Introduction
Liver cancer remains one of the leading causes of
cancer-related mortality. Herbal medicine is a valid source of new
therapeutic agents since an immense chemical diversity is found in
different herb medicines. Some of the active principles from herbal
medicines have been isolated and characterized for cancer drug
development. Herbal medicine offers promise with its complementary
role in the cancer treatment with cancer drugs. Gambogic acid (GA)
is one of the naturally occurring compounds present in a
brownish-to-orange resin called gamboge, which is derived from
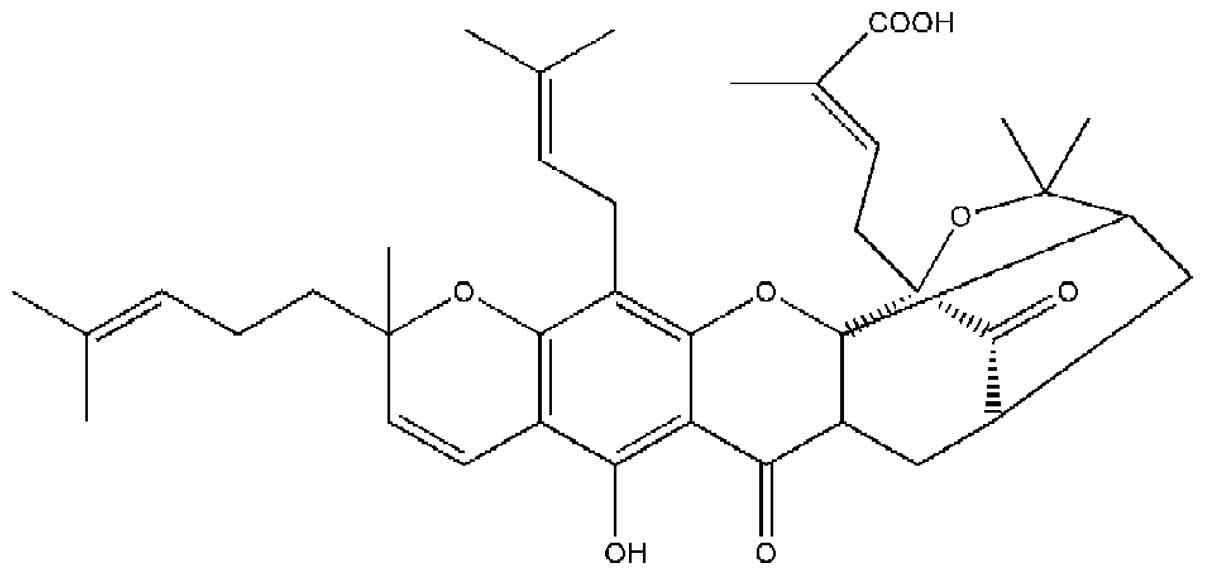
Garcinia hanburyi (Fig. 1).
Garcinia hanburyi has a long history of medicinal use in
Southeast Asia, and it is used as a folk medicine and coloring
agent (1). An improved separation
method for the determination of twelve xanthones in gamboges from
Garcinia hanburyi enabled researchers to purify GA from
Garcinia hanburyi(2).
Previous studies have reported that GA has potent antitumor
activity via induction of reactive oxygen species accumulation
which consequently led to apoptosis of SMMC-7721 cells (3). GA was reported to covalently modify
IκB kinase β subunit to inhibit lipopolysaccharide-induced
activation of NF-κB in macrophages (4). Other studies showed GA mediated the
control of nucleophosmin and nucleoporins in the programmed cell
death of Jurkat cells (5). GA
caused microtubule depolymerization and phosphorylation of c-Jun
N-terminal kinase-1 leading to cell cycle arrest in MCF-7 cells
(6). The induction of apoptosis in
cancer cells is vital in cancer treatment. The present study
explored the antitumor activity of GA in Hep3B and Huh7 human liver
cancer cell lines.
Materials and methods
Chemicals and reagents
Trypsin-EDTA (1X), Dulbecco’s minimal essential
medium (DMEM), Roswell Park Memorial Institute (RPMI)-1640 medium,
penicillin-streptomycin (PS) antibiotic mixture (100X) and
qualified fetal bovine serum (FBS) were purchased from Invitrogen.
Chemicals and reagents were of analytical grade purchased from
Sigma Chemicals, St. Louis, MO, USA. Dimethylsulfoxide (DMSO) was
purchased from Fisher Scientific, USA. Bromophenol Blue, Agarose,
Tris-Base, Boric Acid, NaCl and SDS powder were ordered from USB,
Cleveland, OH, USA. Ethanol was purchased from BDH.
Cell culture and treatment
Hep3B, Huh7 and WRL68 were purchased from ATCC and
cultured according to their protocols. GA was a generous gift from
the Chinese Medicine Laboratory, Hong Kong Jockey Club Institute of
Chinese Medicine, and was isolated by the established method
(2). GA was prepared by dissolving
4 mg of dry GA into 1 ml of DMSO. Cancer cells were seeded in a
96-well plate at the density of 5.0×105 cells/well and
treated with various concentrations of GA for 24 h.
