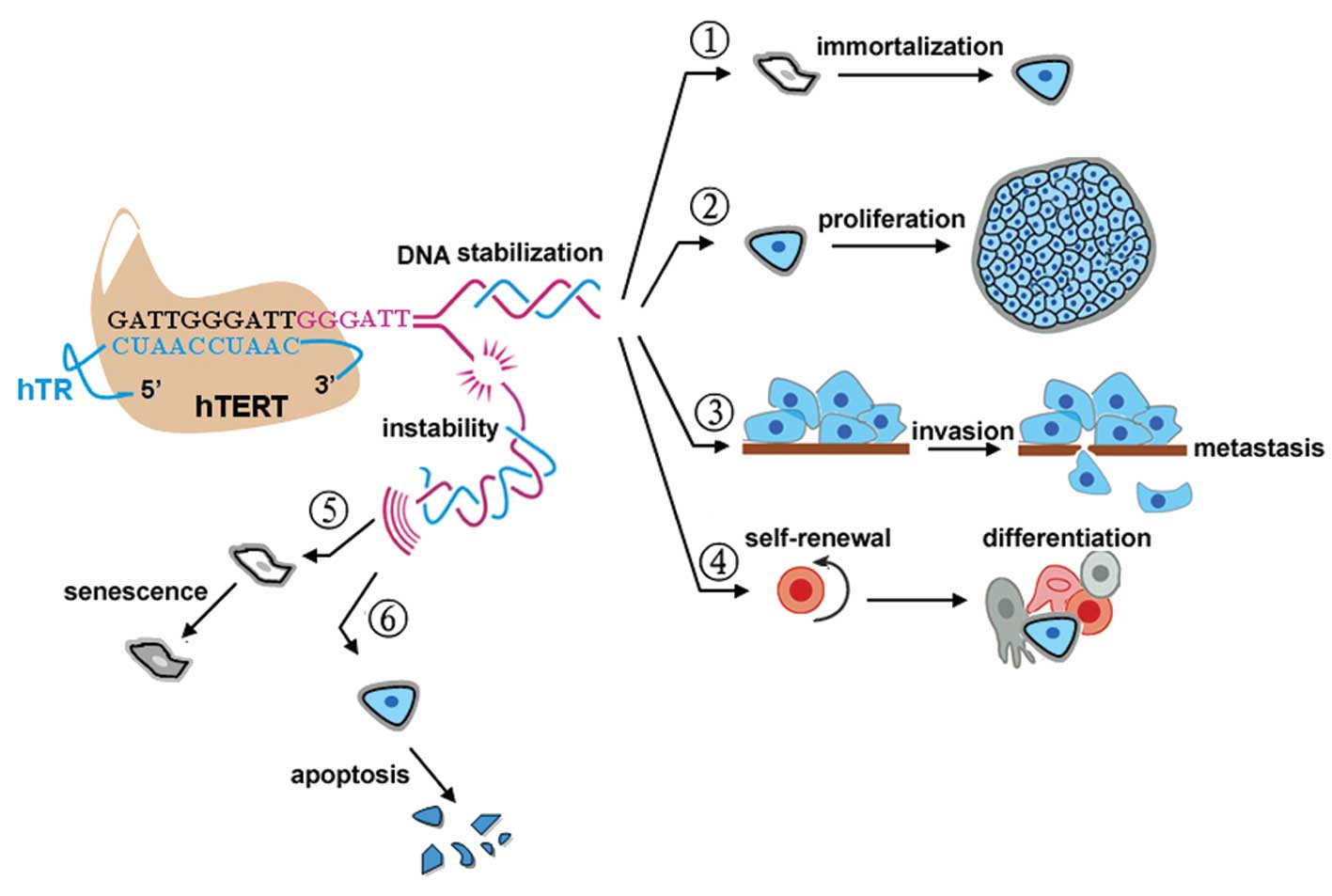
1. Introduction
Telomeres are the physical ends of chromosomes that
are composed of tandemly repeated G-rich DNA sequences in humans
and other vertebrates (1), but
progression through replication cycles will lead to
telomere-dependent pathways of cell cycle arrest, senescence and
mortality (2). To circumvent this
crisis, telomerase, a ribonucleoprotein complex, adds TTAGGG
repeats to the ends of the chromosomes (2). Human telomerase reverse transcriptase
(hTERT) is the catalytic subunit of telomerase and is involved in
the rate-limiting step in the activation of telomerase (3). The presence of hTERT is obligatory for
aberrant cell proliferation and immortalization in most tumors
(>85%) (4) and recent studies
have found that cancer stem cells are also hTERT-positive; however,
hTERT has little or no expression in normal somatic cells (4). Recently, Lü et al observed that
hTERT also plays a key role in the metastatic progression of
gastric cancer (5). This finding
suggests that, although hTERT itself is not an oncogene, hTERT
inhibition in humans appears to be a tumor suppressor mechanism in
both early- and late-stage cancers (6). Moreover, in contrast to hTERT, the
expression of the other two subunits of telomerase, human
telomerase RNA (hTR) and human telomerase associated protein (TP1),
did not parallel telomerase activity (7) and are independent of tumor stage and
histology. Based on the above features, hTERT is therefore
considered an ideal therapeutic target in human cancer (Fig. 1).
Currently, therapy of malignant tumors still
consists mainly of surgery, radiotherapy, chemotherapy and
combinations of these methods (8),
and these approaches for eradicating malignant tumor cells have
their own weaknesses. Recently, promising new hTERT-based therapies
have been developed, such as immunotherapy, suicide gene therapy,
and small-molecule interfering therapy. These treatments are
critical and specific for eradicating many cancer types. Therefore,
hTERT has been chosen as a molecular drug target in human cancer
with a broad therapeutic window.
This review summarizes recent advances in
hTERT-based drug development, and explores the experimental
challenges that need to be overcome to develop hTERT-based
therapies to treat human cancers.
