Introduction
Malignant gliomas are among the most extensively
vascularized human tumors (1,2), which
is further supported by the identification of tumor blood vessel
density as an independent prognostic parameter for human astroglial
tumors (3). The prognosis of
malignant gliomas is still dismal despite aggressive treatment
attempts. Thus, alternative therapy strategies are needed. Since
malignant gliomas are characterized by extensive vascularization
and their proliferation is hallmarked by a distinct proliferative
vascular component (3), it seems to
be a logical consequence to apply antiangiogenic treatment
strategies to malignant gliomas. Recently, a large, randomized
phase II trail of bevacizumab, pan-VEGFR inhibitor cediranib and
other angiogenesis inhibitors was completed (4,5).
Preliminary results confirmed the safety of these agents and showed
a significant increase in the rate of progress-free survival for
patients with malignant gliomas.
There are several angiogenesis inhibitors in the
body, which help to suppress pathologic angiogenesis (6–8).
Alphastatin, an endogenous angiogenesis inhibitor, is a 24-amino
acid peptide derived from the amino terminus of the α chain of
human fibrinogen, and has potent antiangiogenic properties in
vitro and in tumor models (9–11). In
our previous detailed in vitro and in vivo study of
angiogenesis inhibition, we utilized a lentivirus-mediated gene
transfer system that allows local sustained long-term expression of
alphastatin (11) and established
models for the prevention of human glioma tumorigenesis. In this
study, the recombinant alphastatin lentiviruses were able to stably
infect human umbilical vein endothelial cell lines (HUVECs), and
exhibited potent inhibitory effects on HUVEC migration and
differentiation induced by vascular endothelial growth factor
(VEGF) or basic fibroblast growth factor (bFGF). Notably, our data
also showed that the stable expression of alphastatin in HUVECs
evidently inhibited human glioma tumorigenicity by inhibiting
angiogenesis. Furthermore, according to alterations in protein
expression during HUVEC tube formation induced by the activation of
mitogen-activated protein kinase (MAPK) signaling, we determined
that alphastatin inhibited VEGF- or bFGF-induced angiogenesis by
the blocking JNK and ERK phosphorylation pathway (11).
To assess the antitumor effects of secreted protein
alpha-statin and to extend these observations, in the present
study, we chose to establish a treatment model of human glioma to
assess whether alphastatin could be secreted in human glioma cells
transduced by an alphastatin gene delivery system, and
simultaneously evaluated the inhibition efficacy of tumor growth
following treatment with the secreted protein alphastatin, in order
to identify the antitumor mechanism of alphastatin. We report here
that the lentivirus-mediated gene delivery system of alphastatin is
an effective method for the treatment of human malignant glioma of
the brain.
Materials and methods
Cells and reagents
HUVECs, and human glioma cell lines SHG44 and U87
were purchased from Keygen (Yuhuatai, Nanjing, China). Cells were
maintained in DMEM supplemented with 10% FCS, 1% penicillin and 1%
streptomycin, and were grown at 37°C in a humidified incubator with
a gas phase of 5% CO2. Recombinant human VEGF and bFGF
were acquired from PeproTech EC (London, UK). Restriction enzymes
MluI and EcoRI were purchased from New England
Biolabs (Beverly, MA, USA).
Lentiviral vector production and
transduction
Construction of the recombinant lentivirus with
alphastatin was performed as previously described (11,12). A
four-plasmid-based lentiviral expression system was used in our
experiment. Lentiviral shuttle plasmid pWPXL-MOD was a kind gift
from the University of California, San Diego. Here, SpNT4 stands
for the fusion gene containing the human Neurotrophin-4 signal
peptide and pro-region. Briefly, to prepare pseudotyped lentiviral
vectors, lent-SpNT4-Al, 20 μg pWPXL/SpNT4-Al, 12 μg pMDLG/pRRE, 10
μg pRSV/REV and 10 μg pMD2.G were co-transfected into 293T cells,
which were then cultured in 10-cm dishes with Lipofectamine 2000
(Invitrogen, USA). The lentiviral vector titers were estimated by
flow cytometric analyses [fluorescence-activated cell sorting
(FACS)] as described previously (13). SHG44 and U87 cells were transduced
with Lent-GFP and Lent-SpNT4-Al at the multiplicity of infection of
5–10 in the presence of 6 mg/ml polybrene (Sigma-Aldrich, St.
Louis, MO, USA) for 72 h. SHG44-GFP (SHG44-Null),
SHG44-SpNT4-Al-GFP (SHG44-Al), U87-GFP (U87-Null) and
U87-SpNT4-Al-GFP (U87-Al), four types of tranduced cells were
harvested.
Determination of the expression of
secreted protein alphastatin
Based on the identification of secreted protein
alphastatin from HUVECs carrying the alphastatin gene as described
previously (11), we assessed
whether protein alphastatin was secreted from SHG44 and U87 cells
carrying the alphastatin gene in conditioned media using a HUVEC
migration assay. HUVEC migration is inhibited when alphastatin is
secreted in media. The cell migration assay was adapted from
Malinda et al(14) and
involves the use of a 24-well microchemotaxis chamber (AM; Neuro
Probe, Gaithersburg, MD, USA) with 8-μm pore size polycarbonate
membranes (Neuro Probe). SHG44, U87, SHG44-GFP, U87-GFP, SHG44-Al
and U87 cells were, respectively, cultured in the lower chamber for
another 24 h as subconfluent monolayers in DMEM (containing 1%
FCS), and HUVEC cell suspension (1×105 cells/ml) was
then added to the upper chambers and incubated at 37°C for 8 h with
VEGF or bFGF alone (10 ng/ml). Migrating cells adhering to the
undersurface of the membrane were fixed with 4% paraformaldehyde
and stained with hematoxylin and eosin. The migrating cells were
then counted using an optical microscope at ×20 magnification.