Methylthiazoletetrazolium (MTT) solution (5 mg/ml) was added to the
assay mixture and incubated for 4 h. The culture media was removed
prior to addition of DMSO. Each experiment with GA treatment was
repeated three times. By comparing the absorbance of the wells of
cells treated with different concentrations of GA with the control,
the viability of cells after GA treatment was calculated. The
concentration of GA that reduced the cell viability of 50%
(IC50) was recorded.
DNA extraction
The 100 bp DNA ladder was from Fermentas and the
Cell Death Detection ELISA kit was purchased from Roche Applied
Science. The cell samples were processed according to the
manufacturer’s protocols. The supernatant was subsequently removed
and the DNA pellet was allowed to air dry for 15 to 20 min. TE
buffer (20–50 μl) containing 0.2 mg/ml of RNase A was added to the
DNA pellet. The samples were incubated at 37°C for 90 min to
completely dissolve the sample. The DNA solution (2 μl) was added
to 998 μl TE buffer and the concentration was measured using UV
spectrophotometry (DU 650; Beckman-Coulter) with
OD260.
DNA agarose gel electrophoresis
Ten microliters of the dissolved DNA samples were
mixed with 2 μl 6X DNA loading dye. The mixtures were loaded on the
2% agarose gel and run at 75V for 1 h. The DNA bands were examined
under UV illuminator and the gel was photographed for
documentation.
DAPI stain
Changes in cell morphology during apoptosis were
examined by fluorescence microscopy of DAPI-stained cells. The
monolayer of cells was washed in PBS and fixed with 4%
paraformaldehyde for 30 min at room temperature. The fixed cells
were permeabilized with 0.2% Triton X-100 in PBS three times and
incubated with 1 μg/ml of DAPI for 30 min. The cells were washed
with PBS three times. The apoptotic nuclei (intensely stained,
fragmented nuclei and condensed chromatin) were examined under ×400
magnification using a fluorescent microscope with a 340/380 nm
excitation filter for at least two investigations.
Cell death detection with ELISA
ELISA was carried out according to the
manufacturer’s protocol (Invitrogen). The cells were incubated with
different concentrations of GA in a 96-well plate for 24 h prior to
centrifugation at 200 × g for 10 min. The lysis buffer (200 μl) was
added to lyse the cell pellet. The plate was kept at room
temperature for 30 min prior to centrifugation at 200 × g for 10
min. The supernatant (20 μl) was placed in the streptavidin-coated
plate for analysis. The immunoreagent (80 μl) was added to the well
and covered by aluminum foil with shaking at 300 rpm for 2 h. The
solution in each well was removed and the well was rinsed with 250
μl incubation buffer three times. ABTS solution of 100 μl was added
into the wells prior to shaking in the plate shaker at 250 rpm for
color development. After 10–20 min, 100 μl ABTS Stop Solution was
added to stop the reaction and the well was measured at 405 nm.
Absorbance of wells was recorded and the enrichment factor, which
shows the extent of apoptosis in the GA-treated cells, was
calculated by the following expression: (absorbance of the
GA-treated sample-blank)/(absorbance of the vehicle-treated
sample-blank).
Protein expression in GA-induced
cells
The antibodies, anti-p53, anti-Bcl-2, anti-Bax,
anti-caspase-8 and anti-PARP, were purchased from Santa Cruz
Biotechnology, Inc. Anti-cytochrome c, anti-Bid and
anti-GAPDH were purchased from BD Pharmingen. For HRP-secondary
antibodies, anti-mouse and anti-rabbit antibodies were obtained
from Invitrogen. Cells (1×106) were seeded into a 100 mm
dish. Different concentrations of GA were added prior to incubation
for 4, 8, 12 and 24 h. The cell samples were spun at 14,000 × g for
15 min. The supernatant of the samples was collected for protein
concentration determination according to the Bio-Rad Protein Assay
protocol.
Extraction of cytosolic and mitochondrial
protein fraction
Cells were collected and counted prior to the
addition of the cytosolic protein lysis buffer (50 μl). The sample
was vortexed for 30 sec and centrifuged at 14,000 × g for 1 min at
4°C. A volume of 2X SDS sample loading buffer was added to the
supernatant. The whole cell lysis buffer and 2X SDS sample loading
buffer were added to completely lyse the sample.
Protein gel electrophoresis by
SDS-PAGE
The electrophoresis system, Mini-PROTEAN®
II cell from Bio-Rad, was used to perform sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The
resolving gel solution (10%) was set and poured into the gel
casting form. The top of the gel was layered with 50 μl of
isopropanol and the gel was left for 30 min. Stacking gel solution
(4%) was added onto the top of the resolving gel. A 15-tooth comb
was inserted and the gel was allowed to polymerize for 40 min. The
samples mixed with 2X sample loading dye were boiled at 100°C for
10 min. After samples were loaded onto the wells, the gel was run
at constant voltage at 150 V for 1 h in 1X running buffer.