2. Structure, function and regulation of
hTERT
hTERT is encoded by a single copy gene, mapped to
chromosome 5p15,33. The gene consists of 16 exons and 15 introns
spanning more than 37 kb (9). The
protein encoded by hTERT is composed of 1132 amino acid residues
(9) that contain seven functional
motifs conserved among reverse transcriptases (RTs) and one
telomerase specific motif. hTERT belongs to a group of RTs, which
is the key determinant of telomerase activity. Introduction of
hTERT into normal telomerase-negative human cells can prevent entry
into senescence, thereby extending the replicative life span of
these cells. Furthermore, Yu et al observed that, at the
cellular level, transfection of hTERT into U2OS cells, an
hTERT-negative malignant cell line, enabled telomerase activity and
further promoted their invasive and metastatic potential (10). It has been reported that TERT
controls stem cells via transcriptional regulation of a
developmental program converging on the Wnt/μ-catenin signaling
pathway (11). Sequence analysis
revealed that the hTERT promoter also lacks the TATA and CAAT
boxes, but contains binding sites for several transcription
factors, including the oncogenes SP1 and c-Myc (12), and the suppressor genes HER2 and p53
(13). Furthermore, downregulation
of hTERT expression was partially mediated through inhibition of
the DNA methyltransferase and histone acetyltransferase activities
of the hTERT promoter. Treatment with epigallocatechin-3-gallate
(EGCG) and a prodrug of EGCG, which remodel chromatin structures of
the hTERT promoter by reducing the level of acetyl-H3K9, acetyl-H4,
and acetyl-H3 binding to the hTERT promoter and facilitate the
binding of hTERT repressors to the hTERT regulatory region, result
in inhibition of hTERT transcription (14).
Attempts to regulate hTERT were initiated soon after
research into the structure and function of hTERT began; however,
hTERT expression is subject to multiple stages of control. These
factors may be critical regulators of hTERT, not only in cancer
cells but also in normal cells and the combined action of these
regulators will determine the final expression pattern of hTERT.
Therefore, the development of drugs that critically and
specifically target hTERT is essential for cancer treatment.
3. hTERT-based immunotherapy
Identification of hTERT as a good,
universal, tumor-associated antigen
The concept of cancer immunotherapy is based on
manipulating the host immune system to attack the cancer. However,
initial tumor immunotherapy proved disappointing because of the
lack of ‘resistance’ to tumors in many animal cancer models. This
lack of tumor immunogenicity may not arise because the tumor does
not spontaneously express antigens but because the self-antigens
expressed by the tumors do not effectively stimulate naïve or
activated T cells. Therefore, expression in differentiated, healthy
somatic cells resulted in cognate tumor immune escape.
Consequently, it has been a challenge for researchers to identify
ideal tumor-associated antigen (TAA) for the immunotherapy of
various tumors. An ideal TAA should have the following
characteristics: i) expression in most cancers to be broadly
applied to many different types of cancer; ii) restriction to the
tumor to avoid autoimmune reactions; iii) rare expression in
normal, mature tissues so that tolerance is broken; iv) possession
of an irreplaceable role in tumor progression to prevent tumor
variance and deletion; v) inducement of a strong immune response to
repress tumors; and vi) recognition by both major
histocompatibility complex (MHC)-I and -II in a restricted fashion
to elicit a CD4+ and CD8+ T lymphocyte
response (15).
To date, hTERT is the TAA that is most consistent
with the above criteria. Accumulated evidence based on both humans
and mice has shown that the same endogenous hTERT antigen-specific
CTL can induce the efficient lysis of tumor cells of different
histological origins and types (16). hTERT peptide-specific CTLs may
derive from the original T cell pool, and can be primed for
antitumor responses by hTERT antigen induction repeatedly (17). Immune escape by downregulation of
hTERT expression in tumors could also lead to progressive telomere
shortening and tumor death. Therefore, hTERT should be an ideal
tumor-associated antigen (Fig.
2).
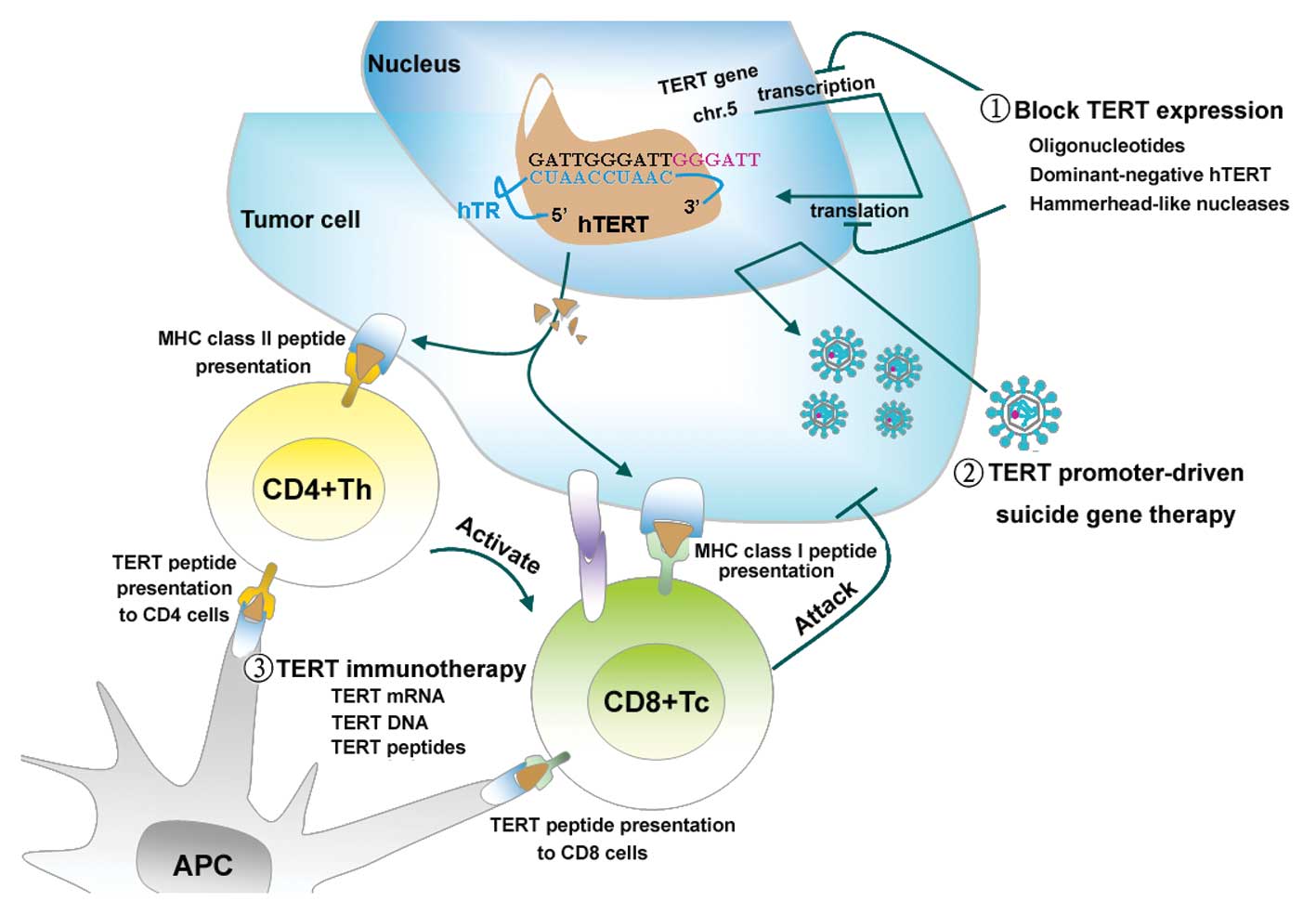 | Figure 2Three hTERT-based strategies for
killing tumor cells. The presence of hTERT, as the rate-limiting
step in the activation of telomerase, is a prerequisite for
carcinogenesis. Recent evidence has shown that the recently
developed hTERT-based therapies are successful in cancer treatment.
