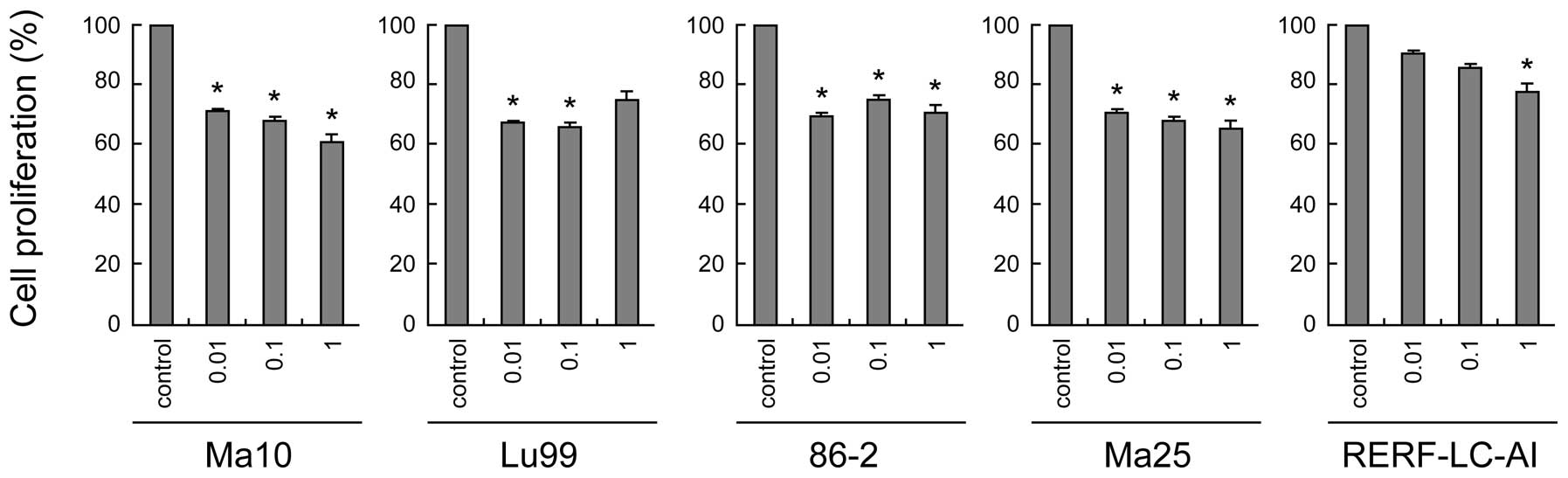
First, we examined the dose-dependent effect of rapamycin on the
proliferation of these cell lines. As shown in Fig. 1, rapamycin generally suppressed the
proliferation of all NSCLC cell lines, although the concentration
required for significant suppression was different in each cell
line. In most cell lines (Ma10, Lu99, 86-2, and Ma25 cells), very
low concentrations (0.01 μM) of rapamycin was sufficient for
significant suppression of cell proliferation, whereas 1 μM of
rapamycin was required in RERF-LC-AI cells. These data suggest that
rapamycin has a modest but obvious growth inhibitory effect on the
growth of NSCLC cell lines that possess p53 mutations.
 | Table IHistological properties and p53
mutation status of non-small cell lung cancer cell lines. |
Table I
Histological properties and p53
mutation status of non-small cell lung cancer cell lines.