Proliferation assay
The MTT assay was performed as previously described
to measure the effects of secreted protein alphastatin on the in
vitro growth of SHG44 and U87 cells (15). Briefly, SHG44, U87, SHG44-GFP,
U87-GFP, SHG44-Al and U87-Al cells were, respectively, seeded into
96-well plates at a density of 3×104 cells/ml in DMEM
for 24, 48, 72 and 96 h. At each time point, a quarter volume of
MTT solution (2 mg MTT/ml in PBS) was added to each well, and the
plates were then incubated for 4 h at 37°C. The medium was
aspirated and the formazan crystals were dissolved in 150 μl of
DMSO buffered at pH 10.5. The absorbance was finally read at 590 nm
using a Dynex ELISA plate reader (Ashford, Middlesex, UK).
Immunofluorescence
The surface expression and localization pattern of
adherens junction proteins was assessed by immunocytochemistry in
VEGF- or bFGF-induced endothelial tube formation as previously
described (16). HUVECs
(2×105 cells/ml) were incubated for 2 h in 24-well
plates coated with 250 μl/well GFR Matrigel, either with or without
20 ng/ml bFGF or VEGF, in conditioned media that were collected
from SHG44-Al or U87-Al cells, each of which was cultured until
subconfluence in DMEM media for 24 h. The medium was then replaced
with DMEM containing 1% FCS for a further 24 h. Cells were then
washed and fixed with 4% (w/v) paraformaldehyde at room temperature
for 10 min and then permeabilized with 0.5% (v/v) Triton and
blocked with 5% (w/v) BSA in PBS for 30 min at room temperature.
They were then incubated with a monoclonal anti-VE-cadherin
antibody (1:100) overnight at 4°C. Cells were then washed before
incubation with FITC-conjugated goat anti-mouse IgG (1:1,000) for 2
h at 37°C. Finally, cells were washed again before being overlaid
with PBS and analyzed using a fluorescence microscope.
Western blot analysis
HUVECs were treated as described in the
Immunofluorescence section. For each phase during tube formation
HUVECs were then washed twice with ice-cold PBS and lysed in lysis
buffer [50 mM Tris-HCl (pH 7.9), 5 mM EDTA, 0.1% SDS, 10% glycerol,
0.2% Triton X-100, 5 μg/ml aprotinin, 1 mM PMSF and one protease
inhibitor cocktail tablet]. Protein concentrations were determined
using a BCA kit (Pierce, Rockford, IL, USA). Each protein lysate
(30 μg) was separated on SDS-PAGE gels and then transferred onto a
polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA).
The blots were incubated overnight at 4°C with primary antibodies
to VE-cadherin and β-actin, followed by a 1-h incubation with
horseradish peroxidase-conjugated secondary antibodies. Proteins
were visualized using an enhanced chemiluminescence detection
system (Amersham Life Science, Little Chalfont, UK).
Tumor growth inhibition
All animal experiments were approved by the
Institutional Animal Care and Use Committee of The Fourth Military
Medical University. All experiments were performed on 6-week-old
Balb/c mice weighing 20 g (n=13) purchased from Jackson Laboratory
(Bar Harbor, ME, USA). Animals were anesthetized with an
intraperitoneal injection of 10% chloral hydrate before
experiments. Then, 2×106 cells/ml (in 100 μl medium) of
viable SHG44 or U87 cells were respectively implanted
subcutaneously. Three days after the tumor cells were implanted
subcutaneously in the mice, recombinant lentNT4/Al
(lentivirus-SpNT4-alphastatin) was injected intratumorally twice
daily, for two days, with 20 μl 3.4×108 TU/ml virus.
Tumor volumes were measured daily using calipers (17), along with body weight and the state
of well-being.
Immunohistochemical analysis
Mice were sacrificed after 16 days. The tumor
tissues were excised, divided in half and fixed overnight in either
10% neutral-buffered formalin or a zinc-based fixative (18), before being embedded in paraffin wax
and sectioned. Formalin-fixed tissue sections were subsequently
stained with anti-CD31, periodic acid-Schiff (PAS) and hematoxylin
to assess microvessel density (MVD). The average vessel count
within three selected fields was then scanned at increased
magnification (x20), and MVD was expressed as the mean percentage
of vessel areas/field from three highly vascularized areas.
Statistical analysis
The data were expressed as the means ± SEM and
representative data from one of the three replicate experiments are
shown. The differences between the groups were determined using
one-way ANOVA followed by the Student's t-test. A P-value of
<0.05 was considered to indicate a statistically significant
difference.
Results
Expression of alphastatin in human glioma
cells
The recombinant lentivirus vectors for long-term
expression of alphastatin were constructed (Fig. 1A). We examined this gene delivery
system in SHG44 and U87 human glioma cell lines. Subsequently,
SHG44 or U87 cells that were transduced with recombinant lentiviral
vectors showed both nuclear and cytoplasmic expression of GFP.
Typically at a multiplicity of infection of 5–10, >96% of
lentivirally transduced SHG44 cells and >98% of U87 cells are
achieved. To confirm that SHG44-Al and U87-Al, but not SHG44-Nul l
and U87-Null cells, expressed biologically active alphastatin, we
next examined the effect of alphastatin expression in an in
vitro HUVEC migration assay, in which SHG44-Al and U87-Al cell
conditioned media resulted in significant inhibition of endothelial
cell migration, whereas conditioned media of SHG44-Null and
U87-Null cells did not inhibit endothelial cell migration (Fig. 1B). Based on these data, and combined
with previous early-stage results (11), we concluded that the lentiviral
vector efficiently transduced alphastatin into the SHG44 and U87
cells, and the SHG44-Al and U87-Al cells secreted active
alphastatin.
