Introduction
As a response to nutrient restriction, autophagy is
an intracellular pathway in which cytosolic elements and even
organelles are enclosed in double-membrane vesicles known as
autophagosomes (1–4). The physiological role of autophagy is
to degrade long-lived proteins and damaged organelles to ensure the
survival of cells during starvation (5). However, autophagy also plays
pathophysiological roles. In cancer, it acts as a protector against
certain cancer treatments and, most importantly, prevents cell
death in a hostile microenvironment. Previous studies have
indicated that autophagy plays a key role in tumor cell resistance
to chemotherapy, and that the inhibition of autophagy can enhance
the cytotoxicity of certain chemotherapeutic agents (6,7). By
contrast, other cancer cells undergo autophagic cell death
following various anticancer therapies (8). As an alternative cell death pathway,
called programmed cell death type II or autophagic cell death has
morphological and biochemical features distinguishing it from
apoptosis and necrosis.
Autophagy is regulated by the autophagy-related
(Atg) genes that control the formation of autophagosomes
(9). Beclin 1, the mammalian
ortholog of yeast Atg6/Vps30, was originally discovered in a yeast
two-hybrid screen as a Bcl-2-interacting protein, and it was the
first human protein shown to be indispensable for autophagy
(10). Beclin 1 is a component of a
complex consisting of the class III phosphatidylinositol 3-kinase
(PI3K) that stimulates autophagy. The interaction between Beclin 1
and its binding partners regulates the initial steps of autophagy.
Beclin 1 also possesses the so-called BH3 domain (amino acids
114–123) that mediates its interaction with Bcl-2 and other close
Bcl-2 homologs, such as Bcl-xL and Mcl-1 (11,12).
The induction of Beclin 1 expression can be observed during
autophagy in various cell types (13).
A number of signaling pathways are involved in the
regulation of autophagy. The serine/threonine kinase mammalian
target of rapamycin complex 1 (mTORC1) is a major negative
regulator of autophagy (14,15).
The key upstream regulators of mTORC1 include the class I PI3K-Akt
pathway and the AMP-activated protein kinase (AMPK) pathway. The
Akt kinase phosphorylates the mTORC1 repressor, tuberous sclerosis
complex 2 (TSC2), thus leading to the activation of mTORC1 and the
subsequent inhibition of the expression and function of Atg
proteins (16). The class I
PI3K/Akt signaling molecules link receptor tyrosine kinases to
mTORC1 activation and thereby repress autophagy in response to a
sufficient amount of growth factors (17). In addition to Akt, one of the main
mTORC1 regulators is AMPK, which maintains energy homeostasis by
inducing autophagy and blocking protein synthesis and cell
proliferation, mainly through the phosphorylation of its downstream
target, raptor, and the consequent inhibition of mTORC1 in response
to starvation and calcium signals (18).
Reactive oxygen species (ROS) consist of a group of
highly reactive molecular forms of oxygen containing unpaired
electrons that are continuously produced as a byproduct of the
mitochondrial respiratory chain in normal cells (19,20).
Due to their high reactivity, certain long half-life ROS can
oxidize cell constituents and damage cell structures and functions.
ROS damage has been shown to cause various types of cell death,
including apoptosis and autophagy (21,22).
Compared with normal cells, tumor cells have long-term high levels
of ROS; therefore, the redox system of tumor cells is more
vulnerable to drugs that can increase ROS levels compared to normal
cells (23,24).
Quinoline compounds are widely used as ‘parental’
compounds to synthesize molecules with medical benefits,
particularly those with anti-malarial and anti-microbial
activities. Certain quinoline-based compounds have shown effective
anticancer activity. Diarylquinoline compounds have recently been
shown to exert anti-proliferative effects on cancer cell lines
(25–27). In our study, nine diarylquinoline
compounds were newly synthesized based on the parental structure of
a new anti-tuberculosis drug, TMC207. These compounds were then
screened for their cytotoxic activity against human tumor cells,
and their mechanisms of action were investigated.
Materials and methods
Compounds and reagents
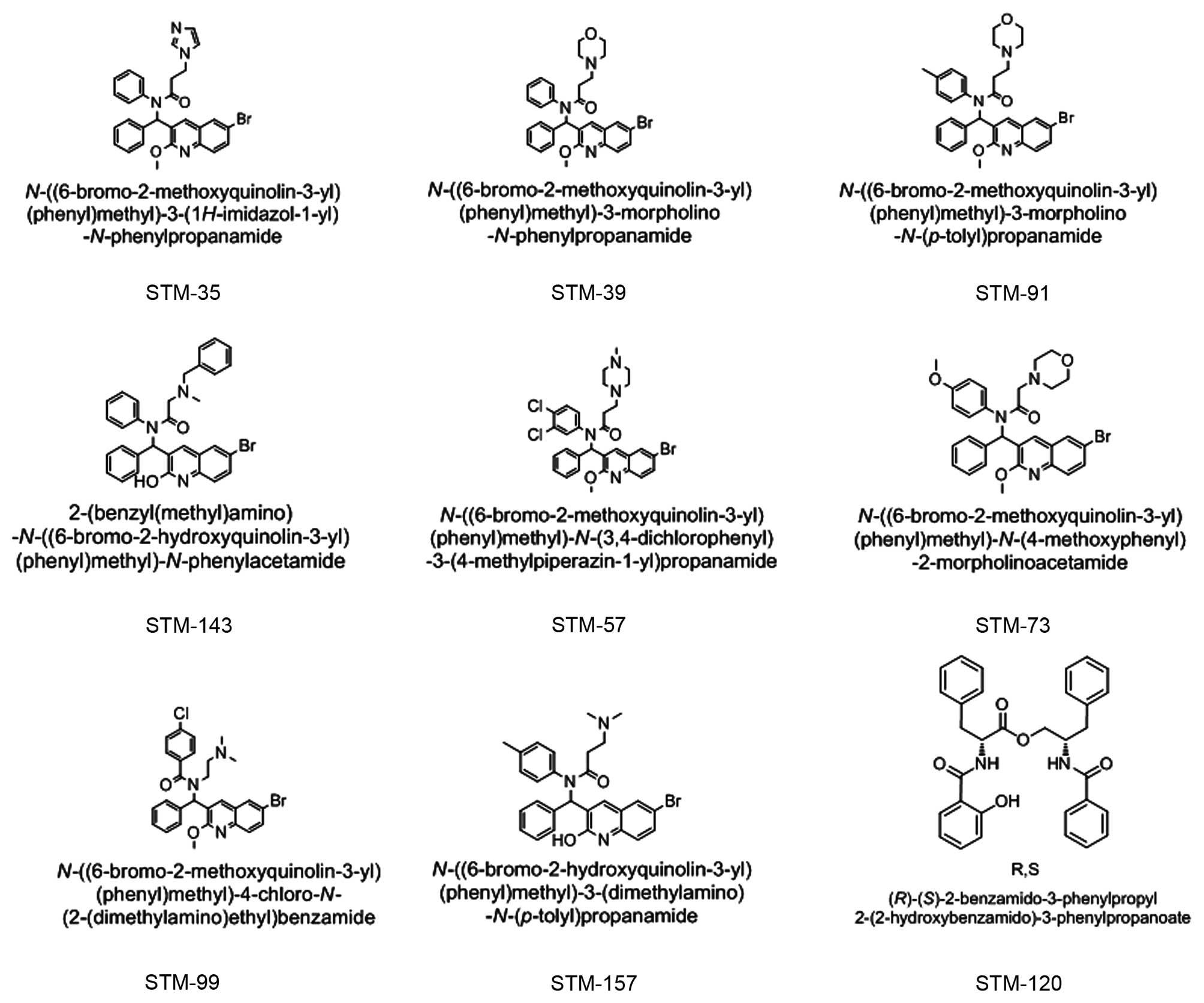
All nine of the new compounds were synthesized by
Shenyang Pharmaceutical University. The chemical structures of
these compounds are presented in Fig.