| Cell line | Histological
sub-type | p53 status |
|---|
| Ma10 | Adenocarcinoma | Mutant |
| Lu99 | Giant cell
carcinoma | Mutant |
| 86-2 | Large cell
carcinoma | Mutant |
| Ma25 | Large cell
carcinoma | Mutant |
| RERF-LC-AI | Squamous cell
carcinoma | Mutant |
Rapamycin induces apoptosis in NSCLC cell
lines with p53 mutation
To evaluate the contribution of apoptosis to the
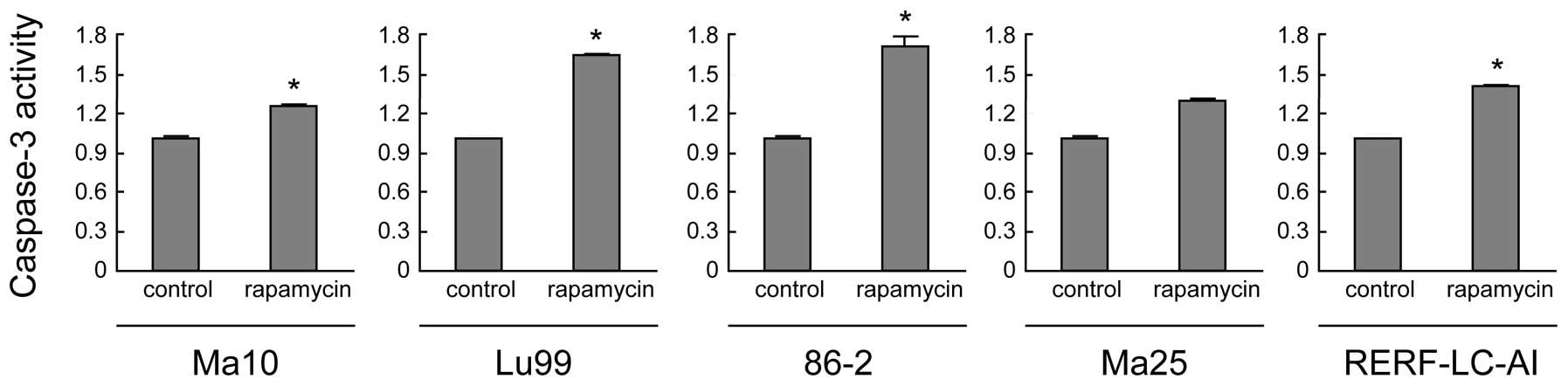
growth-inhibitory effects of rapamycin, we then evaluated the
apoptotic cell death induced by rapamycin. For this, we measured
the activity of caspase-3, which is the most important effector
caspase involved in rapamycin-induced apoptosis. As shown in
Fig. 2, caspase-3 activities were
significantly enhanced by rapamycin treatment of all NSCLC cell
lines, except for Ma25 (P<0.05). These data suggest the
potential ability of rapamycin to induce apoptosis in these lung
cancer cell lines.
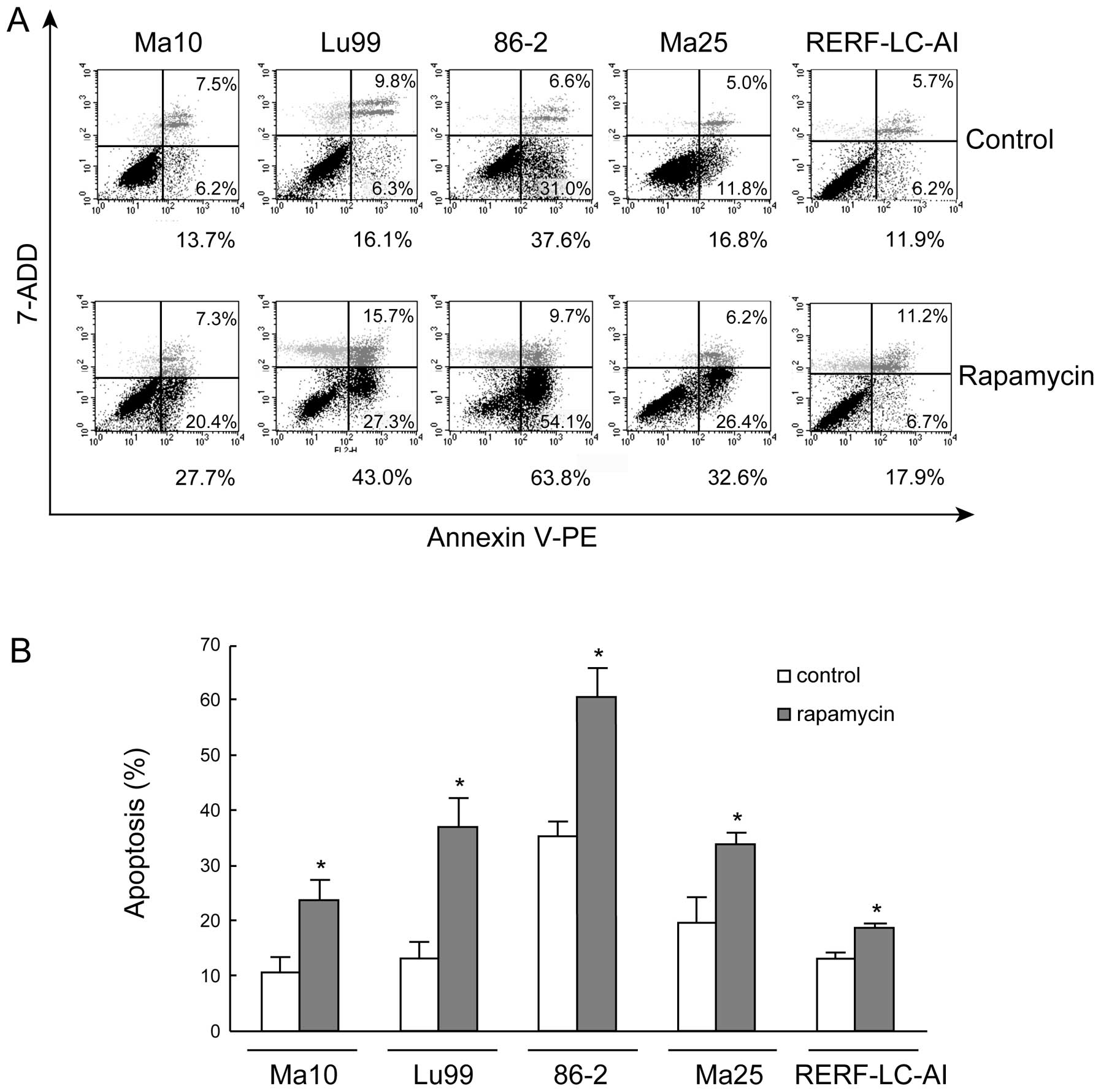
To further document the evidence of
rapamycin-induced apoptosis, we then stained the cells with Annexin
V-PE and the vital dye 7-AAD and analyzed the cells by flow
cytometry after rapamycin treatment. Representative results are
shown in Fig. 3A. In this analysis,
cells that were primarily Annexin V-negative and 7-ADD-negative