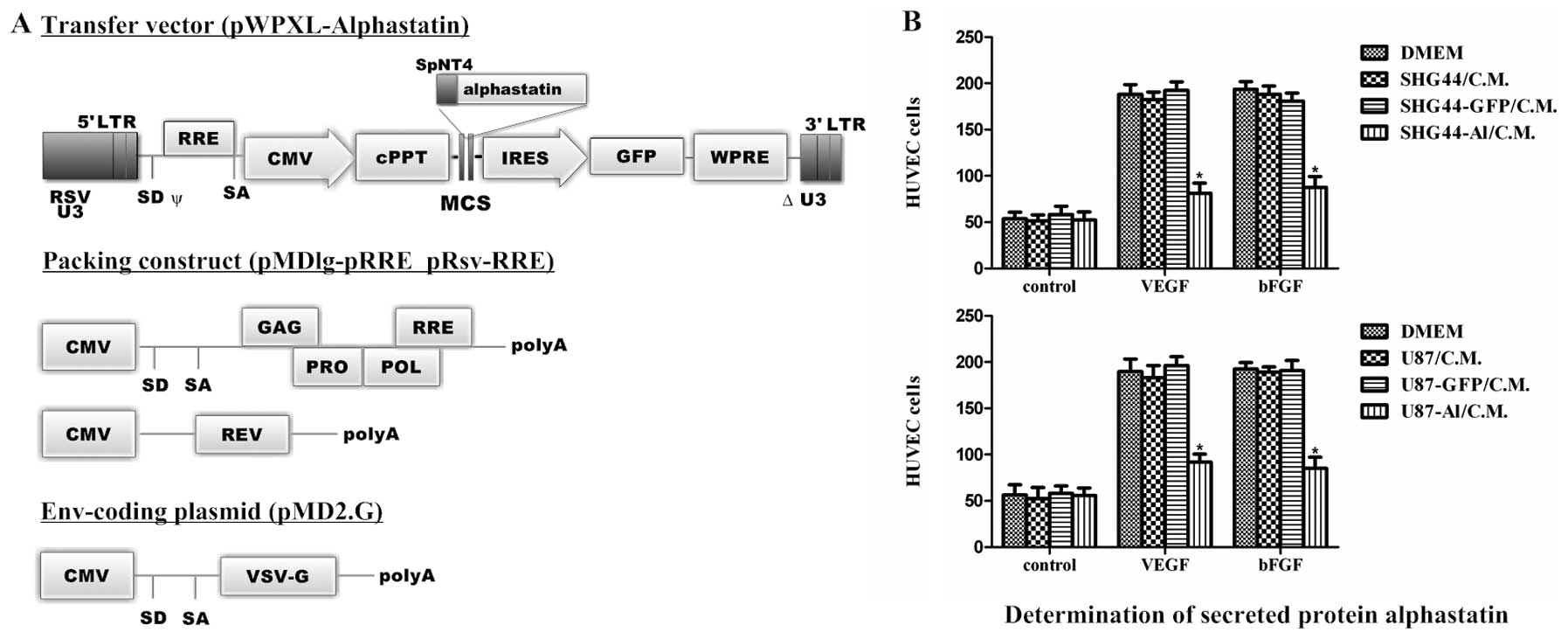 | Figure 1Illustration of the construction and
determination of alphastatin expression. (A) Self-inactivating
lentiviral vectors: The human neurotrophin-4 signal peptide and
pro-region fusion sequences (SpNT4) were cloned into lentiviral
transfer vector plasmid pWPXL/GFP, to construct pWPXL-SpNT4-Al.
Recombinant lentiviral vectors were produced by cotransfection of
the envelope plasmid pMD2.G, packaging plasmid pMDlg-RRE and
pRsv-RRE, and transfer vector plasmid. LTR, long terminal repeat;
RSV U3, U3 region from Rous sarcoma virus; ψ, packaging signal; SD,
splice donor; SA, splice acceptor; RRE, Rev response element; CMV,
cytomegalovirus promoter; MCS, multiple cloning site; IRES,
internal ribosome entry site; GFP, green fluorescent protein marker
gene; ΔU3, self-inactivating deletion in U3 region; polyA,
polyadenylation signal; VSV-G, vesicular stomatitis virus G protein
envelope. (B) HUVEC migration assay was performed in response to
medium or conditioned medium containing or not containing (control)
10 ng/ml VEGF or bFGF, to confirm that the SHG44-Al (upper panel)
and U87-Al cells (lower panel) expressed biologically active
alphastatin. All data shown are means ± SEM. *P<0.002
compared with the respective VEGF or bFGF group. |
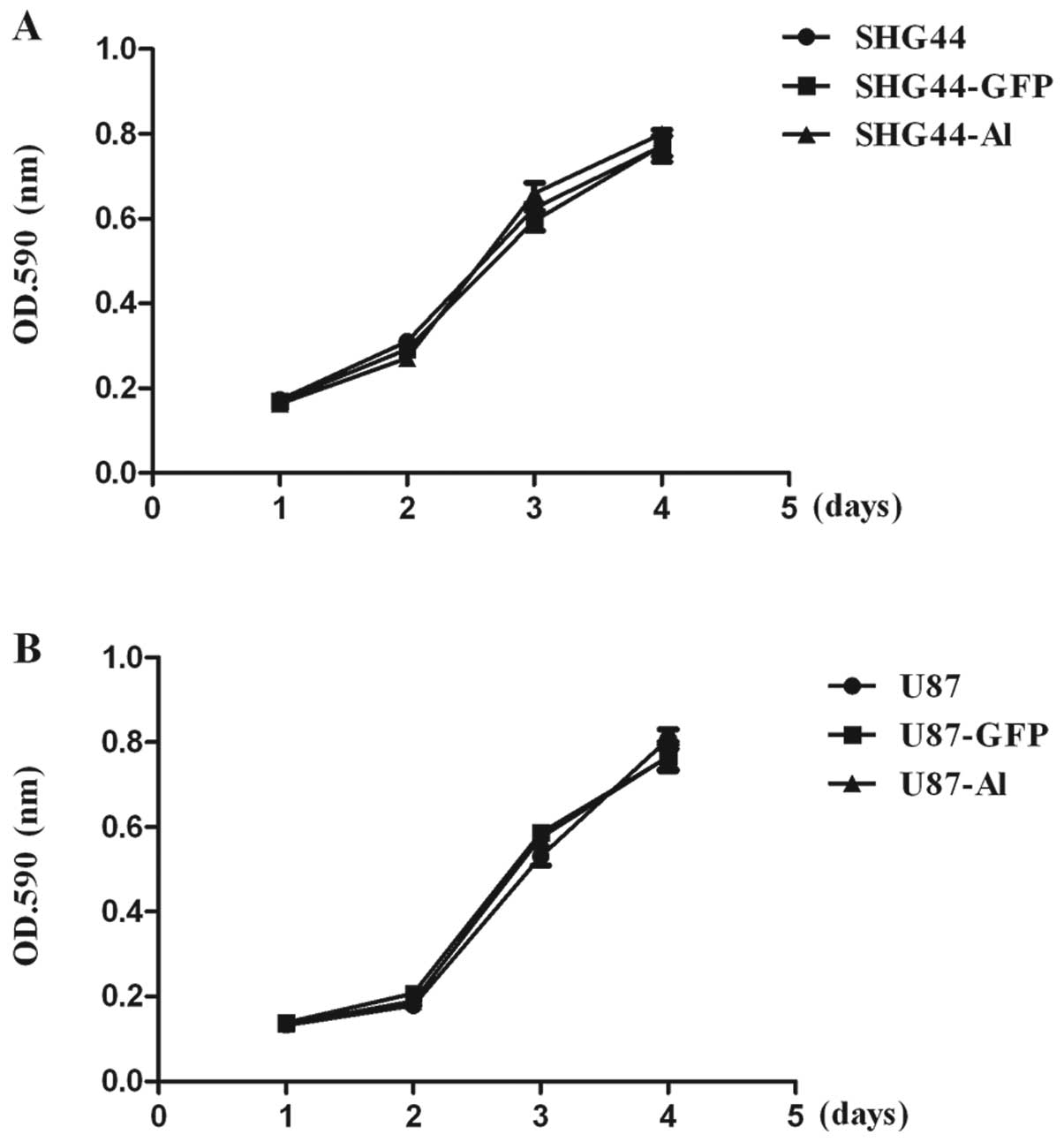