Western blotting
The SDS-PAGE was completed when the dye front (blue
in color) reached the bottom of the gel. The gel was removed and
immersed into transfer buffer. The Whatman 3 MM paper (6
pieces/gel) and PVDF membrane were cut into the same dimension as
the resolving gel. The PVDF membrane was washed with 100% methanol
for 1 min and was immersed into the transfer buffer. The Whatman
papers were soaked into the transfer buffer. A gel sandwich was
assembled with the PVDF membrane and the resolving gel layered
between two stacks of Whatman papers in the semi-dry Trans-Blot
electroblotter (Bio-Rad). Proteins were transferred to the membrane
at constant voltage at 10 V for 1.5 h. The membrane was collected
and rinsed briefly with TBST buffer. Then, the membrane was
immersed into blocking solution with primary antibody for 16 h at
4°C with continuous agitation. The primary antibody used was mouse
monoclonal anti-p53 (1:1,000), anti-BID (1:1,000), anti-Bcl-2
(1:1,000), anti-Bax (1:500), anti-cytochrome c (1:2,000),
anti-caspase-8 (1:1,000), anti-PARP (1:1,000), anti-transferrin
receptor (1:1,000) and anti-GAPDH (1:5,000). The unbound primary
antibody was removed by washing with TBST for 20 min, thrice. The
membrane was immersed into secondary antibody (goat anti-mouse
HRP/mouse anti-rabbit HRP) for 1 h at room temperature with slow
agitation. The excess antibody was removed by washing with TBST
three times (20 min). Millipore Immobilon Western HRP kit (0.5 ml
of each reagent) was applied to the membrane for 3 to 5 min. After
removing excess reagent, protein bands on the membrane were
visualized and recorded on Fuji Super RX film (Fujifilm). Intensity
of the bands was measured using ImageJ program.
Immunoprecipitation
Cells were seeded into 100 mm dishes. After 24 h,
different concentrations of GA (2, 1, 0.5 μM) were added before
incubation for another 24 h. The cells were collected and lysed in
CHAPS buffer. The concentration of the lysate was measured and