1 and include the following: STM-57,
N-((6-bromo-2-methoxyquinolin-3-yl)(phenyl)methl)-N-(3,4-dichlorophenyl)-3-(4-methylpiperazin-
1-yl)propanamide; STM-35, N-((6-bromo-
2-methoxyquinolin-3-yl)(phenyl)methyl)-3-(1H-imidazol-1-yl)-N-phenylpropanamide;
STM-39,
N-((6-bromo-2-methoxyquinolin-3-yl)(phenyl)methyl)-3-morpholino-N-phenylpropanamide;
STM-91, N-((6-bromo-2-methoxyquinolin-
3-yl)(phenyl)methyl)-3-morpholino-N-(p-tolyl) propanamide;
STM-143, 2-(benzyl (methyl) amino)-N-((6-bromo-2-
hydroxyquinolin-3-yl)(phenyl)methyl)-N-phenylacetamide;
STM-73,
N-((6-bromo-2-methoxyquinolin-3-yl)(phenyl)methyl)-N(4-methoxyphenyl)-2-morpholinoacetamide;
STM-99, N-((6-bromo-2-methoxyquinolin-3-yl)(phenyl)
methyl)-4-chloro-N-(2-(dimethylamino)ethyl)benzamide;
STM-157,
N-((6-bromo-2-hydroxyquinolin-3-yl)(phenyl)methyl)-3-(dimethylamino)-N-
(p-tolyl) propanamide; and STM-120,
(R)-(S)-2-benzamido-3-phenylpropyl-2-(2-hydroxybenzamido)-3-phenylpropanoate.
All the compounds were initially dissolved in 100% dimethyl
sulfoxide (DMSO). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) was purchased from Sigma Co.
(Sigma-Aldrich). RPMI-1640, DMEM and McCoy’s 5A were purchased from
Invitrogen.
Cell lines and culture
The human nasopharyngeal carcinoma cell lines, CNE-1
and CNE-2, the human lung adenocarcinoma cell line, A549, the human
acute promyelocytic leukemia cell line, HL-60, and the human
Burkitt’s lymphoma cell line, Raji, were cultured in RPMI with 10%
fetal bovine serum (FBS). The human breast carcinoma cell lines,
MDA-MB-231 and MCF-7, and its related MDR cell line, MCF-7/ADM,
were maintained in DMEM with 10% FBS. The human colon cancer cell
line, HT-29, was cultured in McCoy’s 5A medium with 10% FBS. The
cells were incubated at 37°C in a humidified incubator in 5%
CO2.
Cell growth inhibition test
The cells were plated in a 96-well plate and
cultured in medium with various concentrations of the test
compounds added after 24 h. After an incubation of 68 h, 20 μl of
MTT were added to each well (100 μg/well) and incubated for an
additional 4 h, and the liquid in the wells was then evaporated.
The insoluble formazan produced was dissolved with 200 μl DMSO, and
the optical density was measured with an ELISA reader (Thermo
Labsystems, Espoo, Finland) at wavelengths of 570 and 630 nm. The
experiments were performed in triplicate. From these results, the
percentage of live cells in each well was estimated and plotted
against the drug concentrations as dose-response curves, from which
the IC50 value was derived.
Cell cycle detection
The cells were collected, resuspended in 1 ml
pre-cooled 70% ethanol, and fixed overnight at 4°C. After the
ethanol was removed, 0.5 ml staining solution [50 mg/ml propidium
iodide (PI), 100 mg/ml RNaseA, and 0.2% Triton X-100] was added,
and the cells were incubated at 37°C for 30 min in the dark. The
cell cycle distribution was immediately analyzed with flow
cytometry (Beckman Coulter, Fullerton, CA) at a wavelength of 488
nm.
Annexin V-FITC/PI staining assay
Cells exposed to different treatments were
collected, resuspended in 100 μl PBS, adjusted to approximately
1×106 cells/ml, and transferred to a microfuge tube (0.5
ml). After the addition of 10 μl binding reagent and 1.25 μl
Annexin V-FITC, the cells were incubated at room temperature for 15
min in the dark. The cells were then centrifuged, and the medium
was removed. The cells were resuspended in 0.5 ml cold binding
buffer (10 mM HEPES, 150 mM NaCl, 2.5 mM CaCl2, 1 mM
MgCl2 and 20% BSA, pH 7.4), and 10 μl (PI, 30 mg/ml) was
added. The samples were placed on ice in the dark. Apoptosis was
immediately analyzed with flow cytometry (Beckman Coulter) at a
wavelength of 488 nm.
Confocal microscopy and indirect
immunofluorescence
CNE-2 cells were grown on glass coverslips and
transfected with an expression vector containing the LC3 gene fused
to green fluorescent protein (GFP-LC3). At 24 h after transfection,
the cells were treated with STM-57 and analyzed after an additional
12 h. The cells were fixed with 4% paraformaldehyde in PBS for 30
min at room temperature, and the slides were mounted in anti-fade
solution and stored at 4°C. The coverslips were viewed with a
laser-scanning confocal microscope (Olympus, FV1000).
Electron microscopy
The cells were fixed by immersion in a mixture of
2.5% glutaraldehyde, 2.5% paraformaldehyde and 0.05% picric acid in
0.067 M cacodylate buffer (pH 7.4). Post-fixation was performed in
1% osmium tetroxide followed by an overnight immersion in 0.3%
uranyl acetate dissolved in 50 mM maleate buffer (pH 5.0). Standard
procedures for dehydration and embedding in Epon were used. Thin
sections were further stained with lead citrate and examined with
an electron microscope (Philip, CN10).
Western blot analysis
Lysates were prepared from 4×105 cells by
dissolving cell pellets in 100 μl of lysis buffer [20 mM
Na2PO4 (pH 7.4), 150 mM NaCl, 1% Triton
X-100, 1% aprotinin, 1 mM phenymethysulfonyl fluoride, 10 mg/ml
leupeptin, 100 mM NaF and 2 mM Na3VO4]. The
lysates were centrifuged at 14,000 × g for 20 min, and the
supernatant was collected. The protein content was determined using
the Bio-Rad protein assay. SDS-PAGE sample buffer [10 mM Tris-HCl,
pH 6.8, 2% SDS, 10% glycerol and 0.2 M dithiothreitol (DTT)] was
added to the lysates. A total of 50 μg of protein was loaded in
each well of a 5–12% SDS-PAGE gel. The resolved proteins were
electrophoretically transferred onto PVDF membranes (Roche,
Mannheim, Germany) and blocked with TBS with 5% non-fat milk
overnight at 4°C. The membranes were then incubated with the
primary antibody. After washing, the membranes were incubated with
the secondary anti-mouse/anti-rabbit antibody (1:1,000,
HRP-labeled; Dako) at room temperature for 1 h. After washing, the
bound antibody complex was detected using LumiGLO reagent [Cell
Signaling Technology, Inc. (CST) #7003] and XAR film (Kodak,
XBT-1), as described by the manufacturers. The following primary
antibodies were used: caspase 3 (CST #9662), poly(ADP-ribose)
polymerase (PARP) (sc-7150), glyceraldehyde 3-phosphate
dehydrogenase (sc-47724; both from Santa Cruz Biotechnology, Inc.),
LC3 (NB100-2220; Novus Biologicals), Beclin 1 (CST #3738), Bcl-2
(CST #2870), Bcl-xL (CST #2762), phospho-Akt (ser473; CST #4058),
Akt1 (CST #2967), phospho-mTOR (ser2448; CST #2971) and mTOR (CST
#4517) antibodies.