Effects of alphastatin on the growth of
SHG44 and U87 cells
We previously reported that secreted peptide
alphastatin inhibited angiogenesis by inhibiting HUVEC migration
and tube formation. We further sought to determine whether
alphastatin also inhibits the growth ability of human glioma cells
following treatment. To evaluate the effects of alphastatin
transduction and expression on the growth of SHG44 and U87 cells
in vitro, the relative growth rates of SHG44-GFP, U87-GFP,
SHG44-Al and U87-Al, and parental SHG44 and U87 cells were compared
using MTT methods. As shown in Fig.
2, there was no significant difference between the growth rates
of SHG44, SHG44-GFP and SHG44-Al cells, suggesting that neither the
lentivirus transduction procedure nor the overexpression of either
GFP or alphastatin affected the intrinsic rate of cellular
proliferation in these cells. Furthermore, in contrast, these data
confirmed that secreted protein alphastatin did not effect the
growth of SHG44 and U87 cells.
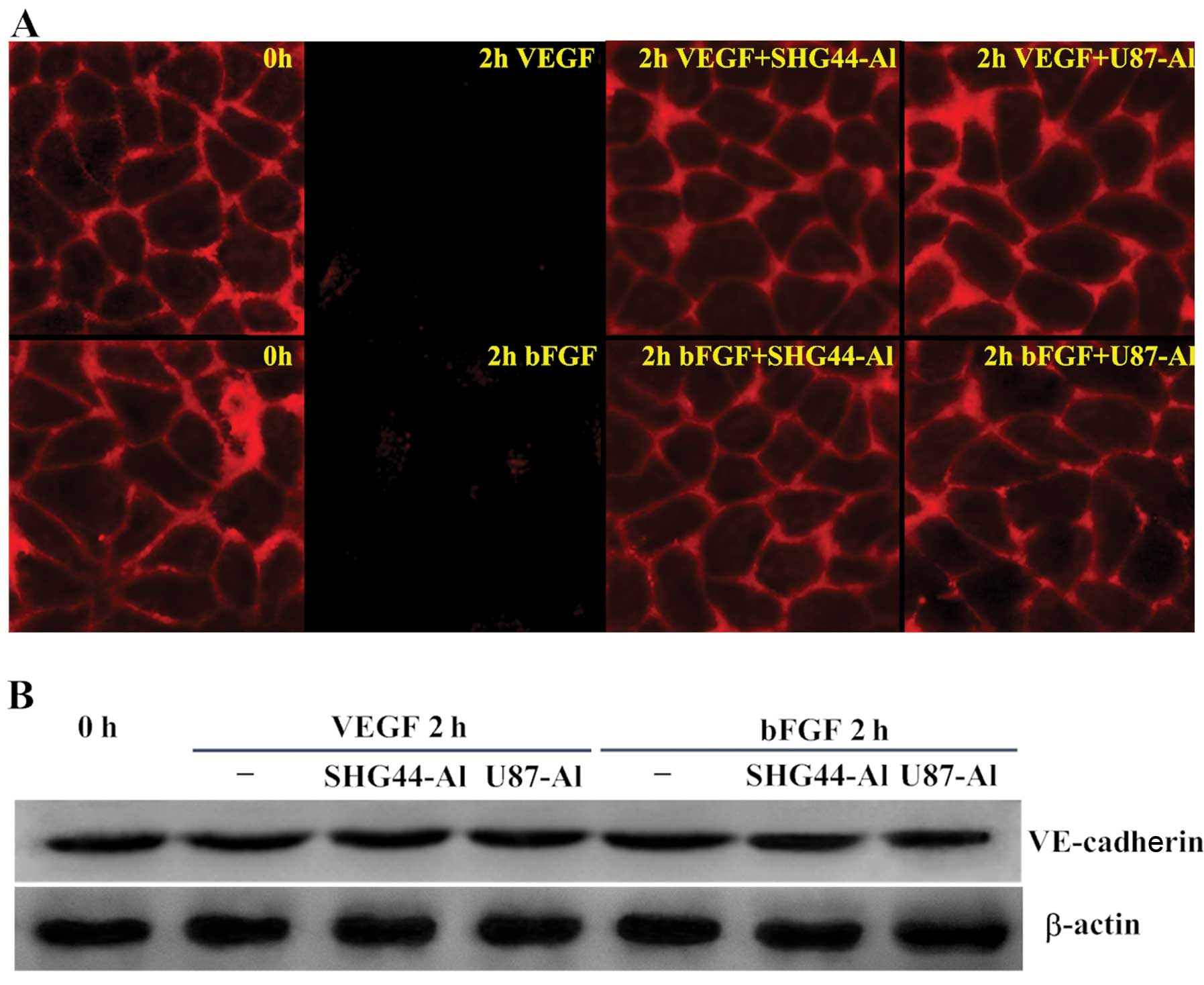
Effects of alphastatin on
VE-cadherin
Since VE-cadherin is crucial for controlling the
state of adherens junctions, which in turn regulate endothelial
cell-cell adhesion, cell motility, morphogenesis and intracellular
signaling pathways, this molecule has many clinical implications.