1, Studies regarding the regulation of hTERT were initiated soon
after research into the transcription, translation, transport
processing of hTERT was begun. These methodologies have the common
goal of downregulating hTERT expression at multiple stages of
biogenesis. Examples of these approaches include antisense
oligonucleotides, dominant-negative hTERT, and hammerhead-like
nucleases. 2, hTERT promoter-driven suicide gene therapy is based
on genes that encode proteins that control the replication of
microbial enzymes or oncolytic viruses that convert a prodrug into
a toxic substance. This therapy is also based on effective delivery
systems, such as liposomes and adenovirus vectors. 3, There is
increasing evidence that the hTERT antigen recognized in both an
MHC-I and -II restricted fashion elicits a CD4+ and
CD8+ T lymphocyte response. In the immune response, DCs
are the most efficient antigen-presenting cells. They have been
used extensively to activate antitumor effector T and B lymphocytes
through pulsing with hTERT RNA, DNA and an adenoviral vector
containing the hTERT gene (Ad-hTERT) to specifically eradicate
autologous hTERT-positive tumor cells. The relative advantages and
disadvantages of these different methodologies are reviewed in this
study. |
Activation of cytotoxic T-lymphocytes by
hTERT-pulsed dendritic cells (DCs)
DCs are the most efficient antigen-presenting cells
(APCs), and they have been extensively used to activate antitumor
effector T and B lymphocytes by presenting TAAs, including
previously unknown epitopes restricted in various MHC-II manners
(18). In addition, they are
capable of promoting the activation of natural killer (NK) cells,
which also have substantial antitumor effects. During the past few
years, many studies have shown that DCs pulsed with hTERT RNA, DNA
and an adenoviral vector containing the hTERT gene (Ad-hTERT)
result in the restoration of telomerase activity and give rise to a
strong CD8+ cytotoxic T-lymphocyte (CTL) response that
specifically eradicates autologous tumor cells (19). Interestingly, DCs transduced with
rAd-hTERT do not induce autoimmunity in normal control because the
hTERT protein found in normal tissues is below the threshold level
found in malignant cells that are recognized and lysed by
hTERT-specific CTLs (19). Another
study demonstrated that human DCs transfected with chimeric
hTERT/lysosome-associated membrane protein (LAMP-1) mRNA are
capable of stimulating a CD4+ T-cell reaction. This
reaction is required to stimulate and sustain an optimal
CD8+ CTL reaction in vivo (20). Moreover, the combination with
adjuvant may induce DC maturation and activation, which may also
enhance the immune response. However, obstacles to the development
of this therapy should be taken into account, including potential
adenovirus toxicity, high level of neutralizing antibodies, the
inability of DCs to migrate to the draining lymph nodes, and
activation or suppression by regulatory T-cells (Treg) (21). Therefore, further research regarding
engineered hTERT-modified DC vaccines must be performed to address
the above problems.
Identification of hTERT-based peptide
epitopes
Peptide-based vaccination against tumors has
progressed significantly based on the observations that
CD8+ CTLs lyse TAA-expressing tumor cells from multiple
tissues and that CD4+ helper T lymphocytes (HTLs) are
also activated by peptides derived from TAAs in the presence of DCs
loaded with cognate TAAs (15). The
I540 (ILAKFLHWL) was the first hTERT immunogenic peptide identified
by epitope prediction from melanoma, and it has entered phase III
clinical trials for melanoma treatment (22). To boost the anti-hTERT immune
response, 38 hTERT peptides have been subsequently identified that
are capable of inducing specific CTLs in vitro or in
vivo (23–33) (Table
I). The K973 peptide (KLFGVLRLK) (Table I) binds tightly to HLA-A3; it is
capable of inducing a specific CTL response in an MHC-restricted
manner, and it can also lyse a range of HLA-A2-positive tumor cell
lines derived from various histological origins (34). HLA-A3, expressed in 15–25% patients,
can expand the range of hTERT-based tumor immunotherapy to more
than 60% of people. The peptide sequences VYAETKHFL and VYGFVRACL
(Table I), derived from hTERT, are
capable of eliciting hTERT peptide-specific CD8+ CTLs
that generate a cytotoxic response against leukemia cells in an
HLA-A24-restricted manner (29).