Measurement of mitochondrial membrane
potential (ΔΨm)
ΔΨm was determined by the retention of the dye
DiOC6(3). Cells were
harvested and washed twice with PBS. After incubation with 50 nM
DiOC6(3) at 37°C for 30
min, the cells were washed again and analyzed by flow
cytometry.
Analysis of intracellular Ca2+
concentration
The changes of intracellular Ca2+
concentration were determined by the fluorescent dye, Fluo-3/AM.
Cells were washed twice with PBS and incubated with 5 μM Fluo-3/AM
at 37°C for 30 min. Subsequently, the cells were washed and
analyzed by flow cytometry.
Measurement of intracellular ROS
Intracellular ROS production was measured by using
the fluorescent dye, dichlorfluorescein-diacetate (DCFH-DA), which
can be converted to DCFH by esterases when the cells take it up.
DCFH reacts with ROS to form a new highly fluorescent compound,
dichlorofluorescein, which can be analyzed by flow cytometry. The
treated cells were incubated with DCFH-DA (10 μM) at 37°C for 1 h,
and then measured by flow cytometry.
Statistic analysis
All the data were obtained from at least three
independent experiments. The comparisons of the various treatment
protocols were performed with a one-way analysis of variance
(ANOVA) using SPSS software to obtain the P-value for comparisons
between treatments. P<0.05 was considered the minimal level of
significance.
Results
Antitumor activity screening of
diarylquinoline compounds
The nine diarylquinoline compounds (Fig. 1) demonstrated anti-proliferative
activity in human nasopharyngeal carcinoma, lung adenocarcinoma,
breast cancer, colon cancer, leukemia and lymphoma cell lines. As
shown in Table I, the
IC50 values ranged from 9.9±0.9 to 123.3±20.4 μM. Among
these compounds, STM-57, STM-35, STM-91, STM-99 and STM-157 showed
the most potent anti-proliferative activity in the cancer cell
lines.
 | Table IAnti-proliferative activity of the
nine newly synthesized diarylquinoline compounds in human cancer
cell lines. |
Table I
Anti-proliferative activity of the
nine newly synthesized diarylquinoline compounds in human cancer
cell lines.
| Cell line |
|---|
|
|
|---|
| Compound | CNE-1 | CNE-2 | A549 | MCF-7 | MCF-7/ADM | MDA-MB-231 | HT-29 | HL-60 | Raji |
|---|
| STM-57 | 10.8±2.2 | 10.2±4.8 | 12.3±2.1 | 10.8±2.9 | 11.9±2.5 | 20.1±1.1 | 29.2±13.0 | 12.4±2.6 | 13.5±2.2 |
| STM-35 | 10.8±2.7 | 21.5±2.4 | 15.6±2.7 | 12.8±2.2 | 17.7±2.5 | - | - | 21.1±2.1 | 22.3±2.0 |
| STM-39 | 23.6±2.6 | 46.1±2.9 | 46.7±3.2 | 47.0±4.2 | 45.7±4.2 | - | - | 45.4±3.5 | 45.5±2.2 |
| STM-73 | 55.8±3.2 | 52.6±5.2 | 67.9±4.4 | 64.5±5.1 | 122.9±11.2 | - | - | 55.3±2.2 | 57.3±3.5 |
| STM-91 | 19.7±2.5 | 23.5±4.2 | 20.4±4.8 | 10.8±2.2 | 17.3±5.3 | 10.7±1.0 | 32.4±5.5 | 24.6±3.4 | 27.5±4.8 |
| STM-99 | 30.9±2.4 | 26.4±4.7 | 21.4±2.4 | 19.1±4.4 | 14.4±4.6 | 18.5±1.1 | 21.0±2.8 | 21.2±4.4 | 21.1±2.2 |
| STM-120 | 32.4±3.1 | 36.2±3.9 | 101.8±5.4 | 30.5±6.4 | 123.3±20.4 | - | - | 53.2±2.4 | 53.4±4.3 |
| STM-143 | 41.3±3.4 | 83.2±4.5 | 91.9±3.7 | 45.6±2.4 | 44.8±4.9 | - | - | 46.8±4.6 | 47.7±3.6 |
| STM-157 | 35.2±4.6 | 26.5±3.6 | 36.9±5.6 | 11.8±0.3 | 34.9±4.0 | 9.9±0.9 | 10.0±2.1 | 34.3±4.1 | 32.2±2.6 |
STM-57-induced caspase-independent cell
death in CNE-2 cells
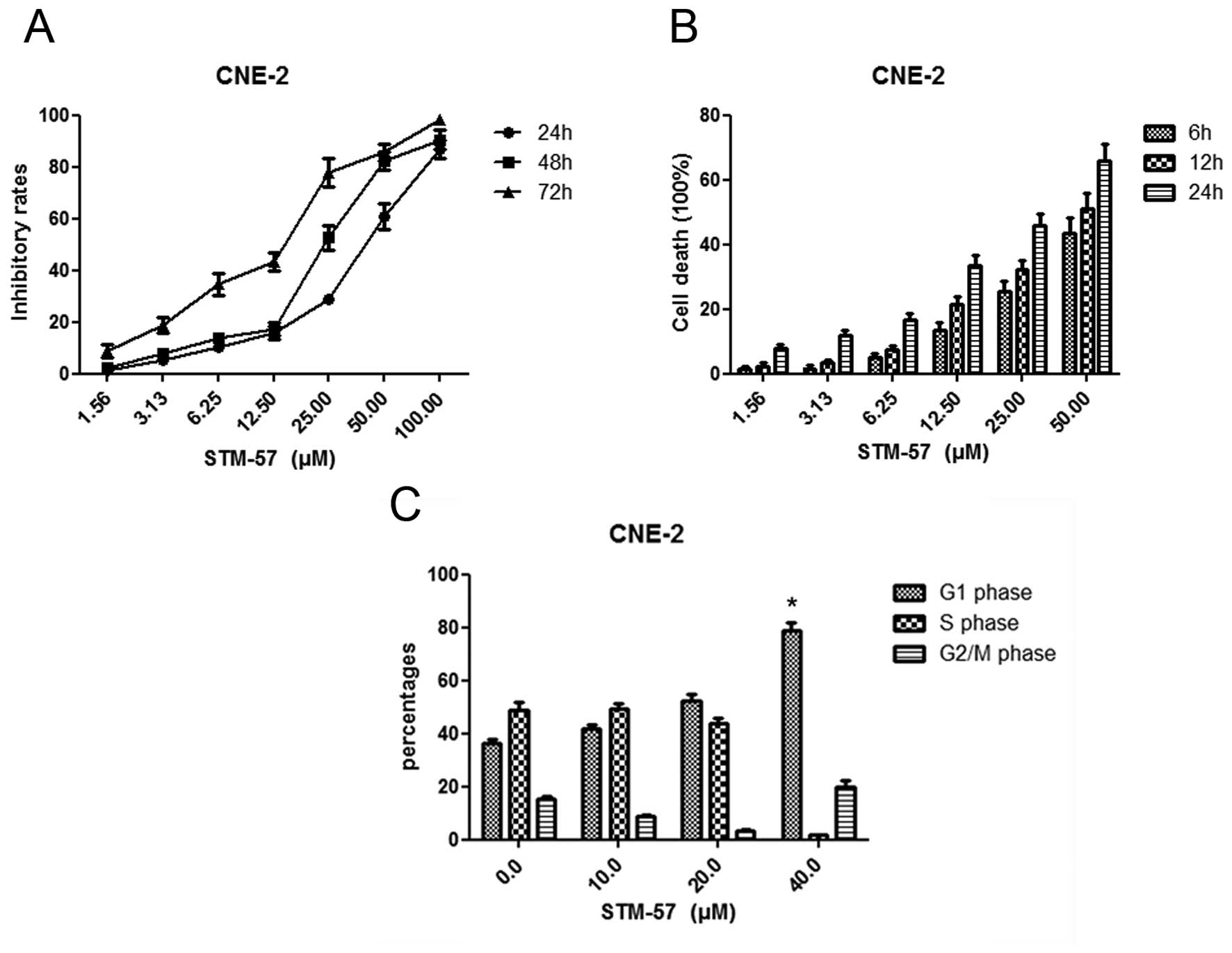
STM-57 resulted in the inhibition of cell growth in
a concentration- and time-dependent manner in CNE-2 cells, and the
IC50 value was 10.2±4.8 μM (Fig. 2A and B). The inhibition of cell
growth may be the result of the induction of apoptosis, autophagy
and/or cell cycle arrest.