To gain insight into one possible molecular mechanism involved in
the inhibition of angiogenesis by alphastatin, we sought to
determine whether alphastatin influences the expression and
distribution of VE-cadherin during HUVEC tube formation.
Immunofluorescence analysis showed that VE-cadherin was localized
along areas of cell-cell contact. However, 1–2 h after stimulation
by VEGF or bFGF, the VE-cadherin fluorescence was greatly reduced,
and even disappeared from most cell-cell contact areas. Whereas,
when cells were treated with SHG44-Al or U87-Al conditioned media
prior to stimulation with VEGF or bFGF, VE-cadherin maintained
localization along areas of cell-cell contact (Fig. 3A). Furthermore, we measured the
total amount of VE-cadherin during HUVEC tube formation using
western blot analysis and found no significant changes in the
levels of total VE-cadherin protein (Fig. 3B). These findings suggest that
alphastatin inhibits the decrease in VE-cadherin induced by VEGF or
bFGF on the surface of HUVECs without any changes in the total
protein of VE-cadherin.
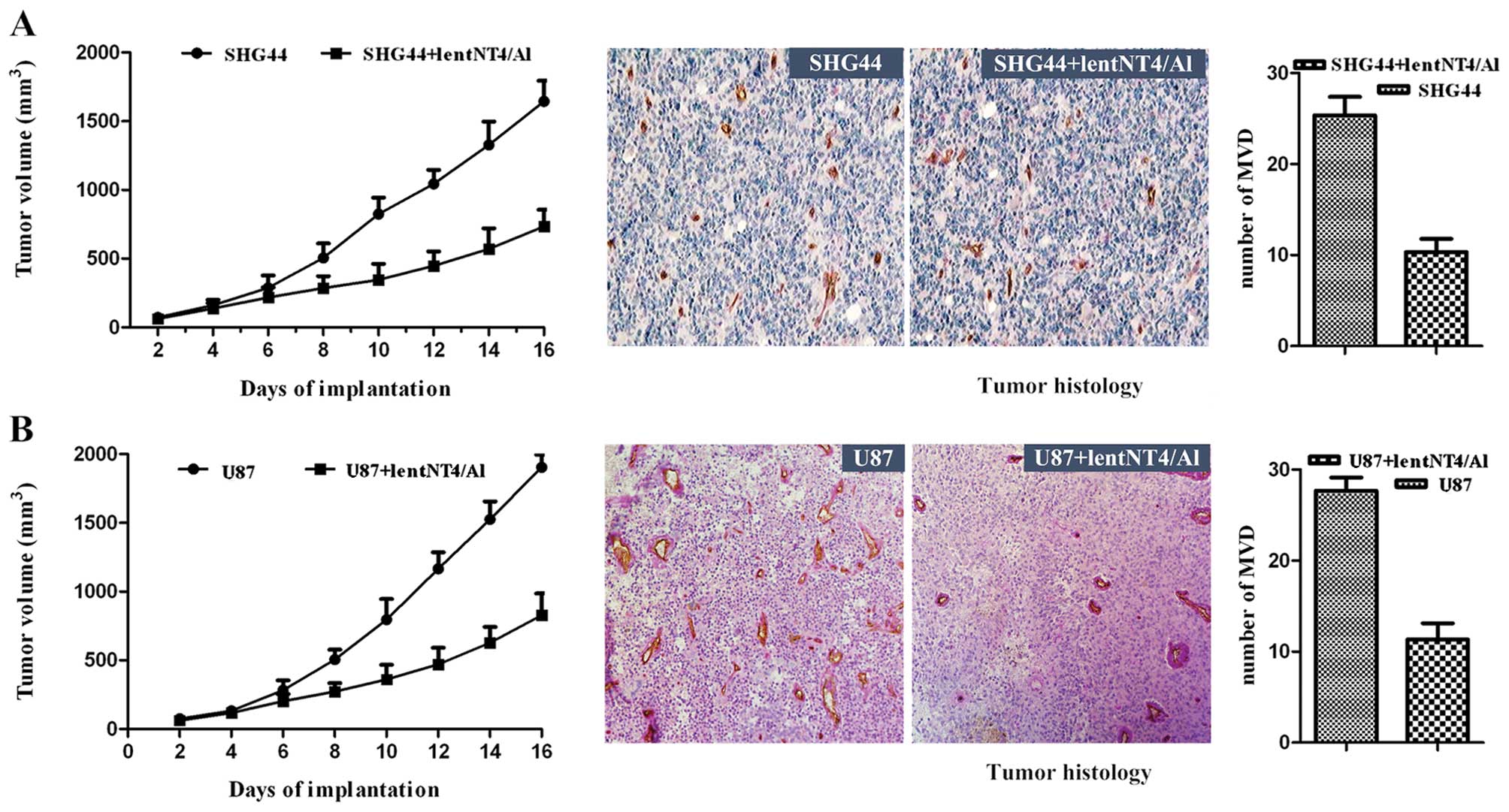
Effects of alphastatin on tumor
growth
We examined the inhibitory effect of secreted
protein alphastatin on tumor growth in vivo. SHG44 or U87
cells were subcutaneously implanted alone in mice. In all mice,
SHG44 tumors grew to a volume between 62 and 1,644 mm3,
and U87 tumors grew to a volume between 63 and 1,904 mm3
after 16 days of implantation. However, SHG44 tumors treated with
lentNT4/Al showed a significantly reduced growth rate, reaching a
final tumor volume of only 734±79 mm3 after 16 days
(P<0.018; Fig. 4A). The growth
rate of U87 tumors was also significantly reduced, reaching a final
volume of only 827±92 mm3 (P<0.022; Fig. 4B). Similar results were obtained in
two repeated experiments. Alphastatin was found to be well
tolerated by the animals, and had no significant effect on body
weight or general well-being (animals were not lethargic, and
intermittent hunching, tremors, or disturbed breathing patterns
were not observed). These data suggest that secreted protein
alphastatin effectively suppresses tumor growth in an established
mouse tumor model.