were viable and not undergoing apoptosis (Fig. 3A, bottom left). Cells that stained
positive for Annexin V and negative for 7-ADD were at an early
stage of apoptosis, in which the cell membrane integrity was
present (Fig. 3A, bottom right).
Cells that stained positive for both Annexin V and 7-ADD were
either at the end stage of apoptosis, in the process of undergoing
necrosis, or already dead (Fig. 3A,
top right). For Ma10, Lu99, 86-2, Ma25, and RERF-LC-AI cells, early
apoptotic cells in controls were 6.2, 6.3, 31, 11.8 and 6.2%,
respectively, and these frequencies represent an intrinsic
apoptotic cell death property of each cell line. After 5 days of 1
μM rapamycin treatment, for Ma10, Lu99, 86-2, and Ma25 cells, the
frequency of apoptotic cells markedly increased to 20.4, 27.3, 54.1
and 26.4%, respectively. In RERF-LC-AI cells, although the increase
of early-stage apoptosis was slight (≤6.7%), end-stage apoptosis
and the percentage of dead cells (Annexin V-positive and
7-ADD-positive cells) were markedly increased from 5.7 to 11.2% by
rapamycin treatment. Quantitative assessment of the percentage of
Annexin V-positive cells from 3 independent experiments showed that
apoptotic cell death was significantly increased by rapamycin
treatment in all NSCLC cell lines (Fig.
3B).