CNE-2 cells were treated with 10, 20 and 40 μM
STM-57 for 12 h, and the cell cycle distribution was analyzed by
flow cytometry. STM-57 (40 μM) significantly induced G1-phase
arrest by 78.8±3.1% compared with 36.2±1.8% in the control group
(Fig. 2C).
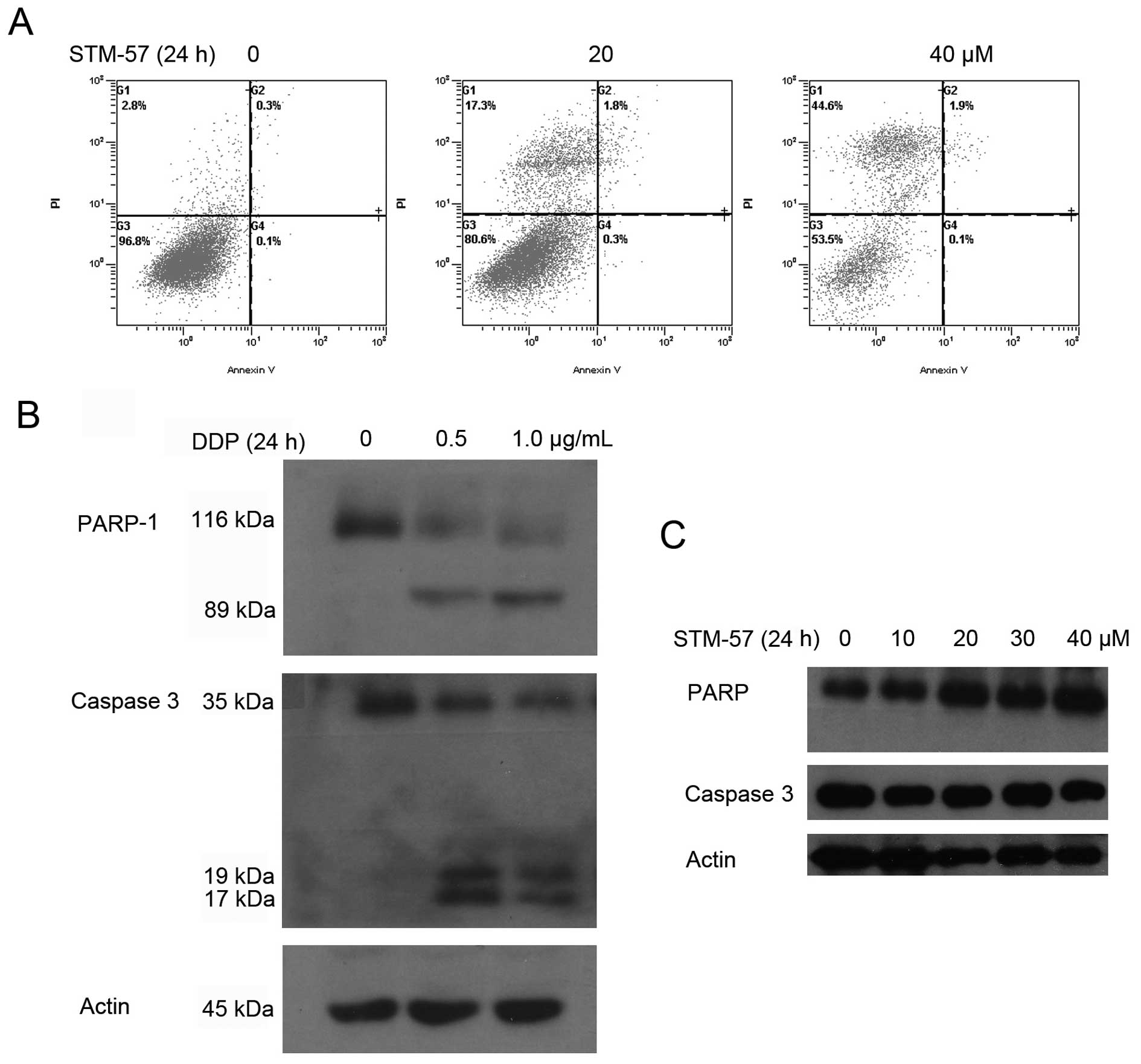
To further confirm that STM-57 mainly induced
non-apoptotic cell death, CNE-2 cells treated with 20 and 40 μM
STM-57 for 24 h were analyzed using the Annexin V-FITC/PI staining
assay. As shown in Fig. 3A, the
highest apoptotic rates were 0.3% in the CNE-2 cells following
treatment. The DNA ladder was not detected after STM-57 treatment
(data not shown). These results strongly suggest that STM-57
induces non-apoptotic cell death in CNE-2 cells.
To determine whether the cell death was
caspase-dependent, we first detected the cleavage of caspase 3. As
a positive control, cisplatin (DDP) induced the cleavage of caspase
3 and PARP in the CNE-2 cells. Pro-caspase 3 was cleaved to yield a
17 kDa fragment and PARP was cleaved to an 89 kDa fragment
following DDP treatment (Fig. 3B).
However, the cleavage of caspase 3 was not observed following
treatment with STM-57. The cleavage of PARP was also undetectable
(Fig. 3C). This result further
confirmed the results that STM-57 induced caspase 3-independent
cell death.