Effects of alphastatin on tumor
histology
Histologic analysis revealed a difference in MVD
between the two implanted xenograft tumor groups. MVD was
determined by the examination of CD31-stained sections at those
sites with the highest number of capillaries and small venules. The
positive expression of CD31 was visualized as brown-yellow or brown
granules in the cytoplasm of the vascular endothelial cells. In the
SHG44 tumors injected with the recombinant lentNT4/A1, the MVD was
significantly reduced (lentNT4/Al + SHG44 vs. SHG44, 10.33±1.453
vs. 25.33±2.028; P<0.039) (Fig.
4A). In U87 tumors injected with the recombinant lentNT4/A1,
the MVD was also significantly reduced (lentNT4/Al + U87 vs. U87,
11.33±1.764 vs. 27.64±1.475; P<0.001) (Fig. 4B). The results suggest that
alphastatin significantly inhibits tumor angiogenesis, and that
secreted protein alphastatin effectively suppresses tumor growth in
an established tumor model, as a result of antiangiogenetic
effects.
Discussion
Alphastatin is an endogenous angiogenesis inhibitor
with potent antiangiogenic properties which inhibits both the
migration and tube formation of human endothelial cells induced by
VEGF or bFGF, and in tumor models by inhibiting tumor growth
through suppressing angiogenesis. Using lentivirus-mediated gene
transfer, we previously found that lentNT4/Al-infected HUVECs were
able to stably secrete alphastatin, which exhibited a potent
inhibitory effect on HUVEC migration and differentiation. We also
explored the inhibitory effects of secreted protein alphastatin on
tumorigenesis in glioma, in which secreted protein alphastatin
significantly suppressed tumorigenesis, since the influence of
alphastatin on the growth of glioma cells and the effects of
alphastatin therapy on established tumors remain unclear. Here, we
demonstrated in SHG44-Al and U87-Al cells and an established
treatment model for human glioma that lentivirus-mediated gene
transfer of alphastatin is a potential therapeutic strategy against
glioma and, alphastatin inhibits tumor growth by blocking
VE-cadherin turnover.
Lentiviral vectors offer the advantage that they can
integrate their cDNA into both dividing and nondividing cells
(19). In addition, VSV-G
pseudotyped lentiviral vectors, which can bind to cell surface
phospholipids (20), have a
potential advantage in achieving efficient gene delivery to
endothelial cells. Furthermore, a third-generation SIN lentiviral
vector, in which the U3 region of the 3′ LTR (including the TATA
box) was deleted, abolishing any LTR promoter activity (21), enhanced the security level of
transgene expression. Accordingly, the NT4 signal peptide and
pro-region sequence were fused in-frame to the 5′ ends of each
coding sequence to ensure exogenous protein expressing in a
secretory manner (22–26), and the lentNT4/Al gene delivery
system was constructed. Moreover, since previous results showed
that secreted protein alphastatin had significantly attained a
saturated concentration of antiangiogenesis in respective
conditioned medium (11), there was
no concern with variation of concentration of secreted protein in
this study.
Since secreted protein alphastatin is sustainably
expressed in HUVEC-Al cells, and in vitro and in glioma
models significantly inhibit angiogenesis and gliomagenesis, we
aimed to ascertain whether alphastatin also remains stably
expressed in SHG44-Al and U87-Al cells. We investigated its
anti-glioma activity involving its efficacy in the treatment of
glioma, and the analysis of the treatment mechanism. In view of the
successful construction of the lentiviral vector containing the
NT4-alphastatin fusion gene and sustainable expression of
alphastatin in HUVEC-Al cells, in this study, we constructed
SHG-44-Al and U87-Al cells, and by morphological observation
confirmed whether alphastatin was secreted in these cells. We
showed that, in an endothelial migration assay, SHG-44-Al and
U87-Al conditioned media had a similar inhibitory effect on
endothelial cell migration, which suggested that alphastatin was
successfully secreted in SHG-44-Al and U87-Al cells. Notably,
although injection of the recombinant lentNT4/Al significantly
inhibited tumor growth, our data also showed that secreted protein
alphastatin did not affect the proliferative ability and migration
of glioma cells in vitro. This may be due to the fact that
alphastatin acts only on endothelial cell angiogenesis, but not on
glioma cells. Taken together, our data suggest that secreted
protein alphastatin is stably expressed in SHG44-Al and U87-Al
cells, leading to growth suppression of glioma by inhibition of
endothelial cell migration and tube formation.
VE-cadherin is a major component of endothelial
adherens junctions necessary for blood vessel integrity and
endothelial cell survival (27,28).
VE-cadherin has emerged as an adhesion molecule that plays
fundamental roles in microvascular permeability and in morphogenic
and proliferative events associated with angiogenesis (29–31).
Since blocking angiogenesis is a common strategy with which to
inhibit tumor growth, several approaches to inhibiting VE-cadherin
function have been suggested as possible antitumor therapies
(32–35). Several studies have demonstrated
that VE-cadherin turnover in endothelial cells suppresses
angiogenesis (16,36–39),
and is closely related to the MAPK pathways (40,41).
Our finding that alphastatin inhibits VE-cadherin downregulation
induced by VEGF or bFGF on the surface membrane of HUVECs without
any changes in total protein of VE-cadherin suggests that VEGF or
bFGF induces VE-cadherin redistribution in endothelial cells, which
is inhibited by alphastatin. Our previous data also indicate that
secreted protein alphastatin inhibits VEGF- or bFGF-induced
angiogenesis by suppressing the JNK and ERK kinase activation
pathways in HUVECs. We previously confirmed that the
antiangiogenetic activity of alphastatin most likely operates at a
post-receptor locus common to the VEGF and bFGF signaling pathways
(9). Thus, our data indicate that
the antiangiogenesis mechanism of alphastatin, by blocking VEGF and
bFGF post-receptor signaling pathways, suppresses JNK and ERK
kinase activation, which lead to VE-cadherin turnover on the
endothelial cell membrane, and then, inhibits endothelial cell
migration and differentiation. Finally, alphastatin not only
inhibits new vessel growth, but more important it significantly
causes regression of existing tumor vessels.