diluted 500 μg protein/500 μl buffer. The anti-Bax 6A7 monoclonal
antibody (2 μg) was added to the diluted lysate and the mixture was
incubated overnight at 4°C. After the incubation, 25 μl of protein
A-agarose gel bead was added and the lysate was incubated for 3 h.
The gel beads were washed three times using the CHAPS buffer and
collected by centrifugation at 14,000 × g for 10 min. The sample
loading dye (50 μl) was added to the beads prior to boiling for 10
min, followed by a centrifugation of 14,000 × g at 4°C. The
supernatant was subjected to western blot analysis using mouse
anti-Bax antibody.
Caspase cascade study of GA-induced
cancer cells
Apo-One™ caspase-3/7 assay kit was purchased from
Promega. Caspase inhibitor z-IETD-fmk and z-LEDH-fmk were from
Calbiochem. Trypan blue was purchased from Invitrogen.
Hemocytometer was obtained from Sigma Chemicals. Caspase-3/7
activity was examined using Apo-One™ Caspase-3/7 Assay kit
(Promega). Cells (Hep3B or Huh7) were seeded in a 96-well plate in
the medium for 24 h followed by GA treatment of the cell lines.
Subsequently, z-DEVD-rhodamine 110 from the assay kit was added
into the well. After 2 h of incubation at room temperature, the
fluorescence intensity of the wells was measured by a fluorescence
plate reader at excitation 492 nm and emission 535 nm.
Results
GA induces apoptosis in hepatocellular
cells
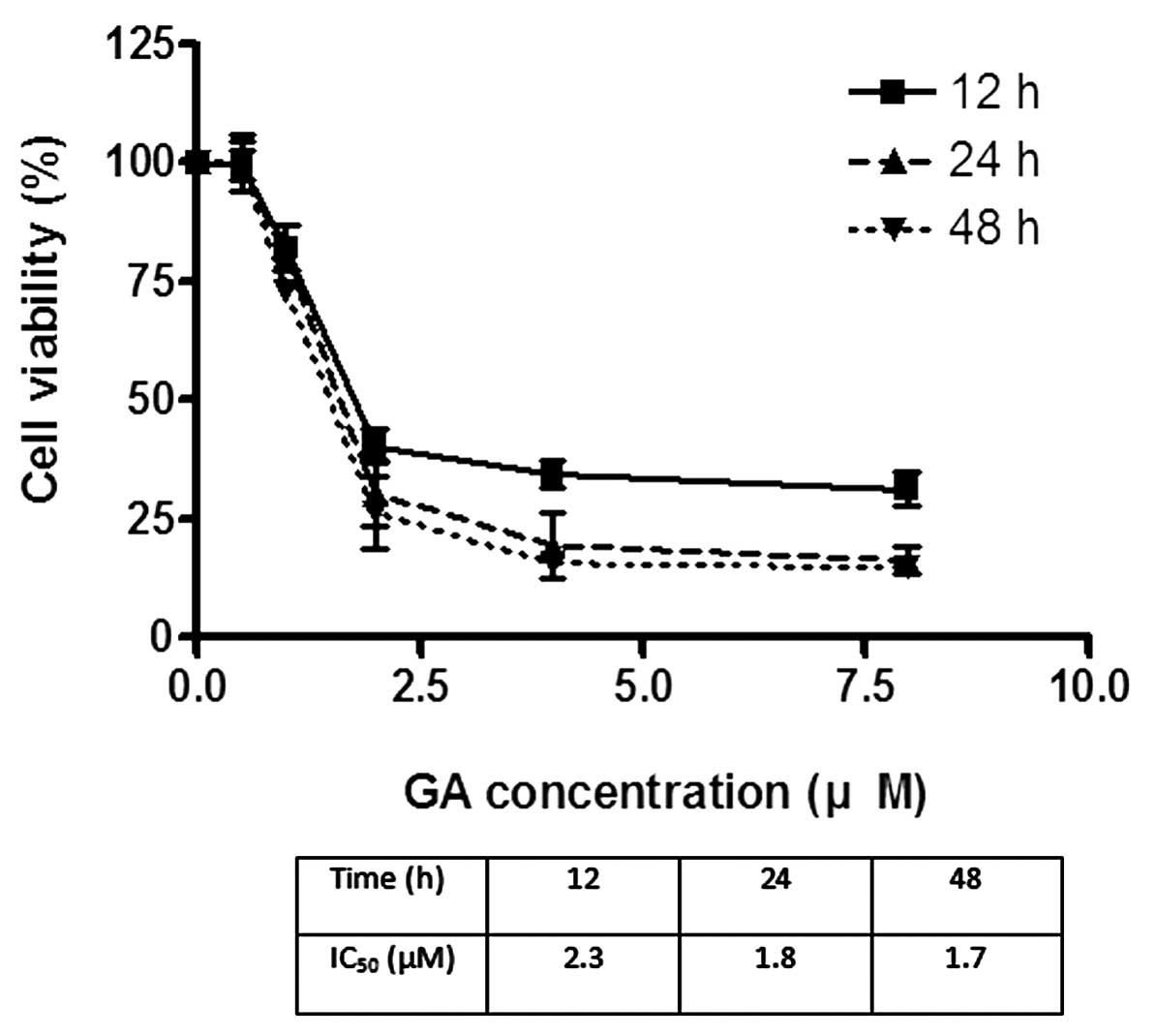
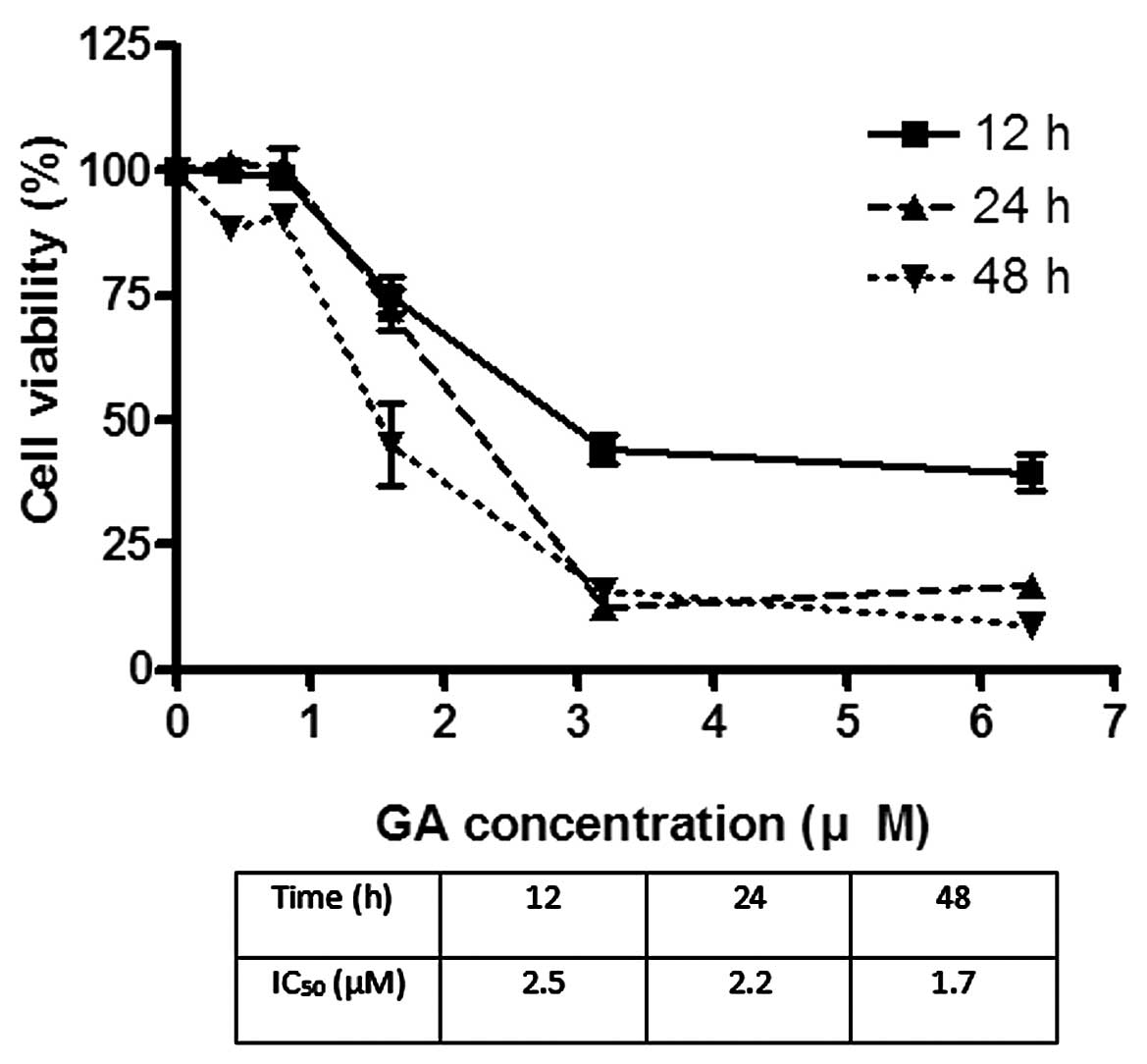
The effects of GA on the viability of Hep3B and Huh7
were investigated using MTT assay. GA effectively reduced the
viability of cancer cells after incubation for 24 h. The
IC50 of GA for Hep3B was 1.8 and 2.2 μM for Huh7
(Figs. 2 and 3). After treatment of cells with GA, cell
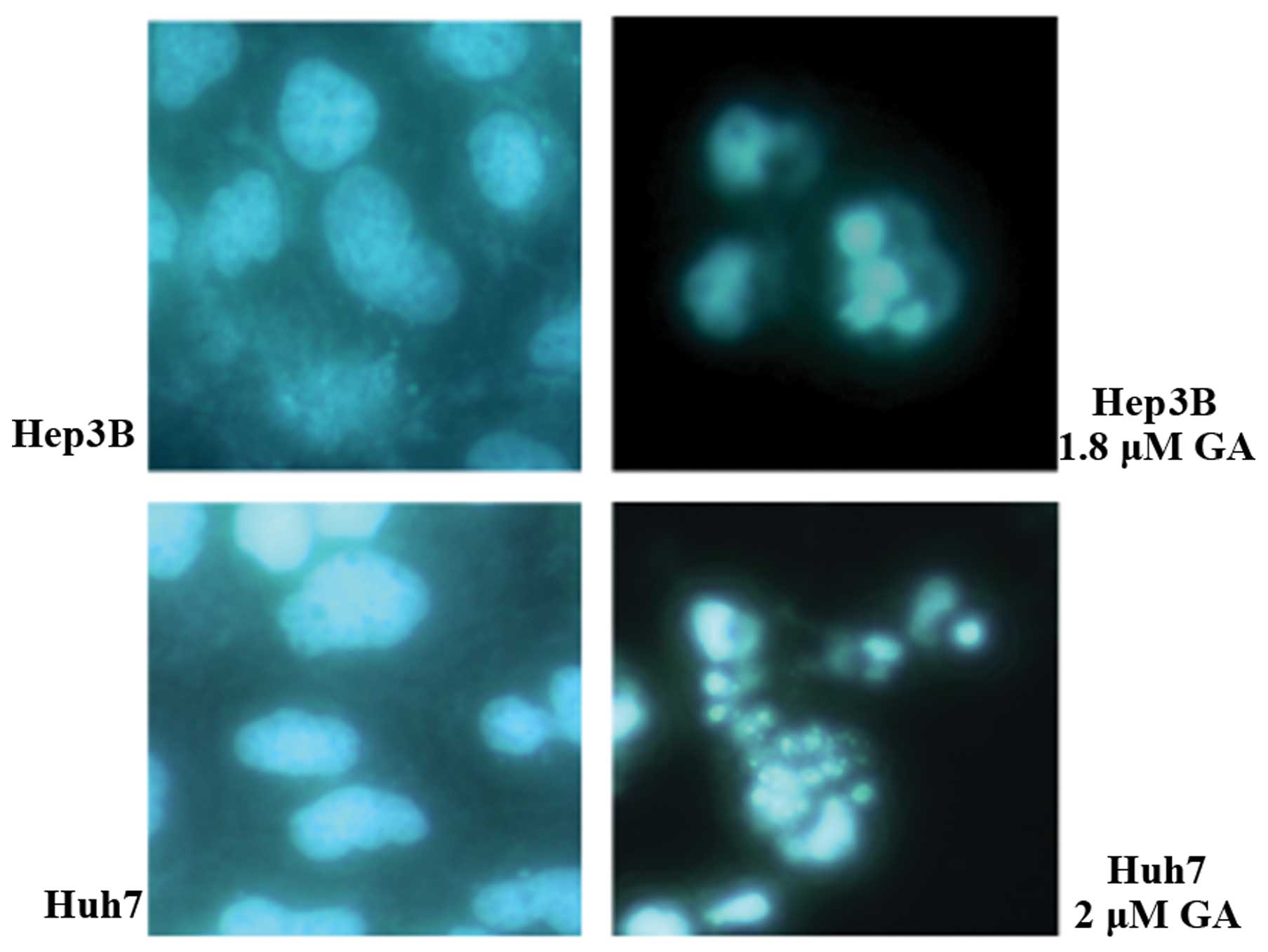
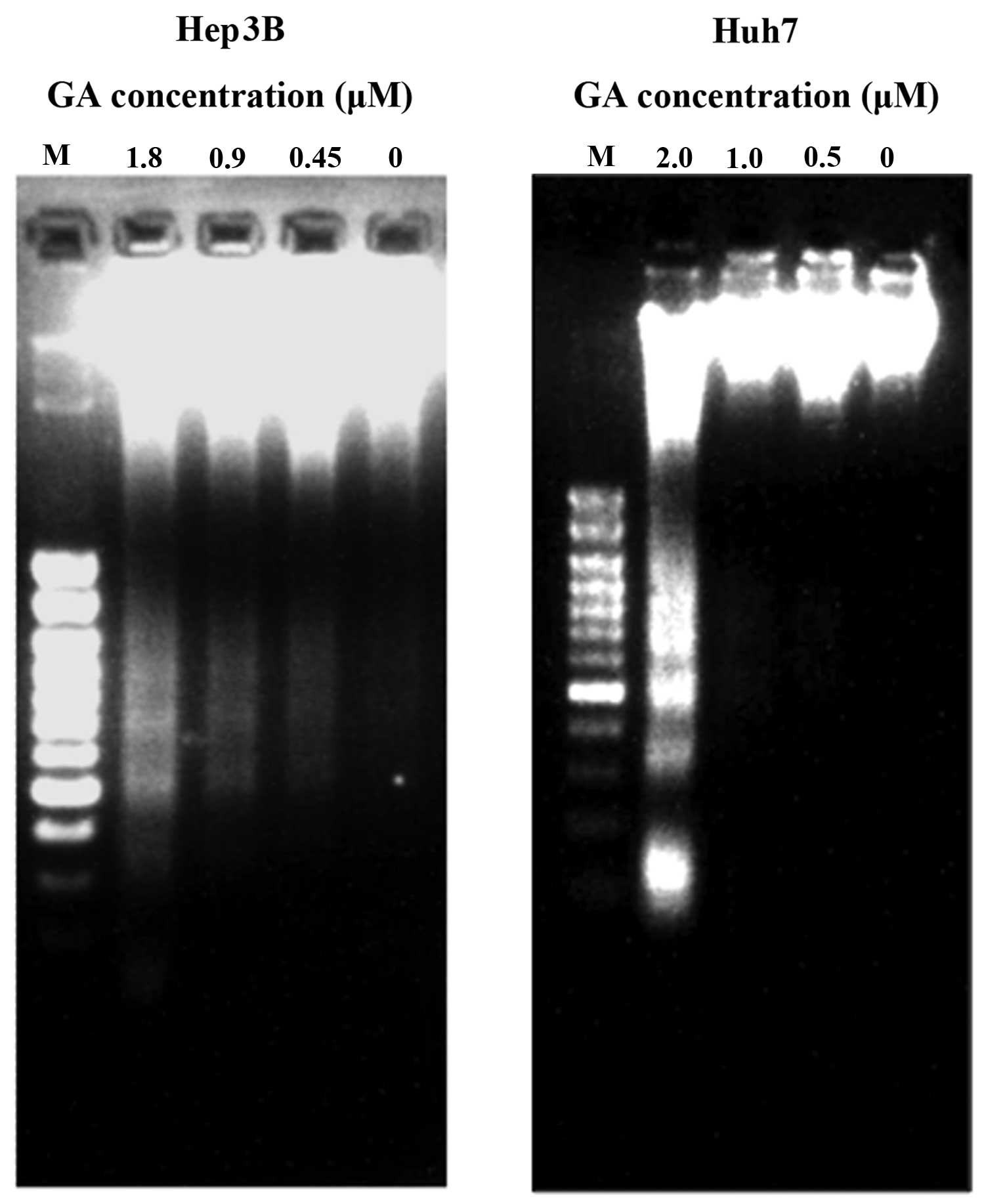
shrinkage and budding were recorded. DNA fragmentation and
condensation were visualized by the DAPI staining investigation
using fluorescence microscopy (Figs.