STM-57 induces cell autophagy-associated
cell death in CNE-2 cells
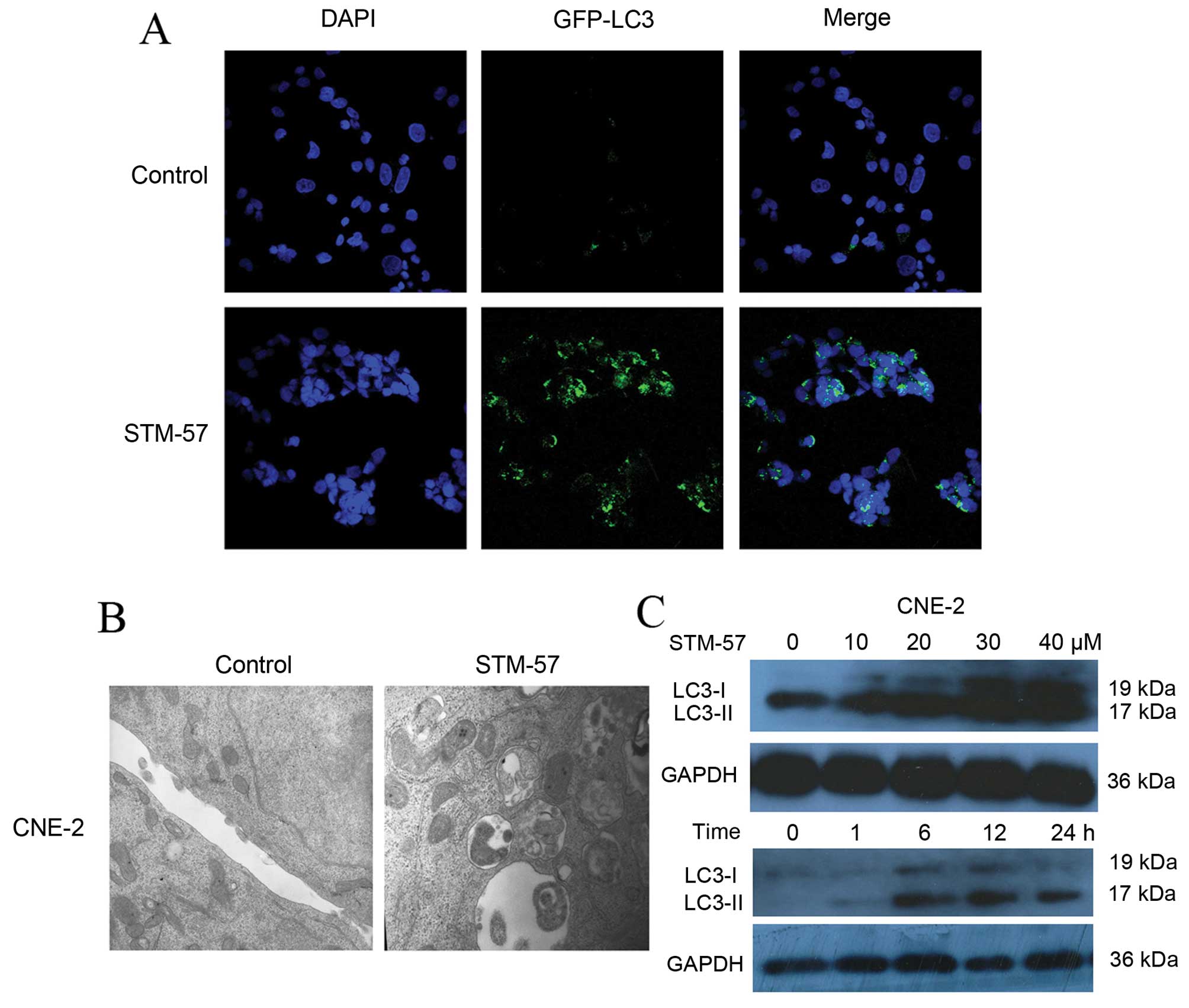
STM-57 induces the formation of
autophagosomes
To observe autophagy, CNE-2 cells were stained with
monodansylcadaverine (MDC) (data not shown) or transfected with
GFP-LC3. Following treatment with STM-57, a significant
accumulation of GFP-LC3 was observed, indicating the association of
GFP-LC3 with autophagosomal membranes following the induction of
autophagy (Fig. 4A). The
ultrastructural analysis of the STM-57-treated CNE-2 cells using
electron microscopy showed large, ragged cytomembranes and swollen
mitochondria in the autophagic vacuoles in contrast to the control
cells, in which no such vacuoles were detected (Fig. 4B). These results suggest that STM-57
induces the formation of autophagosomes.
STM-57 induces LC3-II expression in
CNE-2 cells
The membrane-associated light chain 3 protein (LC3,
Atg8) is a key marker of autophagy. After the induction of
autophagy, LC3-I is converted to LC3-II, which is most likely
conjugated to phosphatidylethanolamine (PE) and tightly bound to
the autophagosomal membranes, forming ring-shaped structures in the
cytoplasm. The amount of PE-conjugated LC3 (LC3-II) correlates well
with the number of autophagosomes. After STM-57 treatment for the
indicated times, LC3 was detected with immunoblot analysis. The
results indicated that the expression of LC3-II was gradually
upregulated (Fig. 4C). These data
confirmed the results of GFP-LC3 transfection and indicated that
STM-57 induced autophagy in CNE-2 cells.
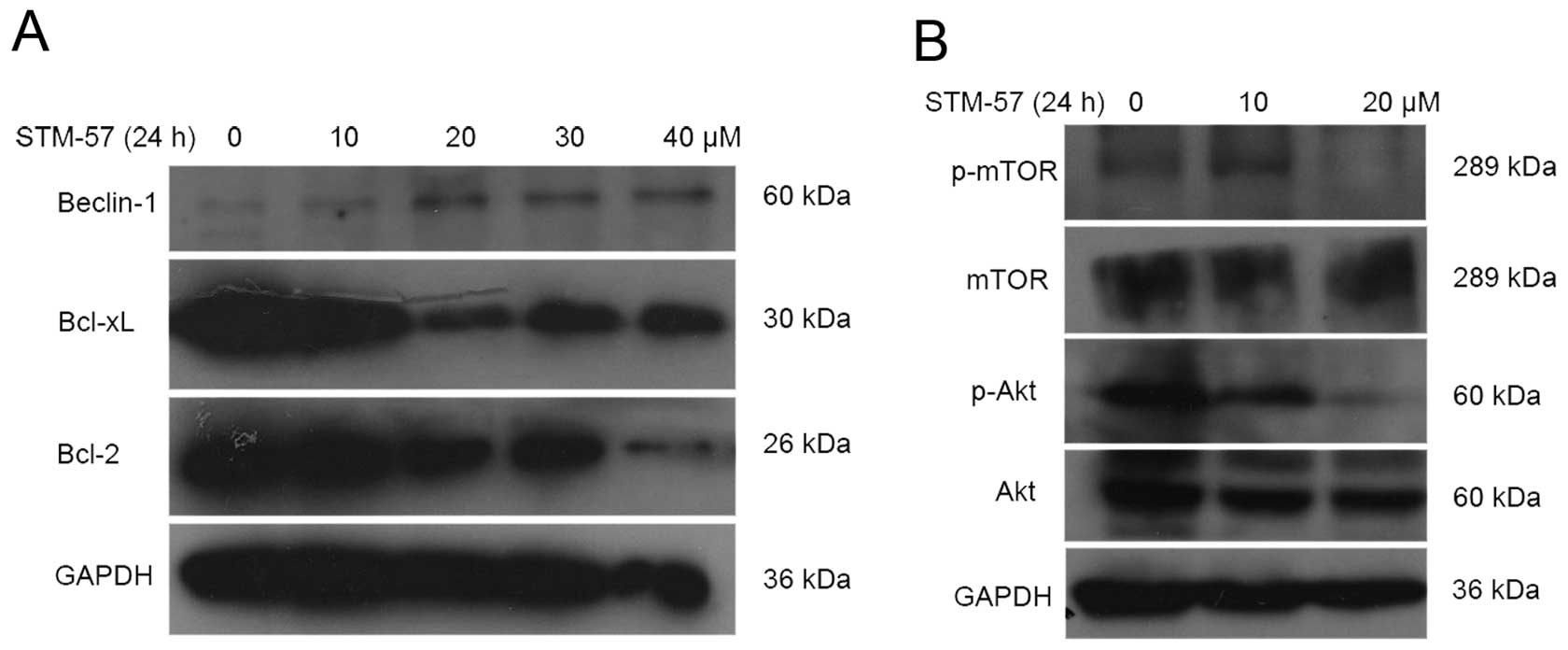
STM-57 inhibits Bcl-2 and Bcl-xL
expression in CNE-2 cells
Anti-apoptotic Bcl-2 family proteins, such as Bcl-2
and Bcl-xL, are well known for their inhibition of autophagy. Bcl-2
and Bcl-xL suppress autophagy by binding to the Beclin 1 protein,
which is required for the initiation of autophagosomal formation
during autophagy. STM-57 inhibited the expression of Bcl-2 and
Bcl-xL in CNE-2 cells, whereas the expression of Beclin 1 was
increased following STM-57 treatment for 24 h (Fig. 5A).