HLA-A24 is the most common allele and is present in more than 60%
of Japanese and in nearly 20% of Europeans. These data strongly
suggest that hTERT-based specific CTL responses for cancer
immunotherapy can occur in most parts of the world. CD4+
Th cells exert helper activity for initiating, regulating, and
maintaining the CD8+ CTL response. Schroers et al
identified two MHC-II restricted antigen peptides from hTERT, the
L766 epitope (LTDLQPYMRQFVAHL) which incorporates to HLA-DR4, DR11,
and DR15 and the R672 epitope (RPGLLGASVLGLDDI) which incorporates
to HLA-DR1, DR7 and DR15 (35,36)
(Table I). These results illustrate
that the binding of hTERT peptides is promiscuous; one Th epitope
may be present within the different binding grooves of a single
MHC-II molecule, and this attribute plays a role in the induction
of a broader immune reaction. To enhance their ability to bind the
MHC molecule, the majority of low-affinity peptides are modified
with a tyrosine in the first position. This modification makes them
highly immunogenic, and they can then induce a potent immune
response for cancer treatment (26,36).
 | Table IhTERT antigenic peptides identified
for tumor immunity. |
Table I
hTERT antigenic peptides identified
for tumor immunity.
| Epitope | Sequence | Position | MHC | Cell line/in
vivo | CD4/CD8 | Refs. |
|---|
| M1 | MPRAPRCRA | 1–9 | HLA-B7 | +/+M | −/+ | (29) |
| R30 | RLGPQGWR | 30–37 | HLA-A2 | +/+M | −/+ | (26) |
| A68 | APSFRQVSCL | 68–77 | HLA-B7 | +/+M | −/+ | (34) |
| A167 | AYQVCGPPL | 167–175 | HLA-A24 | +/+H,M | −/+ | (28) |
| R277 | RPAEEATSL | 277–285 | HLA-B7 | +/+M | −/+ | (29) |
| V324 | VYAETKHFL | 324–332 | HLA-A24 | +/− | −/+ | (29) |
| Y325 | YLEPACAKY | 325–333 | HLA-A1 | +/+M | −/+ | (27) |
| R342 | RPSFLLSSL | 342–350 | HLA-B7 | +/+M | −/+ | (29) |
| R351 | RPSLTGARRL | 351–360 | HLA-B7 | +/+M | −/+ | (29) |
| Y386 |
YWQMRPLFLELLGNH | 386–400 | HLA-DP | +/− | +/− | (32) |
| D444 | DPRRLVQLL | 444–452 | HLA-B7 | +/+M | −/+ | (30) |
| V461 | VYGFVRACL | 461–469 | HLA-A24 | +/− | −/+ | (29) |
| F464 | FVRACLRRL | 464–472 | HLA-B7 | +/+M | −/+ | (30) |
| I540 | ILAKFLHWL | 540–548 | HLA-A2 | +/+H,M | −/+ | .(23) |
| L541 |
LAKFLHWLMSVYVVE | 541–555 | HLA-DP | +/− | +/− | (31) |
| L555 | LLRSFFYN | 555–563 | HLA-A2 | +/+M | −/+ | (33) |
| R572 | RLFFYRKSV | 572–580 | HLA-A2 | +/+M | −/+ | (24) |
| L573 |
LFFYRKSVWSKLQSI | 573–584 | HLA-DP | +/− | +/− | (31) |
| E611 |
EARPALLTSRLRFIPK | 611–626 | HLA-DR, DQ, DP | −/+H | +/− | (31) |
| R613 |
RPALLTSRLRFIPKP | 613–627 | HLA-DP | +/− | +/− | (31) |
| D637 | DYVVGARTF | 637–645 | HLA-A24 | +/+H,M | −/+ | (28) |
| A660 | ALFSVLNYERARRPGLLGA
SVLGLDDIHRA | 660–689 | HLA-A2, DR | +/− | +/+ | (32) |
| S663 |
SVLNYERARRPGLLG | 663–677 | HLA- DR | +/- | +/− | (32) |
| R672 |
RPGLLGASVLGLDDI | 672–686 | HLA-DR1, 7, 15 | +/+M | +/− | (35) |
| P673 |
PGLLGASVLGLDDIH | 673–687 | HLA-A2, DR | +/− | +/+ | (32) |
| G674 | GLLGASVLGL | 674–683 | HLA-A2 | +/− | −/+ | (32) |
| L766 |
LTDLQPYMRQFVAHL | 766–780 | HLA-DR1, 7, 15 | +/+M | +/− | (36) |
| C845 | CYGDMENKL | 845–853 | HLA-A24 | +/+H,M | −/+ | (28) |
| R865 | RLVDDFLLV | 865–873 | HLA-A2 | +/+H,M | −/+ | (23) |
| K973 | KLFGVLRLK | 973–981 | HLA-A2, A3 | +/− | −/+ | (34) |
| D988 | DLQVNSLQTV | 988–997 | HLA-A2 | +/+M | −/+ | (25) |
| T1088 | TYVPLLGSL | 1088–1096 | HLA-A24 | +/+H,M | −/+ | (28) |
| L1107 | LPGTTLTAL | 1107–1115 | HLA-B7 | +/+M | −/+ | (30) |
| L1123 | LPSDFKTIL | 1123–1131 | HLA-B7 | +/+M | −/+ | (30) |
| Y572a | YLFFYRKSV | 572–580 | HLA-A2 | +/+M,H | −/+ | (25) |
| Y988a | YLQVNSLQTV | 988–997 | HLA-A2 | +/+M | −/+ | (25) |
| R38a | RLGPQGWRV | 30–38 | HLA-A2 | +/+M,H | −/+ | (26) |
Applications of hTERT-based peptides for
cancer vaccines
It has not yet been determined whether these hTERT
single epitope vaccines mediate an immunodominant response. This
uncertainty may be due to their low molecular weight, rapid
decline, weak immunogenicity, and their ineffective processing and
presentation on cancer cells (37).
To overcome the above problems, various carrier proteins and fusion
proteins were used as vectors for delivering T-cell epitopes, while
there is a risk that foreign proteins with high molecular weights
will induce the immune response, rather than the target
polypeptides. Currently, single-epitope peptide vaccines are based
on HLA-restricted peptide predictive algorithms (38) and are used in patients with a
particular HLA type, leading to a narrow therapeutic window,
however, vaccinations with full-length mRNA encoding defined
antigens or with multi-epitope, superimposed antigens have many
known and unknown MHC-restricted epitopes.
hTERT peptides are processed and presented on the
surface of DCs in the form of multiple-epitope that produce
multiple CTL cell clones to induce an effective antitumor response
using the MHC-I and -II pathways and avoid immune escape due to the
loss of a single HLA allele. More attractively, the multiple
antigenic peptide (MAP) system based on the core matrix lysine
being coupled with four or eight strips of epitope monomer to form
a branch-like structure, represents a unique way to generate
anti-peptide antibodies (37).