4 and 5). DNA fragmentation was
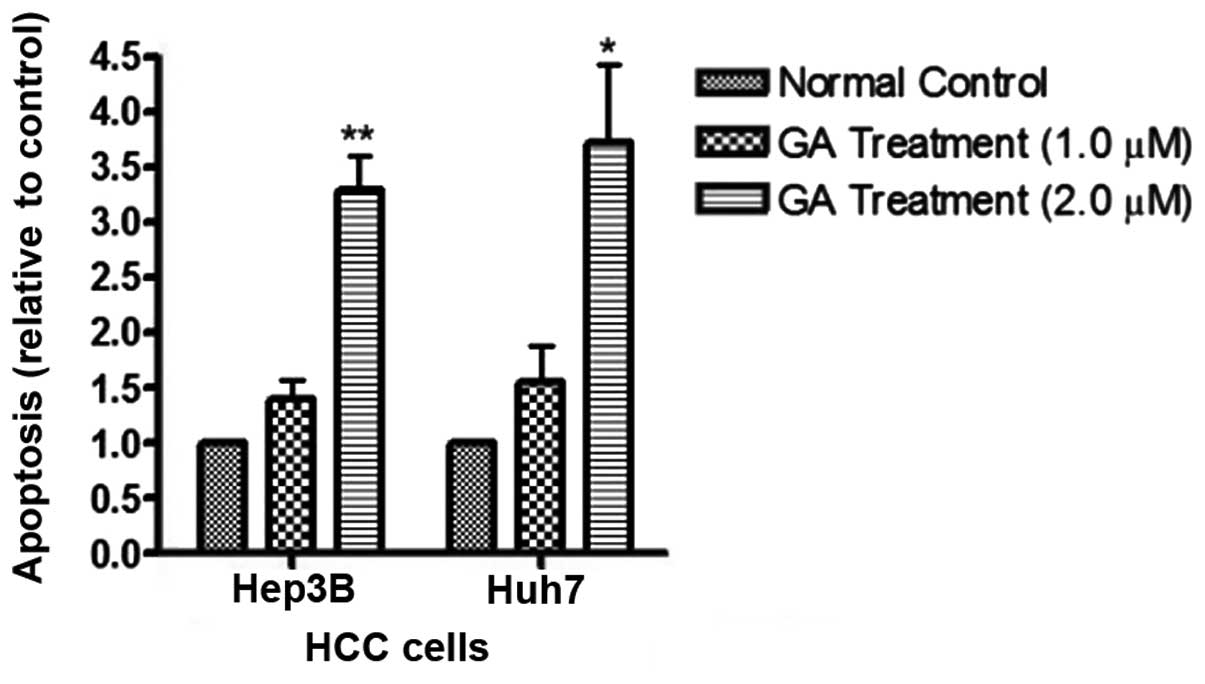
detected after GA treatment in HCC cell lines (Fig. 5). The amount of cytosolic DNA
fragment was determined with cell ELISA kit (Fig. 6). The results showed that the amount
of DNA in the cytosol significantly increased after addition of GA.
Apoptosis is characterized by the occurrence of cell shrinkage and
membrane blebbing (7). In addition,
chromatin condensation and DNA cleavage shown by DAPI staining
(Fig. 4) also indicated apoptosis
(8). To avoid ambiguity, DNA
fragmentation, another hallmark of apoptosis, was examined after
the treatment of GA on the cell lines for 24 h. The result of
agarose gel electrophoresis showed the presence of DNA ladders
(Fig. 5). During apoptosis, mono-