STM-57 inhibits the Akt/mTOR pathway
in CNE-2 cells
The Akt/mTOR pathway is the main regulator of
autophagy. After STM-57 treatment for 24 h, the phosphorylation of
Akt was inhibited in the CNE-2 cells (Fig. 5B). mTOR is the downstream target of
the Akt pathway and plays a key role in autophagy. After STM-57
treatment, the phosphorylation of mTOR was significantly decreased
in the CNE-2 cells. These results suggested that the Akt pathway
was inhibited by STM-57 in the CNE-2 cells.
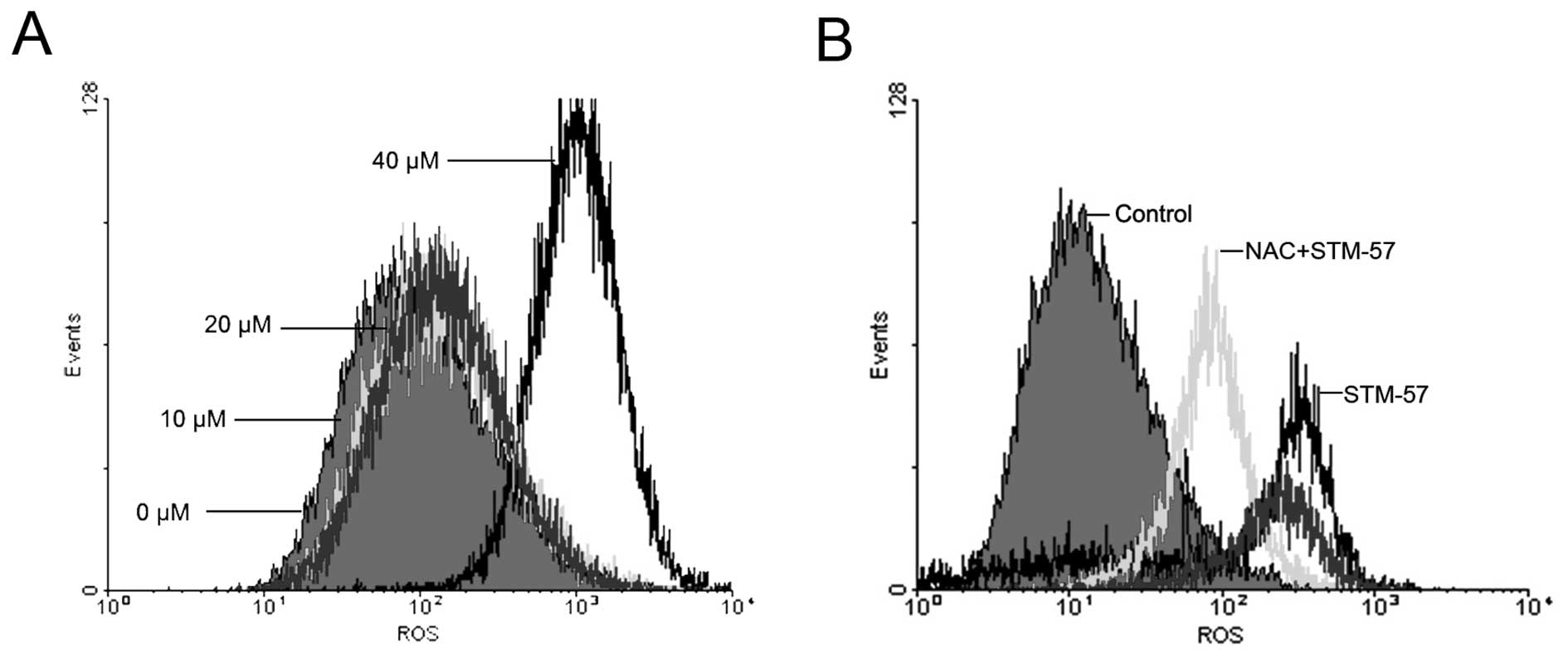
STM-57 induces an increase in ROS
levels in CNE-2 cells
When the CNE-2 cells were treated with 10, 20 or 40
μM STM-57 for 24 h, ROS levels were significantly increased in a
concentration-dependent manner (Fig.
6A). ROS levels were determined by flow cytometry and expressed
in units of mean fluorescence intensity. In the CNE-2 cells, the
ROS level was 124±37.1 after 40 μM STM-57 treatment for 12 h,
whereas the ROS level in the control group was 16.4±2.1. When the
CNE-2 cells were pre-treated with 100 μM N-acetylcysteine (NAC) for
1 h and treated with STM-57 for 12 h, the flow cytometry results
showed that NAC partly prevented the increase in ROS levels induced
by STM-57 (Fig. 6B). These results
suggest that STM-57 induces caspase-independent cell death in CNE-2
cells via ROS accumulation.