Rapamycin suppresses Bcl-2 expression and
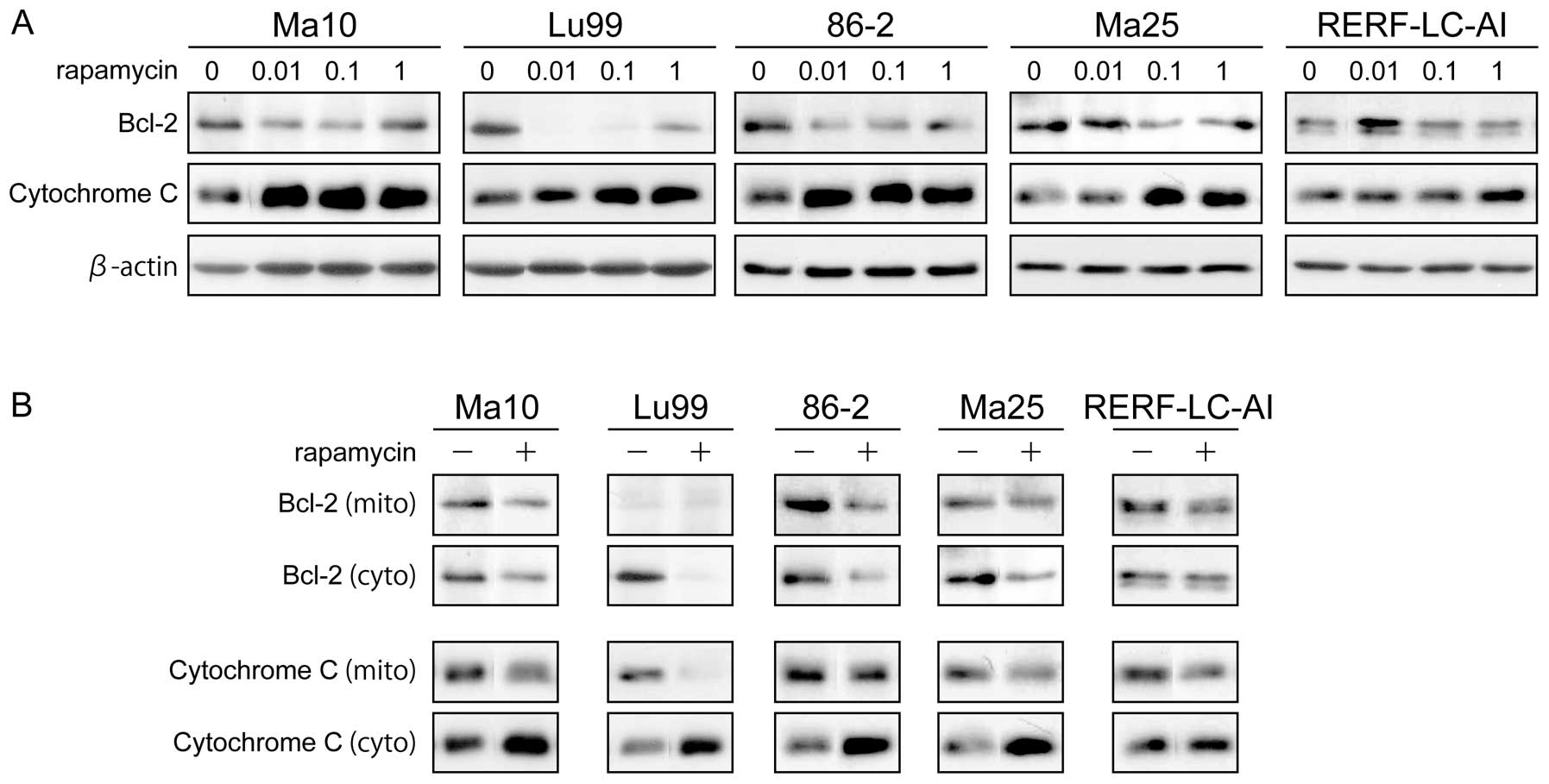
promotes mitochondrial cytochrome c release
To explore the mechanism by which rapamycin induces
apoptosis in p53-mutated NSCLC cell lines, we examined the
mitochondrial pathway of apoptosis. For this, we tested the
expressions of 2 representative regulators of apoptosis in this
pathway the, anti-apoptotic protein Bcl-2 and cytochrome c in the
cytoplasm. As shown in Fig. 4A, the
expression of Bcl-2 was markedly downregulated, and the expression
of cytochrome c in cytoplasm was markedly upregulated by rapamycin
treatment in a dose-dependent manner in all NSCLC cell lines. These
data suggest that rapamycin induces apoptosis through Bcl-2 and
cytochrome c.
Because Bcl-2 acts to prevent the release of
cytochrome c in mitochondria (28),
we further examined the effect of rapamycin for these 2 proteins at
the mitochondrial level using mitochondrial and cytosolic
fractions. As shown in Fig. 4B,
after treatment with 1 μM rapamycin for 48 h, Bcl-2 levels in both
mitochondrial and cytosolic fractions were decreased. Accordingly,
the cytochrome c levels in the cytosol were significantly increased
in all cell lines, while cytochrome c in mitochondrial fraction
decreased moderately to significantly. These data suggest that the
mechanism that rapamycin utilizes to reduce Bcl-2 expression levels
in the cytoplasm, accompanied by decreased levels of Bcl-2 in
mitochondria, leads to the release of cytochrome c from
mitochondria, and possible caspase activation.