Theoretically, the MAP structure not only strengthens the
specificity of the peptide chain structure and increases the
molecular weight of the epitope peptides but also induces a high
titer, high affinity antibody. Moreover, the Th epitopes are also
added to the MAP system to improve vaccine immunogenicity and
enhance CTL responses in an effective way. Recently, it has been
reported that the synthetic dendritic tandem multiple antigenic
hTERT epitope peptides consisting of I540 in a HLA-A02-restricted
manner, V461 in a HLA-A24-restricted manner, and L766 in a
HLA-DR-restricted manner are capable of inducing powerful antitumor
responses in SW480/A549 cells expressing HLA-A2, HepG2/SMMC-7721
cells expressing HLA-A24, and SKOV3 cells negative for HLA-A2/A24
(39). The results also showed that
the immunogenicity of the MAPs was better than a simple combination
of the three individual peptides.
4. hTERT-based suicide gene therapy
Cancer gene therapy depends on an effective vector
system and highly specific molecular targets. Currently, vector
systems consist of non-viral vectors, such as naked DNA injections
and liposomes, and viral vectors, such as adenovirus vectors. This
methodology is based on genes that encode proteins that control the
replication of microbial enzymes or oncolytic viruses that convert
a prodrug into a toxic substance (40) (Fig.
2). Because the enzymatic or oncolytic virus systems can
activate prodrugs only in the infected tumor cells, only the
suicide gene-positive cells can be inhibited and even killed, while
leaving normal somatic cells undamaged (40). Because of these advantages,
adenovirus vector systems are commonly used in suicide gene
therapy. The first generation of adenoviral vectors used to
establish E1/E3-deletion constructs were not replication-competent,
in order to be biologically safe. The increased interest in the
field is expected because of the augmentation of transgene
expression from the tumor-specific promoter without loss of target
specificity. Jacob et al constructed Ad/TRAIL-F/RGD, the
human tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL) gene under the transcriptional control of hTERT promoters.
That report also demonstrated that treatment with Ad/TRAIL-F/RGD
resulted in the apoptosis of human pancreatic and colon cancer
cells in vitro, as well as in vitro suppression of
tumor growth in an orthotopic implantation tumor model in the
pancreas of mice, while having a minimal effect on normal cells
(41). However, because the first
generation of adenoviral vectors is not replication-competent, the
treatment effect is confined only to the initially infected tumor
cells, limiting the long-term therapeutic effect. Another concern
is that the hTERT promoter activity with regard to targeted cancer
gene therapy is low in stem cells (42), and the question remains as to
whether this methodology can be applied to cancer stem cell
therapy.
Although suicide gene therapy has made progress, the
preclinical experimental results are still unsatisfactory. This
method still cannot compensate for the shortcomings of
replication-defective viruses in vivo, even if it is
mediating strong bystander effects that affect not only the tumor
cells transduced with the gene but also neighboring tumor cells.
Therefore, conditionally replicating adenoviruses are emerging as a
promising modality for cancer treatment. A novel replicative
adenoviral vector, AdhTERTp-E1A, in which the hTERT promoter
controls the selective replication of the vector in various tumor
cells was reported. This attribute, combined with its lack of
toxicity, can essentially promote oncolytic therapy as an antitumor
treatment. Compared to the replication-defective virus,
conditionally replicating adenoviruses under control of the hTERT
promoter reduce tumor cell proliferation, play a more effective
role in inhibiting tumor progress, are as safe as the former, and
have no apparent side effects. Moveover, different hTERT promoters
are used at different research institutions, various strategies
have been devised to improve hTERT promoter activity. Several
binding sites for transcription factors, including the
transcription activating factors Myc and SP1, have been cloned into
the adenoviral vector, and a series of constructs containing the
E-box or TATA box upstream of the transcriptional start site have
been cloned into the binding site for transcriptional activation of
the hTERT gene (43). In addition,
a single bicistronic adenoviral vector expressing pro-apoptotic
genes under control of the hTERT promoter was constructed using the
inducible gene expression system, providing a promising new
systemic delivery vehicle for oncolytic adenoviruses. The novel
vector utilizes the hTERT promoter to drive the expression of the
transactivator that binds to the tetracycline-responsive element
without tetracycline. This effect in turn causes the expression of
the tumor-specific Bax gene in various types of tumors (44).
hTERT-based suicide gene therapy may rapidly kill
hTERT-positive cells in tumors versus normal cells. However,
efficient delivery of gene therapy to tumors throughout the human
body is a major challenge, and immunological reactions may limit
the dosing of the vector system.
5. hTERT-based agents that block hTERT
expression and biogenesis
Although recent studies have screened different
therapies targeting hTERT, an effective and specific agent used in
clinical trials has not currently been found. During the past few
years, various hTERT-based agents, including antisense
oligonucleotides, overexpression of dominant-negative hTERT, and
hammerhead-like nucleases, have been described (Fig. 2). Indeed, antisense oligonucleotide
methodologies such as RNA interference were found efficient for
hTERT gene silencing. The RNA interference is a natural process in
which gene expression is silenced by small interfering RNAs (siRNA)
that are complementary to the messenger RNA in eukaryotic cells.
These siRNAs become incorporated into the RNA induced silencing
complex (RISC) through a cleavage mechanism (45). Liu et al demonstrated that
dendrimer-mediated shRNA against hTERT led to a marked reduction of
hTERT expression in human oral cancer cells and mouse tumor
xenografts (46). Gandellini et
al showed that hTERT-specific siRNAs effectively impaired tumor
cell growth and induced a variable degree of programmed cell death
(47). Additionally, microRNAs,
endogenous small RNAs, are hypothesized to be involved in the
regulation of the hTERT gene (48).
However, the relative efficacy and specificity of siRNAs needs to
be carefully assessed for cell-type-dependent global effects and
positional effects that may limit target accessibility by the
siRNAs.
Hammerhead and hairpin ribozymes are attractive
tools due to their small molecular size and the ease with which it
is possible to design these molecules for use in the gene therapy
field. Various studies have been performed regarding the use of
hammerhead and hairpin ribozymes for cancer therapy. Hao et
al designed a hammerhead anti-hTERT ribozyme and found that is
exhibited both growth suppression and rapid apoptosis effects on an
hTERT-positive carcinoma (49).