In conclusion, in our study of antiangiogenesis of
alphastatin, we describe the anti-glioma therapeutic strategy
involving lentivirus-mediated gene transfer of alphastatin and its
role in the inhibition of tumorigenesis. We have demonstrated that
in vivo, a subcutaneous injection of recombined lentiNT4-Al
resulted in the significantly growth inhibition of glioma in
autocrine manner (HUVEC-Al cells) or in paracrine manner (in
SHG-44-Al and U87-Al cells), and in vitro, offering a
molecular explanation for the mechanism of the effect of
alphastatin against glioma involving VE-cadherin. This study is the
first report on an anti-glioma therapeutic strategy involving
lentivirus-mediated gene transfer of alphastatin.
Acknowledgements
We thank Quan-Ying Wang, Guang-Xiao Yang (Xi'an
Huaguang Bioengineering Co.) for their expert technical assistance
concerning the construction of the recombined plasmid. We are also
grateful to Dr Cunjie Li and Huishou Wang for their help during the
experiments. This study was supported by the Postdoctoral Science
Foundation of China (no. 201150M1522) and the National Natural
Science Foundation of China (no. 30672162).
References
|
1
|
Tuettenberg J, Friedel C and Vajkoczy P:
Angiogenesis in malignant glioma - a target for antitumor therapy?
Crit Rev Oncol Hematol. 59:181–193. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jain RK, di Tomaso E, Duda DG, Loeffler
JS, Sorensen AG and Batchelor TT: Angiogenesis in brain tumours.
Nat Rev Neurosci. 8:610–622. 2007. View
Article : Google Scholar
|
|
3
|
Vajkoczy P and Menger MD: Vascular
microenvironment in gliomas. J Neurooncol. 50:99–108. 2000.
View Article : Google Scholar
|
|
4
|
Norden AD, Young GS, Setayesh K,
Muzikansky A, Klufas R, Ross GL, Ciampa AS, Ebbeling LG, Levy B,
Drappatz J, Kesari S and Wen PY: Bevacizumab for recurrent
malignant gliomas: efficacy, toxicity, and patterns of recurrence.
Neurology. 70:779–787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Batchelor TT, Duda DG, di Tomaso E,
Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J,
Hochberg FH, Benner T, Louis DN, Cohen KS, Chea H, Exarhopoulos A,
Loeffler JS, Moses MA, Ivy P, Sorensen AG, Wen PY and Jain RK:
Phase II study of cediranib, an oral pan-vascular endothelial
growth factor receptor tyrosine kinase inhibitor, in patients with
recurrent glioblastoma. J Clin Oncol. 28:2817–2823. 2010.
View Article : Google Scholar
|
|
6
|
Nyberg P, Xie L and Kalluri R: Endogenous
inhibitors of angiogenesis. Cancer Res. 65:3967–3979. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Folkman J: Endogenous angiogenesis
inhibitors. APMIS. 112:496–507. 2004. View Article : Google Scholar
|
|
8
|
Cao Y: Endogenous angiogenesis inhibitors
and their therapeutic implications. Int J Biochem Cell Biol.
33:357–369. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Staton CA, Brown NJ, Rodgers GR, Corke KP,
Tazzyman S, Underwood JC and Lewis CE: Alphastatin, a 24-amino acid
fragment of human fibrinogen, is a potent new inhibitor of
activated endothelial cells in vitro and in vivo. Blood.
103:601–606. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Qiu S, Song L, Yan Q and Yang G:
Secretory expression of p53(N15)-Ant following lentivirus-mediated
gene transfer induces cell death in human cancer cells. Cancer
Invest. 26:28–34. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo SW, Che HM and Li WZ: Anti-tumor
effect of lentivirus-mediated gene transfer of alphastatin on human
glioma. Cancer Sci. 102:1038–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiaojiang T, Jinsong Z, Jiansheng W,
Chengen P, Guangxiao Y and Quanying W: Adeno-associated virus
harboring fusion gene NT4-ant-shepherdin induces cell death in
human lung cancer cells. Cancer Invest. 28:465–471. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marr RA, Rockenstein E, Mukherjee A, Kindy
MS, Hersh LB, Gage FH, Verma IM and Masliah E: Neprilysin gene
transfer reduces human amyloid pathology in transgenic mice. J
Neurosci. 23:1992–1996. 2003.PubMed/NCBI
|
|
14
|
Malinda KM, Ponce L, Kleinman HK,
Shackelton LM and Millis AJ: Gp38k, a protein synthesized by
vascular smooth muscle cells, stimulates directional migration of
human umbilical vein endothelial cells. Exp Cell Res. 250:168–173.
1999. View Article : Google Scholar
|
|
15
|
Liu J, Kolath J, Anderson J, Kolar C,
Lawson TA, Talmadge J and Gmeiner WH: Positive interaction between
5-FU and FdUMP[10] in the inhibition of human colorectal tumor cell
proliferation. Antisense Nucleic Acid Drug Dev. 9:481–486.
1999.PubMed/NCBI
|
|
16
|
Wright TJ, Leach L, Shaw PE and Jones P:
Dynamics of vascular endothelial-cadherin and beta-catenin
localization by vascular endothelial growth factor-induced
angiogenesis in human umbilical vein cells. Exp Cell Res.