Discussion
In the present study, we demonstrated the
rapamycin-induced apoptosis in NSCLC cell lines with p53
mutation. Mechanistically, we showed that rapamycin suppresses the
expression level of Bcl-2, which leads to an increased release of
cytochrome c from mitochondria and subsequent activation of caspase
cascades. These findings suggest that the mTOR inhibitor possesses
a novel mechanism for executing p53-independent apoptosis in
NSCLC cell lines.
Apoptosis induced by the inhibition of the PI3K/AKT
pathway have been well documented, whereas the ability of mTOR
inhibition to induce apoptosis and its potential mechanisms have
not been consistently reported. AKT has been reported to
phosphorylate and inhibit the function of the pro-apoptotic
proteins Bad (29) and Bax
(30), which subsequently execute
anti-apoptotic effects in cancer cells. Therefore, it is widely
accepted that the inhibition of AKT induces apoptosis in various
cancer cells. Conversely, regarding the inhibition of mTOR, the
reported results are inconsistent across cell types, and the
mechanisms in NSCLC cells remain unknown. For example, rapamycin
has been reported to induce a cellular stress response
characterized by rapid and sustained activation of the apoptosis
signal-regulating kinase 1 (ASK1), leading to elevated
phosphorylation of c-Jun and apoptosis in p53-mutated
rhabdomyosarcoma cells (19). The
relationship between cell cycle regulation and apoptosis has also
been investigated in several studies. Aguirre et al reported
that rapamycin blocks cell cycle progression in the G1/S phase by
downregulating cyclin D1 and CDK4, followed by increased expression
of caspase-3 and increased apoptotic cell death in a p53
wild-type ovarian cancer cell line (17). In contrast, Huang et al
reported continued G1 progression and S-phase entry resulting in a
rapamycin-induced apoptosis in p53-mutated rhabdomyosarcoma
cell line (18). In the
mitochondria, the key site of the apoptosis initiation, upregulated
expression of the pro-apoptotic protein Bax, and downregulation of
the anti-apoptotic protein Bcl-XL by rapamycin have been described
in small cell lung cancer (SCLC) cells (31), JN-DSRCT-1 cells (14), and HCC cells (15). Regarding Bcl-2, a well-recognized
and important anti-apoptotic regulator of the mitochondrial
pathway, downregulation by rapamycin, was described in SCLC
(31), HCC (15), anaplastic large cell lymphoma
(16), and rheumatoid synovial
cells (32). Our data indicating
that rapamycin downregulates Bcl-2 in NSCLC cell lines is
consistent with the aforementioned studies in other types of
cancer, with supporting evidence derived from the concomitant
release of cytochrome c from mitochondria.
Our data showing that rapamycin can induce apoptosis
in p53-mutated NSCLC cell lines is consistent with previous
studies reporting that rapamycin induces apoptosis in
p53-mutated cancer cells but not in p53 wild-type
cells (18). The only prior study
testing the effect of rapamycin on NSCLC cells did not detect
rapamycin-induced apoptosis in NSCLC cell lines with wild-type
p53(21). One possible
explanation for this discrepancy is that certain p53
wild-type tumors harbor mutations that can suppress apoptosis
downstream of p53, and rapamycin cannot affect the
anti-apoptotic function of these downstream effector molecules.
Another possibility is that the anti-apoptotic property of cancer
cells becomes dependent on proto-oncogene Bcl-2, but not on
p53, in p53 mutated cancer cells. Therefore, the
downregulation of Bcl-2 by rapamycin effectively induces apoptosis
for these cell lines. In either instance, since the p53
tumor suppressor gene is mutated in half of all cancer cells and is
indirectly inactivated in many others, our finding that rapamycin
induces apoptosis independently from p53 is an important
property, warranting further examination of its tumor specificity
as an anticancer drug.
One limitation of this study, similar to prior
studies that have reported on the downregulation of Bcl-2 by
rapamycin (15,16,31,32),
is that the mechanism by which rapamycin downregulates Bcl-2 has
not been elucidated. Regarding the remaining Bcl-2 family member
proteins, Tirado et al reported that the anti-apoptotic
protein Bcl-XL was downregulated by the inhibition of cap-dependent
translation caused by the inhibitory action of rapamycin on mTOR
(14). The pro-apoptotic protein
Bax was upregulated by the action of rapamycin which prevents Bax
degradation by mTOR-independent proteasome in tumor cells that have
undetectable levels of Bcl-2 (14).