Apoptosis of the cancer cells was not accompanied by telomere
shortening, leaving no time for detecting replicative senescence.
The induction of rapid apoptosis of cancer cells through the
anti-hTERT ribozyme was via a direct mechanism rather than the
telomere shortening-senescence-apoptosis pathway.
Telomerase inhibition in tumor cells using a
dominant-negative hTERT mutant causes telomere shortening and tumor
suppression. Sachsinger et al demonstrated that ectopic
expression of a dominant-negative murine TERT mutant in renal tumor
cells resulted in telomere shortening and telomerase inactivation
(50).
Ribozymes, small interfering RNAs targeting TERT
mRNA, and gene therapies using overexpressing mutant TERT showed
good activity in some model systems. In addition to the ease of
screening these potent candidate agents, another advantage is that
these drugs directly and rapidly block hTERT expression and
biogenesis. Specifically, antisense oligonucleotides are not
removed from the cell by the efflux mechanisms responsible for the
multidrug resistance of cancers (6). However, several obstacles such as
effective transduction of these agents into cells without
degradation and the avoidance of target-off effects for safety
reasons need to be overcome before these anticancer therapies can
be used for treatment.
6. Conclusions and prospects
Targeting hTERT is a promising approach in cancer
treatment; however, there are many more opportunities and
challenges ahead. Although all anti-hTERT therapies force telomere
crisis, resulting in gradual cell apoptosis; however, tumors
continue to grow during the telomere-shortening time. The prognosis
of patients with cancer varies and may be dependent on telomerase
activity, telomere length, and even the microenvironment of the
tumor mass that can produce certain signals that decrease the
effect of anti-cancer therapies (6). Furthermore, we need to note that the
alternative lengthening of telomeres (ALT) mechanism represents a
telomerase-independent, recombination-based marker, referred to as
ALT-associated tumor-type, to maintain telomere length (51). Therefore, targeting hTERT most
likely needs to be coupled with other traditional therapeutic
modalities to create new treatment strategies that lead to wider
and more long-lasting response in cancer patients.
Currently, we remain at the early stages of clinical
development of hTERT as a cancer target. The current hTERT-based
drugs in preclinical and clinical trials are most likely not the
best hTERT-based drugs that we can ultimately develop. The
development of hTERT-based therapy provides an important new
strategy for anticancer treatment; however, preclinical trials are
needed to determine the dosage, time course of treatment,
indications, and possible side effects of this promising anticancer
treatment before entering into clinical use (15). hTERT-based therapies will be an
inevitable part of cancer treatments in the near future.
Acknowledgements
This study was supported by the Natural Science
Foundation of China (No. 81071845), the Chongqing Science Fund for
Distinguished Young Scholars (CSTC, 2009BA5045) and the National
Program for Key Basic Research Projects of China (973 Program, No.
2010CB529406).
Abbreviations:
|
ALT
|
alternative lengthening of
telomeres
|
|
APC
|
antigen-presenting cells
|
|
CTLs
|
cytotoxic T lymphocytes
|
|
DCs
|
dendritic cells
|
|
EGCG
|
epigallocatechin-3-gallate
|
|
HLA
|
human leukocyte antigen
|
|
HTL
|
helper T lymphocytes
|
|
hTERT
|
human telomerase reverse
transcriptase
|
|
LAMP-1
|
lysosome-associated membrane
protein
|
|
MAP
|
multiple antigenic peptide
|
|
MART-1
|
melanoma antigen recognized by T
cell-1
|
|
MHC
|
major histocompatibility complex
|
|
RISC
|
RNA induced silencing complex
|
|
RTs
|
reverse transcriptases
|
|
siRNA
|
small interfering RNAs
|
|
TAAs
|
tumor-associated antigens
|
|
TRAIL
|
tumor necrosis factor-related
apoptosis-inducing ligand
|
|
Treg
|
regulatory T cell
|
References
|
1
|
Cohen SB, Graham ME, Lovrecz GO, Bache N,
Robinson PJ and Reddel RR: Protein composition of catalytically
active human telomerase from immortal cells. Science.
315:1850–1853. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Campisi J, Kim SH, Lim CS and Rubio M:
Cellular senescence, cancer and aging: the telomere connection. Exp
Gerontol. 36:1619–1637. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zvereva MI, Shcherbakova DM and Dontsova
OA: Telomerase: structure, functions, and activity regulation.
Biochemistry. 75:1563–1583. 2010.PubMed/NCBI
|
|
4
|
Raptis S and Bapat B: Genetic instability
in human tumors. EXS. 303–320. 2006.
|
|
5
|
Lü MH, Deng JQ, Cao YL, Fang DC, Zhang Y
and Yang SM: Prognostic role of telomerase activity in gastric
adenocarcinoma: a meta-analysis. Exp Ther Med. 3:728–734.
2012.PubMed/NCBI
|
|
6
|
Harley CB: Telomerase and cancer
therapeutics. Nat Rev Cancer. 8:167–179. 2008. View Article : Google Scholar
|
|
7
|
Onoda N, Ogisawa K, Ishikawa T, Takenaka
C, Tahara H, Inaba M, Takashima T and Hirakawa K: Telomerase
activation and expression of its catalytic subunits in benign and
malignant tumors of the parathyroid. Surg Today. 34:389–393. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sorbe B, Bohr L, Karlsson L and Bermark B:
Combined external and intracavitary irradiation in treatment of
advanced cervical carcinomas: predictive factors for local tumor
control and early recurrences. Int J Oncol. 36:371–378. 2010.
|
|
9
|
Shay JW and Wright WE: Implications of
mapping the human telomerase gene (hTERT) as the most distal gene
on chromosome 5p. Neoplasia. 2:195–196. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu ST, Chen L, Wang HJ, Tang XD, Fang DC
and Yang SM: hTERT promotes the invasion of telomerase-negative
tumor cells in vitro. Int J Oncol. 35:329–336.
2009.PubMed/NCBI
|
|
11
|
Park JI, Venteicher AS, Hong JY, Choi J,
Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, et al:
Telomerase modulates Wnt signalling by association with target gene
chromatin. Nature. 460:66–72. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kanzawa T, Komata T, Kyo S, Germano IM,
Kondo Y and Kondo S: Down-regulation of telomerase activity in
malignant glioma cells by p27KIP1. Int J Oncol.