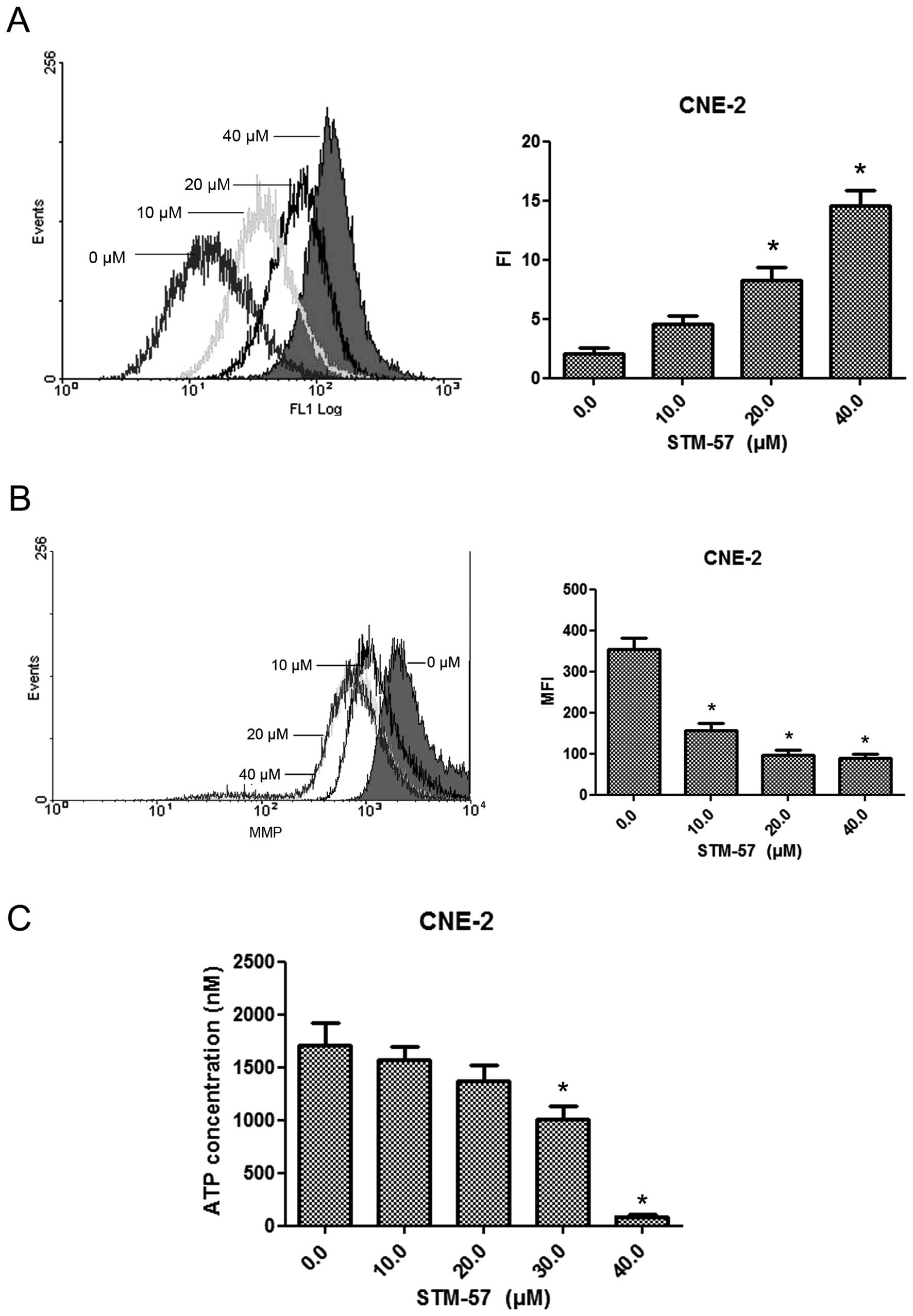
STM-57 affects intracellular calcium
concentrations, ΔΨm and ATP concentration
When the CNE-2 cells were treated with 10, 20 or 40
μM STM-57 for 12 h, the intracellular calcium concentrations were
significantly increased, whereas the ATP level and the ΔΨm were
decreased (Fig. 7). These results
suggest that STM-57 induces caspase-independent cell death in CNE-2
cells via a mitochondrial dysfunction pathway, including ATP
depletion, permeability of the mitochondrial membrane and
intracellular calcium influx.
Discussion
The results of this study demonstrate that
diaryquinoline compounds exert broad-spectrum anticancer activity
in human cancer cell lines. STM-57 induces caspase-independent cell
death though autophagy and a mitochondrial-mediated pathway in
human cancer cell lines.
Autophagy plays a key role in regulating important
cellular functions, such as survival during starvation and the
control of infecting pathogens. Numerous multifaceted links between
autophagy and cancer have emerged (28). Although autophagy has been reported
as a pro-survival response, in certain instances, the cytotoxicity
of cytokines and chemicals in some cell types is mediated by the
induction of autophagy. Targeting selected protein kinases involved
in the regulation of autophagy using small molecule inhibitors may
be a feasible approach in cancer therapy. Previously, imatinib has
been shown to induce autophagy in multi-drug-resistant Kaposi’s
sarcoma and glioblastoma cells (29). The histone deacetylase (HDAC)
inhibitor, suberoylanilide hydroxamic acid (SAHA), has also been
reported to induce autophagy in HeLa cells independent of caspase
activation and apoptosis (30).
mTOR inhibitors have been reported to induce autophagic cell death
and sensitize various tumor cells to radiation therapy (31,32).
It has been reported that quinoline compounds exert potential
cytotoxic effects against breast cancer, prostate cancer and
colorectal adenocarcinoma (25–27).
Therefore, we screened nine newly synthesized diarylquinoline
compounds for their cytotoxic effects against cancer cell lines.
STM-57 is one of the most potent compounds; it induces
non-apoptotic cell death in CNE-2 cells. We further investigated
whether the induction of autophagy is required for STM-57-mediated
cell death. Autophagic cells enlarge without permeabilization of
their plasma membrane, convert LC3-I to LC-II, show punctate
cytoplasmic LC3 translocation and develop autophagosomes. In this
study, CNE-2 cells exhibited morphological and biochemical features
characteristic of cells undergoing autophagy, not apoptosis,
following STM-57 treatment.
Anti-apoptotic Bcl-2 family proteins, such as Bcl-2
and Bcl-xL, are frequently overexpressed in cancers (33–35).
These proteins inhibit apoptosis by binding to Bax or Bak. Bcl-2
and Bcl-xL are also well known for their anti-autophagic abilities.
Bcl-2 and Bcl-xL suppress autophagy by binding to the Beclin 1
protein, which is required for the initiation of autophagosomal
formation in autophagy (36,37,12).
In our study, STM-57 inhibited the expression of Bcl-2 and Bcl-xL
in CNE-2 cells, whereas the expression of Beclin 1 was increased
following STM-57 treatment. Bcl-2 and Bcl-xL inhibit autophagy by
sequestering Beclin 1 from hVps34 and reducing the affinity of the
UV radiation resistance-associated gene protein (UVRAG) for Beclin
l (38,39). Since Bcl-2 and Bcl-xL are localized
to the surface of mitochondria, we wished to determine the effect
of STM-57 on ΔΨm. Mitochondrial damage plays an important role in
apoptosis and autophagy. One hypothesis is that cells respond to
mitochondrial damage in a gradual fashion: when only a few
mitochondria are damaged, autophagy occurs and the mitochondria are
degraded; when more mitochondria are damaged, apoptosis is induced,
and the cells die (40,41). We observed that STM-57 decreased ΔΨm
and ATP levels in CNE-2 cells, suggesting that STM-57 induces
autophagy via a mitochondrial dysfunction pathway. Høyer-Hansen
et al(42) emphasized the
important role of Ca2+ in the formation of
autophagosomes, and Ca2+ homeostasis and signaling are
modulated though Bcl-2 in macro-autophagy. We also observed that
STM-57 increased the intracellular calcium concentration in CNE-2
cells, suggesting that STM-57-induced autophagy is associated with
an increase in calcium influx. Taken together, these data suggest
that STM-57 induces autophagic cell death in CNE-2 cells though a
mitochondrial dysfunction pathway, including ATP depletion,
permeability of the mitochondrial membrane and intracellular
calcium influx.
mTORC1 plays a key role in the repression of
autophagy through the integration of different signal inputs. The
Akt phosphorylation of TSC2 destabilizes TSC2 and disrupts its
interaction with TSC1, thus abolishing the negative regulatory
effect of the TSC2/TSC1 complex on mTORC1 (43,44).
In our study, STM-57 inhibited the phosphorylation of Akt in CNE-2
cells. The phosphorylation of mTOR was also dramatically decreased
following STM-57 treatment. These results suggest that STM-57
induces autophagy though the inhibition of the Akt/mTOR pathway in
CNE-2 cells.
ROS are a class of single-electron reduction
products of oxygen, including singlet oxygen, hydroxyl radicals,
superoxide and hydrogen peroxide. It has been estimated that 2% of
the oxygen consumed by the mitochondria is converted to ROS
(20). The generation of ROS is
closely associated with tumorigenesis and treatment. An abnormal
increase in ROS levels can promote tumorigenesis. However, an
excessive increase in ROS levels can induce the apoptosis and
autophagic death of tumor cells, resulting in cancer cell growth
inhibition (45,46). In the present study, we found that
the treatment of CNE-2 cells with STM-57 led to the production of
ROS, and the ROS scavenger, NAC, significantly prevented
STM-57-induced cell death, suggesting that ROS accumulation is
involved in STM-57-induced autophagy.