Likewise, the expression of Bcl-2 family member proteins are known
to be tightly regulated by transcriptional and post-transcriptional
modifications (33), and further
efforts to elucidate the as yet unknown mechanisms of rapamycin
regulation of Bcl-2 expression is needed.
Our results suggested several potential therapeutic
approaches for treating NSCLC with rapamycin. First, because our
data indicate that the inhibition of mTOR induces apoptosis in both
a PI3K- and AKT-independent manner, the development of a dual
inhibitor of PI3K and mTOR (34),
such as NVP-BEZ235 (35), may be
more attractive alternative agents to inducing apoptosis in
p53-mutated cancer cells than rapamycin alone. Second, since
Bcl-2 has been revealed as the target of rapamycin-induced
apoptosis of NSCLC cell lines, Bcl-2 could be a novel molecular
marker to select for cells sensitive to rapamycin-induced
apoptosis. Further studies are needed to address these issues.
In conclusion, rapamycin has potential activity to
induce apoptosis in p53-mutated NSCLC cell lines, through
downregulation of Bcl-2 followed by cytochrome c release from
mitochondria. These findings provide new insights into the possible
antitumor mechanisms of rapamycin and may be useful in directing
future dual inhibition therapy targeting the PI3K/AKT pathway, as
well as a novel biological marker for rapamycin sensitivity in lung
cancer.
Acknowledgements
This study was supported by grants-in-aid for
Scientific Research (C) 21590994 (to H. Chikumi and E. Shimizu) and
22590863 (to E. Shimizu and H. Chikumi) from the Ministry of
Education, Science, and Culture, Sports, Science and Technology,
Japan.
References
|
1
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar
|
|
2
|
Delbaldo C, Michiels S, Rolland E, et al:
Second or third additional chemotherapy drug for non-small cell
lung cancer in patients with advanced disease. Cochrane Database
Syst Rev. 17:CD0045692007.
|
|
3
|
Hanahan D: The hallmarks of cancer. Cell.
100:57–70. 2000. View Article : Google Scholar
|
|
4
|
Vignot S, Faivre S, Aguirre D and Raymond
E: mTOR-targeted therapy of cancer with rapamycin derivatives. Ann
Oncol. 16:525–537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shaw RJ and Cantley LC: Ras, PI (3) K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sabers CJ, Martin MM, Brunn GJ, et al:
Isolation of a protein target of the FKBP12-rapamycin complex in
mammalian cells. J Biol Chem. 270:815–822. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lorenz MC and Heitman J: TOR mutations
confer rapamycin resistance by preventing interaction with
FKBP12-rapamycin. J Biol Chem. 270:27531–27537. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Balsara BR, Pei J, Mitsuuchi Y, et al:
Frequent activation of AKT in non-small cell lung carcinomas and
preneoplastic bronchial lesions. Carcinogenesis. 25:2053–2059.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wislez M, Spencer ML, Izzo JG, et al:
Inhibition of mammalian target of rapamycin reverses alveolar
epithelial neoplasia induced by oncogenic K-ras. Cancer Res.
65:3226–3235. 2005.PubMed/NCBI
|
|
11
|
Marinov M, Fischer B and Arcaro A:
Targeting mTOR signaling in lung cancer. Crit Rev Oncol Hematol.
63:172–182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gridelli C, Maione P and Rossi A: The
potential role of mTOR inhibitors in non-small cell lung cancer.
Oncologist. 13:139–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Muthukkumar S, RaMesh TM and Bondada S:
Rapamycin, a potent immunosuppressive drug, causes programmed cell
death in B lymphoma cells. Transplantation. 60:264–270. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tirado OM, Mateo-Lozano S and Notario V:
Rapamycin induces apoptosis of JN-DSRCT-1 cells by increasing the
Bax: Bcl-xL ratio through concurrent mechanisms dependent and
independent of its mTOR inhibitory activity. Oncogene.