23:1703–1708. 2003.PubMed/NCBI
|
|
13
|
Papanikolaou V, Iliopoulos D, Dimou I,
Dubos S, Tsougos I, Theodorou K, Kitsiou-Tzeli S and Tsezou A: The
involvement of HER2 and p53 status in the regulation of telomerase
in irradiated breast cancer cells. Int J Oncol. 35:1141–1149.
2009.PubMed/NCBI
|
|
14
|
Meeran SM, Patel SN, Chan TH and
Tollefsbol TO: A novel prodrug of epigallocatechin-3-gallate:
differential epigenetic hTERT repression in human breast cancer
cells. Cancer Prev Res. 4:1243–1254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang YF, Tang XD, Gao JH, Fang DC and
Yang SM: Heparanase: a universal immunotherapeutic target in human
cancers. Drug Discov Today. 16:412–417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amarnath SM, Dyer CE, Ramesh A, Iwuagwu O,
Drew PJ and Greenman J: In vitro quantification of the
cytotoxic T lymphocyte response against human telomerase reverse
transcriptase in breast cancer. Int J Oncol. 25:211–217. 2004.
|
|
17
|
Titu LV, Loveday RL, Madden LA, Cawkwell
L, Monson JR and Greenman J: Cytotoxic T-cell immunity against
telomerase reverse transcriptase in colorectal cancer patients.
Oncol Rep. 12:871–876. 2004.PubMed/NCBI
|
|
18
|
Naito K, Ueda Y, Itoh T, Fuji N, Shimizu
K, Yano Y, Yamamoto Y, Imura K, Kohara J, Iwamoto A, et al: Mature
dendritic cells generated from patient-derived peripheral blood
monocytes in one-step culture using streptococcal preparation
OK-432 exert an enhanced antigen-presenting capacity. Int J Oncol.
28:1481–1489. 2006.
|
|
19
|
Chen L, Liang GP, Tang XD, Chen T, Cai YG,
Fang DC, Yu ST, Luo YH and Yang SM: In vitro anti-tumor immune
response induced by dendritic cells transfected with hTERT
recombinant adenovirus. Biochem Biophys Res Commun. 351:927–934.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su Z, Vieweg J, Weizer AZ, Dahm P, Yancey
D, Turaga V, Higgins J, Boczkowski D, Gilboa E and Dannull J:
Enhanced induction of telomerase-specific CD4(+) T cells using
dendritic cells transfected with RNA encoding a chimeric gene
product. Cancer Res. 62:5041–5048. 2002.PubMed/NCBI
|
|
21
|
Chen M, Chen G, Deng S, Liu X, Hutton GJ
and Hong J: IFN-beta induces the proliferation of
CD4+CD25+Foxp3+ regulatory T cells
through upregulation of GITRL on dendritic cells in the treatment
of multiple sclerosis. J Neuroimmunol. 242:39–46. 2012.
|
|
22
|
Liu JP, Chen W, Schwarer AP and Li H:
Telomerase in cancer immunotherapy. Biochim Biophys Acta.
1805:35–42. 2010.PubMed/NCBI
|
|
23
|
Minev B, Hipp J, Firat H, Schmidt JD,
Langlade-Demoyen P and Zanetti M: Cytotoxic T cell immunity against
telomerase reverse transcriptase in humans. Proc Natl Acad Sci USA.
97:4796–4801. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hernandez J, Garcia-Pons F, Lone YC, Firat
H, Schmidt JD, Langlade-Demoyen P and Zanetti M: Identification of
a human telomerase reverse transcriptase peptide of low affinity
for HLA A2.1 that induces cytotoxic T lymphocytes and mediates
lysis of tumor cells. Proc Natl Acad Sci USA. 99:12275–12280. 2002.
View Article : Google Scholar
|
|
25
|
Scardino A, Gross DA, Alves P, Schultze
JL, Graff-Dubois S, Faure O, Tourdot S, Chouaib S, Nadler LM,
Lemonnier FA, et al: HER-2/neu and hTERT cryptic epitopes as novel
targets for broad spectrum tumor immunotherapy. J Immunol.
168:5900–5906. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thorn M, Wang M, Kloverpris H, Schmidt EG,
Fomsgaard A, Wenandy L, Berntsen A, Brunak S, Buus S and Claesson
MH: Identification of a new hTERT-derived HLA-A*0201
restricted, naturally processed CTL epitope. Cancer Immunol
Immunother. 56:1755–1763. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schreurs MW, Hermsen MA, Geltink RI,
Scholten KB, Brink AA, Kueter EW, Tijssen M, Meijer CJ, Ylstra B,
Meijer GA and Hooijberg E: Genomic stability and functional
activity may be lost in telomerase-transduced human CD8+
T lymphocytes. Blood. 106:2663–2670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizukoshi E, Nakamoto Y, Marukawa Y, Arai
K, Yamashita T, Tsuji H, Kuzushima K, Takiguchi M and Kaneko S:
Cytotoxic T cell responses to human telomerase reverse
transcriptase in patients with hepatocellular carcinoma.
Hepatology. 43:1284–1294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Adotevi O, Mollier K, Neuveut C, Cardinaud
S, Boulanger E, Mignen B, Fridman WH, Zanetti M, Charneau P,
Tartour E, et al: Immunogenic HLA-B*0702-restricted
epitopes derived from human telomerase reverse transcriptase that
elicit antitumor cytotoxic T-cell responses. Clin Cancer Res.
12:3158–3167. 2006.PubMed/NCBI
|
|
30
|
Cortez-Gonzalez X, Sidney J, Adotevi O,
Sette A, Millard F, Lemonnier F, Langlade-Demoyen P and Zanetti M:
Immunogenic HLA-B7-restricted peptides of hTRT. Int Immunol.