In conclusion, the current study demonstrates that
STM-57 kills CNE-2 cells via the induction of autophagy. The
inhibition of Bcl-2 expression and the Akt/mTOR pathway plays a key
role in STM-57-mediated autophagy. STM-57 induces autophagic cell
death though a mitochondrial dysfunction pathway, including ATP
depletion, permeability of the mitochondrial membrane and
intracellular calcium influx. Our study also provides evidence that
STM-57 warrants further investigation as a potential anticancer
agent.
Acknowledgements
This study was supported by the National Eleventh
Five-Year major technology project (Grant no. 2008ZX09312-002).
References
|
1
|
Wang CW and Klionsky DJ: The molecular
mechanism of autophagy. Mol Med. 9:65–76. 2003.
|
|
2
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanism and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Høyer-Hansen M and Jäättelä M:
AMP-activated protein kinase: a universal regulator of autophagy?
Autophagy. 3:381–383. 2007.PubMed/NCBI
|
|
4
|
Meijer AJ and Codogno P: Signalling and
autophagy regulation in health, aging and disease. Mol Aspects Med.
27:411–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abedin MJ, Wang D, McDonnell MA, Lehmann U
and Kelekar A: Autophagy delays apoptotic death in breast cancer
cells following DNA damage. Cell Death Differ. 14:500–510. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cuervo AM: Autophagy: in sickness and in
health. Trends Cell Biol. 14:70–77. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohsumi Y: Molecular dissection of
autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol.
2:211–216. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang XH, Yu J, Brown K and Levine B:
Beclin 1 contains a leucine-rich nuclear export signal that is
required for its autophagy and tumor suppressor function. Cancer
Res. 61:3443–3449. 2001.PubMed/NCBI
|
|
12
|
Maiuri MC, Le Toumelin G, Criollo A, Rain
JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K,
Tavernarakis N, Hickman JA, Geneste O and Kroemer G: Functional and
physical interaction between Bcl-X(L) and a BH3-like domain in
Beclin-1. EMBO J. 26:2527–2539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chu CT, Zhu J and Dagda R: Beclin
1-independent pathway of damage-induced mitophagy and autophagic
stress: implications for neurodegeneration and cell death.
Autophagy. 3:663–666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hay N: The Akt-mTOR tango and its
relevance to cancer. Cancer Cell. 8:179–183. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shaw RJ: LKB1 and AMP-activated protein
kinase control of mTOR signalling and growth. Acta Physiol (Oxf).
196:65–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Riley PA: Free radicals in biology:
oxidative stress and the effects of ionizing radiation. Int J
Radiat Biol. 65:27–33. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Laurent A, Nicco C, Chereau C, Goulvestre
C, Alexandre J, Alves A, Levy E, Goldwasser F, Panis Y, Soubrane O,
Weil B and Batteux F: Controlling tumor growth by modulating
endogenous production of reactive oxygen species. Cancer Res.
65:948–956. 2005.PubMed/NCBI
|
|
22
|
Valencia A and Morán J: Reactive oxygen
species induce different cell death mechanisms in cultured neurons.
Free Radic Biol Med. 36:1112–1125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Trachootham D, Zhou Y, Zhang H, Demizu Y,
Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J and
Huang P: Selective killing of oncogenically transformed cells
through a ROS mediated mechanism by beta-phenylethyl
isothiocyanate. Cancer Cell. 10:241–252. 2006. View Article : Google Scholar
|
|
24
|
Trachootham D, Zhang H, Zhang W, Feng L,
Du M, Zhou Y, Chen Z, Pelicano H, Plunkett W, Wierda WG, Keating MJ
and Huang P: Effective elimination of fludarabine-resistant CLL
cells by PEITC through a redox-mediated mechanism. Blood.
112:1912–1922. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thapa P, Karki R, Yoo HY, Park PH, Lee E,
Jeon KH, Na Y, Cho WJ, Kwon Y and Lee ES:
2,4-Diaryl-5,6-dihydro-1,10-phenanthroline and
2,4-diaryl-5,6-dihydrothieno[2,3-h] quinoline derivatives for
topoisomerase I and II inhibitory activity, cytotoxicity, and
structure-activity relationship study. Bioorg Chem. 40:67–78.
2012.PubMed/NCBI
|
|
26
|
Muñoz A, Sojo F, Arenas DR, Kouznetsov VV
and Arvelo F: Cytotoxic effects of new
trans-2,4-diaryl-r-3-methyl-1,2,3,4-tetrahydroquinolines and their
interaction with antitumoral drugs gemcitabine and paclitaxel on
cellular lines of human breast cancer. Chem Biol Interact.
189:215–221. 2011.
|
|
27
|
Metwally K, Aly O, Aly E, Banerjee A,
Ravindra R and Bane S: Synthesis and biological activity of
2,5-diaryl-3-methylpyrimido[4,5-c]quinolin-1(2H)-one derivatives.
Bioorg Med Chem. 15:2434–2440. 2007.PubMed/NCBI
|
|
28
|
Liu B, Cheng Y, Liu Q, Bao JK and Yang JM:
Autophagic pathways as new targets for cancer drug development.
Acta Pharmacol Sin. 31:1154–1164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ertmer A, Huber V, Gilch S, Yoshimori T,
Erfle V, Duyster J, Elsässer HP and Schätzl HM: The anticancer drug
imatinib induces cellular autophagy. Leukemia. 21:936–942.
2007.PubMed/NCBI
|
|
30
|
Marks PA: Discovery and development of
SAHA as an anticancer agent. Oncogene. 26:1351–1356. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Younes A: Therapeutic activity of
mTOR-inhibitors in mantle cell lymphoma: clues but no clear
answers. Autophagy. 4:707–709. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha
YI and Lu B: Autophagy upregulation by inhibitors of caspase-3 and
mTOR enhances radiotherapy in a mouse model of lung cancer.
Autophagy. 4:659–668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sasi N, Hwang M, Jaboin J, Csiki I and Lu
B: Regulated cell death pathways: new twists in modulation of BCL2
family function. Mol Cancer Ther. 8:1421–1429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Walensky LD: BCL-2 in the crosshairs:
tipping the balance of life and death. Cell Death Differ.
13:1339–1350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Noble CG, Dong JM, Manser E and Song H:
Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J Biol
Chem. 283:26274–26282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rodriguez-Enriquez S, He L and Lemasters
JJ: Role of mitochondrial permeability transition pores in
mitochondrial autophagy. Int J Biochem Cell Biol. 36:2463–2472.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, Mathiasen IS and Jäättelä M: Control of
macroautophagy by calcium, calmodulin-dependent kinase kinase-beta,
and Bcl-2. Mol Cell. 25:193–205. 2007.PubMed/NCBI
|
|
43
|
Manning BD, Tee AR, Logsdon MN, Blenis J
and Cantley LC: Identification of the tuberous sclerosis complex-2
tumor suppressor gene product tuberin as a target of the
phosphoinositide 3-kinase/akt pathway. Mol Cell. 10:151–162. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Inoki K, Li Y, Zhu T, Wu J and Guan KL:
TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR
signaling. Nat Cell Biol. 4:648–657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schumacker PT: Reactive oxygen species in
cancer cells: live by the sword, die by the sword. Cancer Cell.
10:175–176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pan JS, Hong MZ and Ren JL: Reactive
oxygen species: a double-edged sword in oncogenesis. World J
Gastroenterol. 15:1702–1707. 2009. View Article : Google Scholar : PubMed/NCBI
|