24:3348–3357. 2005. View Article : Google Scholar
|
|
15
|
Zhang JF, Liu JJ, Lu MQ, et al: Rapamycin
inhibits cell growth by induction of apoptosis on hepatocellular
carcinoma cells in vitro. Transpl Immunol. 17:162–168. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vega F, Medeiros LJ, Leventaki V, et al:
Activation of mammalian target of rapamycin signaling pathway
contributes to tumor cell survival in anaplastic lymphoma
kinase-positive anaplastic large cell lymphoma. Cancer Res.
66:6589–6597. 2006. View Article : Google Scholar
|
|
17
|
Aguirre D, Boya P, Bellet D, et al: Bcl-2
and CCND1/CDK4 expression levels predict the cellular effects of
mTOR inhibitors in human ovarian carcinoma. Apoptosis. 9:797–805.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang S, Liu LN, Hosoi H, Dilling MB,
Shikata T and Houghton PJ: p53/p21CIP1 cooperate in
enforcing rapamycin-induced G1 arrest and determine the cellular
response to rapamycin. Cancer Res. 61:3373–3381. 2001.
|
|
19
|
Huang S, Shu L, Easton J, et al:
Inhibition of mammalian target of rapamycin activates apoptosis
signal-regulating kinase 1 signaling by suppressing protein
phosphatase 5 activity. J Biol Chem. 279:36490–36496. 2004.
View Article : Google Scholar
|
|
20
|
Calastretti A, Rancati F, Ceriani MC,
Asnaghi L, Canti G and Nicolin A: Rapamycin increases the cellular
concentration of the BCL-2 protein and exerts an anti-apoptotic
effect. Eur J Cancer. 37:2121–2128. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boffa DJ, Luan F, Thomas D, et al:
Rapamycin inhibits the growth and metastatic progression of
non-small cell lung cancer. Clin Cancer Res. 10:293–300. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buck E, Eyzaguirre A, Brown E, et al:
Rapamycin synergizes with the epidermal growth factor receptor
inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and
breast tumors. Mol Cancer Ther. 5:2676–2684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kishimoto M, Kohno T, Okudela K, et al:
Mutations and deletions of the CBP gene in human lung cancer. Clin
Cancer Res. 11:512–519. 2005.PubMed/NCBI
|
|
24
|
Okabe T, Okamoto I, Tamura K, et al:
Differential constitutive activation of the epidermal growthfactor
receptor in non-small cell lung cancer cells bearing EGFR gene
mutation and amplification. Cancer Res. 67:2046–2053. 2007.
View Article : Google Scholar
|
|
25
|
Mori T, Okamoto H, Takahashi N, Ueda R and
Okamoto T: Aberrant overexpression of 53BP2 mRNA in lung cancer
cell lines. FEBS Lett. 465:124–128. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park MJ, Shimizu K, Nakano T, et al:
Pathogenetic and biologic significance of TP14ARF alterations in
nonsmall cell lung carcinoma. Cancer Genet Cytogenet. 141:5–13.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nagai Y and Miyazawa H: Genetic
heterogeneity of the epidermal growth factor receptor in non-small
cell lung cancer cell lines revealed by a rapid and sensitive
detection system, the peptide nucleic acid-locked nucleic acid PCR
clamp. Cancer Res. 65:7276–7282. 2005. View Article : Google Scholar
|
|
28
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
del Peso L, Gonzalez-Garcia M, Page C,
Herrera R and Nunez G: Interleukin-3-induced phosphorylation of BAD
through the protein kinase Akt. Science. 278:687–689.
1997.PubMed/NCBI
|
|
30
|
Gardai SJ, Hildeman DA, Frankel SK, et al:
Phosphorylation of Bax Ser184 by Akt regulates its activity and
apoptosis in neutrophils. J Biol Chem. 279:21085–21095. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wangpaichitr M, Wu C, You M, et al:
Inhibition of mTOR restores cisplatin sensitivity through
down-regulation of growth and anti-apoptotic proteins. Eur J
Pharmacol. 591:124–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Migita K, Eguchi K, Ichinose Y, et al:
Effects of rapamycin on apoptosis of rheumatoid synovial cells.
Clin Exp Immunol. 108:199–203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang YJ, Duan Y and Zheng XF: Targeting
the mTOR kinase domain: the second generation of mTOR inhibitors.
Drug Discov Today. 16:325–331. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Serra V, Markman B, Scaltriti M, et al:
NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and
inhibits the growth of cancer cells with activating PI3K mutations.
Cancer Res. 68:8022–8030. 2008. View Article : Google Scholar : PubMed/NCBI
|