280:159–168. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grosios K, Holwell SE, McGown AT, Pettit
GR and Bibby MC: In vivo and in vitro evaluation of combretastatin
A-4 and its sodium phosphate prodrug. Br J Cancer. 81:1318–1327.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Beckstead JH: A simple technique for
preservation of fixation-sensitive antigens in paraffin-embedded
tissues. J Histochem Cytochem. 42:1127–1134. 1994. View Article : Google Scholar
|
|
19
|
Naldini L, Blömer U, Gallay P, Ory D,
Mulligan R, Gage FH, Verma IM and Trono D: In vivo gene delivery
and stable transduction of nondividing cells by a lentiviral
vector. Science. 272:263–267. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schlegel R, Tralka TS, Willingham MC and
Pastan I: Inhibition of VSV binding and infectivity by
phosphatidylserine: is phosphatidylserine a VSV-binding site? Cell.
32:639–646. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zufferey R, Dull T, Mandel RJ, Bukovsky A,
Quiroz D, Naldini L and Trono D: Self-inactivating lentivirus
vector for safe and efficient in vivo gene delivery. J Virol.
72:9873–9880. 1998.PubMed/NCBI
|
|
22
|
Nagai K, Oubridge C, Kuglstatter A,
Menichelli E, Isel C and Jovine L: Structure, function and
evolution of the signal recognition particle. EMBO J. 22:3479–3485.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Müller JP, Bron S, Venema G and van Dijl
JM: Chaperone-like activities of the CsaA protein of Bacillus
subtilis. Microbiology. 146:77–88. 2000.PubMed/NCBI
|
|
24
|
Tjalsma H, Antelmann H, Jongbloed JD,
Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax
WJ, Kuipers OP, Bron S, Hecker M and van Dijl JM: Proteomics of
protein secretion by Bacillus subtilis: separating the
‘secrets’ of the secretome. Microbiol Mol Biol Rev. 68:207–233.
2004. View Article : Google Scholar
|
|
25
|
Zanen G, Antelmann H, Meima R, Jongbloed
JD, Kolkman M, Hecker M, van Dijl JM and Quax WJ: Proteomic
dissection of potential signal recognition particle dependence in
protein secretion by Bacillus subtilis. Proteomics.
6:3636–3648. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan G, Egles C, Sun Y, Minichiello L,
Renger JJ, Klein R, Liu G and Jaenisch R: Knocking the NT4 gene
into the BDNF locus rescues BDNF-deficient mice and reveals
distinct NT4 and BDNF activities. Nat Neurosci. 3:350–357. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Corada M, Mariotti M, Thurston G, Smith K,
Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro
A, Ruco L, McDonald DM, Ward PA and Dejana E: Vascular
endothelial-cadherin is an important determinant of microvascular
integrity in vivo. Proc Natl Acad Sci USA. 96:9815–9820. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carmeliet P, Lampugnani MG, Moons L,
Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R,
Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens
D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A,
Poelmann R, Lupu F, Herbert JM, Collen D and Dejana E: Targeted
deficiency or cytosolic truncation of the VE-cadherin gene in mice
impairs VEGF-mediated endothelial survival and angiogenesis. Cell.
98:147–157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dejana E, Bazzoni G and Lampugnani MG:
Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp
Cell Res. 252:13–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dejana E, Spagnuolo R and Bazzoni G:
Interendothelial junctions and their role in the control of
angiogenesis, vascular permeability and leukocyte transmigration.
Thromb Haemost. 86:308–315. 2001.PubMed/NCBI
|
|
31
|
Stevens T, Garcia JG, Shasby DM,
Bhattacharya J and Malik AB: Mechanisms regulating endothelial cell
barrier function. Am J Physiol Lung Cell Mol Physiol.
279:L419–L422. 2000.PubMed/NCBI
|
|
32
|
Liao F, Li Y, O'Connor W, Zanetta L, Bassi
R, Santiago A, Overholser J, Hooper A, Mignatti P, Dejana E,
Hicklin DJ and Bohlen P: Monoclonal antibody to vascular
endothelial-cadherin is a potent inhibitor of angiogenesis, tumor
growth, and metastasis. Cancer Res. 60:6805–6810. 2000.PubMed/NCBI
|
|
33
|
Liao F, Doody JF, Overholser J, Finnerty
B, Bassi R, Wu Y, Dejana E, Kussie P, Bohlen P and Hicklin DJ:
Selective targeting of angiogenic tumor vasculature by vascular
endothelial-cadherin antibody inhibits tumor growth without
affecting vascular permeability. Cancer Res. 62:2567–2575.
2002.
|
|
34
|
Corada M, Zanetta L, Orsenigo F, Breviario
F, Lampugnani MG, Bernasconi S, Liao F, Hicklin DJ, Bohlen P and
Dejana E: A monoclonal antibody to vascular endothelial-cadherin
inhibits tumor angiogenesis without side effects on endothelial
permeability. Blood. 100:905–911. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pierce M, Wang C, Stump M and Kamb A:
Overexpression of the beta-catenin binding domain of cadherin
selectively kills colorectal cancer cells. Int J Cancer.
107:229–237. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abraham S, Yeo M, Montero-Balaguer M,
Paterson H, Dejana E, Marshall CJ and Mavria G:
VE-Cadherin-mediated cell-cell interaction suppresses sprouting via
signaling to MLC2 phosphorylation. Curr Biol. 19:668–674. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hatanaka K, Simons M and Murakami M:
Phosphorylation of VE-cadherin controls endothelial phenotypes via
p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ
Physiol. 300:H162–H172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fukuhra S, Sakurai A, Yamagishi A, Sako K
and Mochizuki N: Vascular endothelial cadherin-mediated cell-cell
adhesion regulated by a small GTPase, Rap1. J Biochem Mol Biol.
39:132–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiao K, Garner J, Buckley KM, Vincent PA,
Chiasson CM, Dejana E, Faundez V and Kowalczyk AP: p120-Catenin
regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol
Cell. 16:5141–5151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu JC, Yan HC, Chen WT, Chen WH, Wang CJ,
Chi YC and Kao WY: JNK signaling pathway is required for
bFGF-mediated surface cadherin downregulation on HUVEC. Exp Cell
Res. 314:421–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grazia Lampugnani M, Zanetti A, Corada M,
Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A,
Kemler R, Daniel TO and Dejana E: Contact inhibition of
VEGF-induced proliferation requires vascular endothelial cadherin,
beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol.
161:793–804. 2003.PubMed/NCBI
|