18:1707–1718. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bernardeau K, Kerzhero J, Fortun A,
Moreau-Aubry A, Favry E, Echasserieau K, Tartour E, Maillere B and
Lang F: A simple competitive assay to determine peptide affinity
for HLA class II molecules: a useful tool for epitope prediction. J
Immunol Methods. 371:97–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Suso EM, Dueland S, Rasmussen AM, Vetrhus
T, Aamdal S, Kvalheim G and Gaudernack G: hTERT mRNA dendritic cell
vaccination: complete response in a pancreatic cancer patient
associated with response against several hTERT epitopes. Cancer
Immunol Immunother. 60:809–818. 2011. View Article : Google Scholar
|
|
33
|
Wang J, Yu L, Li J, Deng R and Wang X:
Characterization of a human telomerase reverse transcriptase
sequence containing two antigenic epitopes with high affinity for
human leucocyte antigen. Biotechnol Appl Biochem. 48:93–99. 2007.
View Article : Google Scholar
|
|
34
|
Vonderheide RH, Anderson KS, Hahn WC,
Butler MO, Schultze JL and Nadler LM: Characterization of
HLA-A3-restricted cytotoxic T lymphocytes reactive against the
widely expressed tumor antigen telomerase. Clin Cancer Res.
7:3343–3348. 2001.PubMed/NCBI
|
|
35
|
Schroers R, Huang XF, Hammer J, Zhang J
and Chen SY: Identification of HLA DR7-restricted epitopes from
human telomerase reverse transcriptase recognized by
CD4+ T-helper cells. Cancer Res. 62:2600–2605.
2002.PubMed/NCBI
|
|
36
|
Schroers R, Shen L, Rollins L, Rooney CM,
Slawin K, Sonderstrup G, Huang XF and Chen SY: Human telomerase
reverse transcriptase-specific T-helper responses induced by
promiscuous major histocompatibility complex class II-restricted
epitopes. Clin Cancer Res. 9:4743–4755. 2003.
|
|
37
|
Tang XD, Wang GZ, Guo J, Lu MH, Li C, Li
N, Chao YL, Li CZ, Wu YY, Hu CJ, Fang DC and Yang SM: Multiple
antigenic peptides based on H-2Kb-restricted CTL epitopes from
murine heparanase induce a potent antitumor immune response in
vivo. Mol Cancer Ther. 11:1183–1192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parker KC, Bednarek MA and Coligan JE:
Scheme for ranking potential HLA-A2 binding peptides based on
independent binding of individual peptide side-chains. J Immunol.
152:163–175. 1994.PubMed/NCBI
|
|
39
|
Niu BL, Du HM, Shen HP, Lian ZR, Li JZ,
Lai X, Wei SD, Zou LQ and Gong JP: Myeloid dendritic cells loaded
with dendritic tandem multiple antigenic telomerase reverse
transcriptase (hTERT) epitope peptides: a potentially promising
tumor vaccine. Vaccine. 30:3395–3404. 2012. View Article : Google Scholar
|
|
40
|
Murofushi Y, Nagano S, Kamizono J,
Takahashi T, Fujiwara H, Komiya S, Matsuishi T and Kosai K: Cell
cycle-specific changes in hTERT promoter activity in normal and
cancerous cells in adenoviral gene therapy: a promising implication
of telomerase-dependent targeted cancer gene therapy. Int J Oncol.
29:681–688. 2006.
|
|
41
|
Jacob D, Davis J, Zhu H, Zhang L, Teraishi
F, Wu S, Marini FC III and Fang B: Suppressing orthotopic
pancreatic tumor growth with a fiber-modified adenovector
expressing the TRAIL gene from the human telomerase reverse
transcriptase promoter. Clin Cancer Res. 10:3535–3541. 2004.
View Article : Google Scholar
|
|
42
|
Kyo S and Inoue M: Complex regulatory
mechanisms of telomerase activity in normal and cancer cells: how
can we apply them for cancer therapy? Oncogene. 21:688–697. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iliopoulos D, Satra M, Drakaki A,
Poultsides GA and Tsezou A: Epigenetic regulation of hTERT promoter
in hepatocellular carcinomas. Int J Oncol. 34:391–399.
2009.PubMed/NCBI
|
|
44
|
Gu J, Zhang L, Huang X, Lin T, Yin M, Xu
K, Ji L, Roth JA and Fang B: A novel single tetracycline-regulative
adenoviral vector for tumor-specific Bax gene expression and cell
killing in vitro and in vivo. Oncogene. 21:4757–4764. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao P, Wang C, Fu Z, You Y, Cheng Y, Lu
X, Lu A, Liu N, Pu P, Kang C, Salford LG and Fan X: Lentiviral
vector mediated siRNA knock-down of hTERT results in diminished
capacity in invasiveness and in vivo growth of human glioma cells
in a telomere length-independent manner. Int J Oncol. 31:361–368.
2007.
|
|
46
|
Liu X, Huang H, Wang J, Wang C, Wang M,
Zhang B and Pan C: Dendrimers-delivered short hairpin RNA targeting
hTERT inhibits oral cancer cell growth in vitro and in vivo.
Biochem Pharmacol. 82:17–23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gandellini P, Folini M, Bandiera R, De
Cesare M, Binda M, Veronese S, Daidone MG, Zunino F and Zaffaroni
N: Down-regulation of human telomerase reverse transcriptase
through specific activation of RNAi pathway quickly results in
cancer cell growth impairment. Biochem Pharmacol. 73:1703–1714.
2007. View Article : Google Scholar
|
|
48
|
Kota SK and Balasubramanian S: Cancer
therapy via modulation of micro RNA levels: a promising future.
Drug Discov Today. 15:733–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hao ZM, Luo JY, Cheng J, Li L, He D, Wang
QY and Yang GX: Intensive inhibition of hTERT expression by a
ribozyme induces rapid apoptosis of cancer cells through a telomere
length-independent pathway. Cancer Biol Ther. 4:1098–1103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sachsinger J, Gonzalez-Suarez E, Samper E,
Heicappell R, Muller M and Blasco MA: Telomerase inhibition in
RenCa, a murine tumor cell line with short telomeres, by
overexpression of a dominant negative mTERT mutant, reveals
fundamental differences in telomerase regulation between human and
murine cells. Cancer Res. 61:5580–5586. 2001.
|
|
51
|
Cesare AJ and Reddel RR: Alternative
lengthening of telomeres: models, mechanisms and implications. Nat
Rev Genet. 11:319–330. 2010. View Article : Google Scholar : PubMed/NCBI
|