and oligo-nucleosomes are released into the cytoplasm before the
plasma membrane breaks down (9,10). The
cytoplasmic content of the tested cells was collected and the
amount of released DNA was detected by the ELISA kit. The results
showed that following GA treatment, the levels of cytoplasmic DNA
in HCC cells was markedly elevated (Fig. 7). These results provide evidence
that GA mediated cell death through apoptosis.
Caspase cascade studies in GA-induced
apoptosis
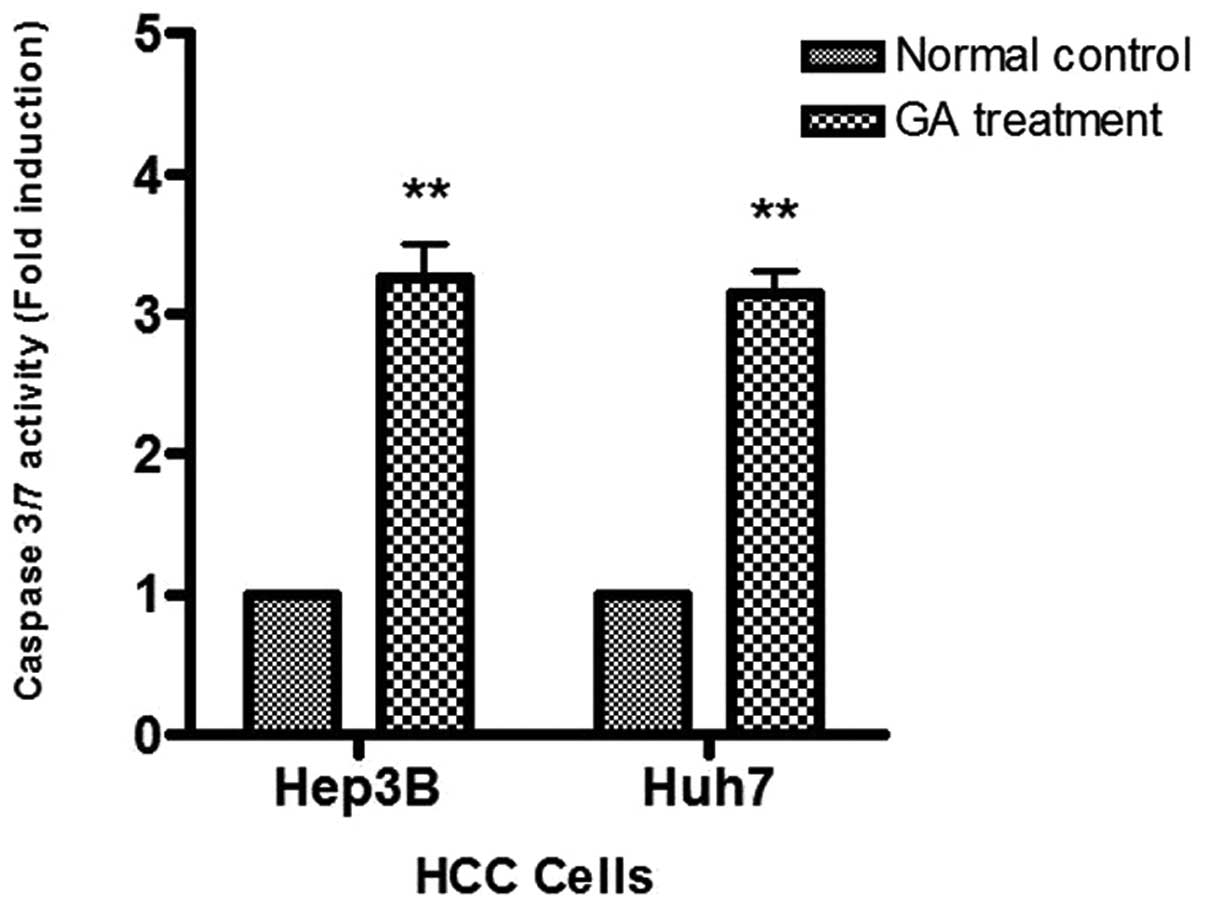
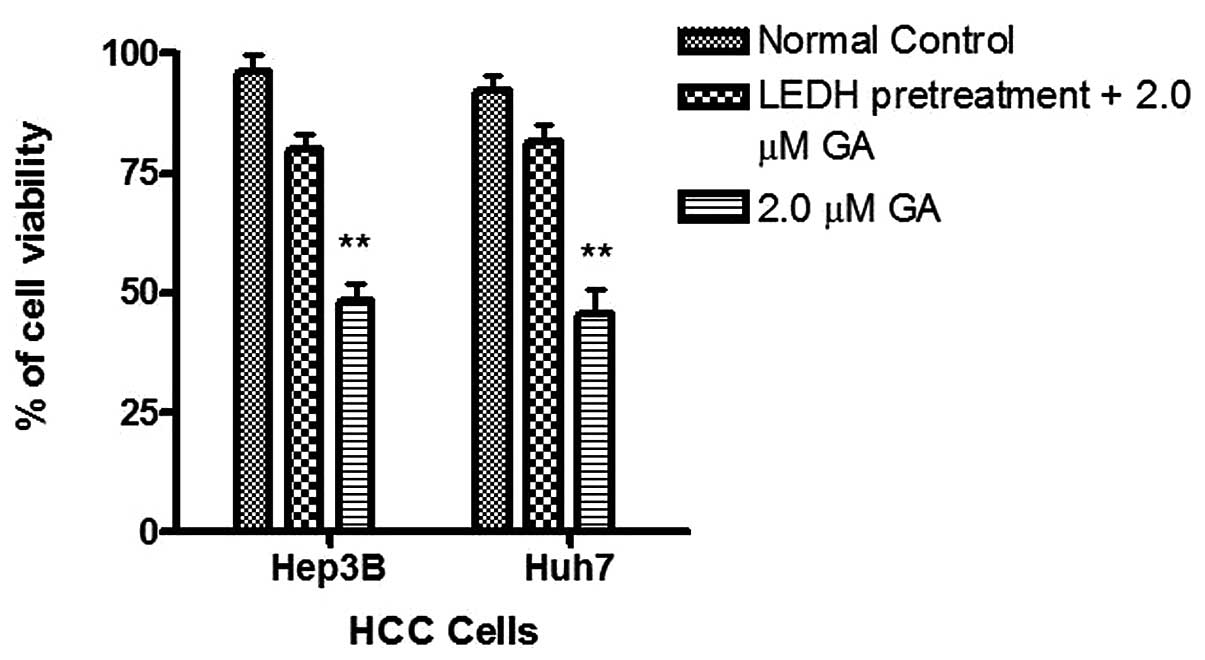
To study caspase activity in the GA-induced
apoptosis, Apo-One™ Caspase-3/7 Assay was used. An increase of
caspase-3/7 activity was found in the two HCC cells after GA
treatment (Fig. 7). The activation
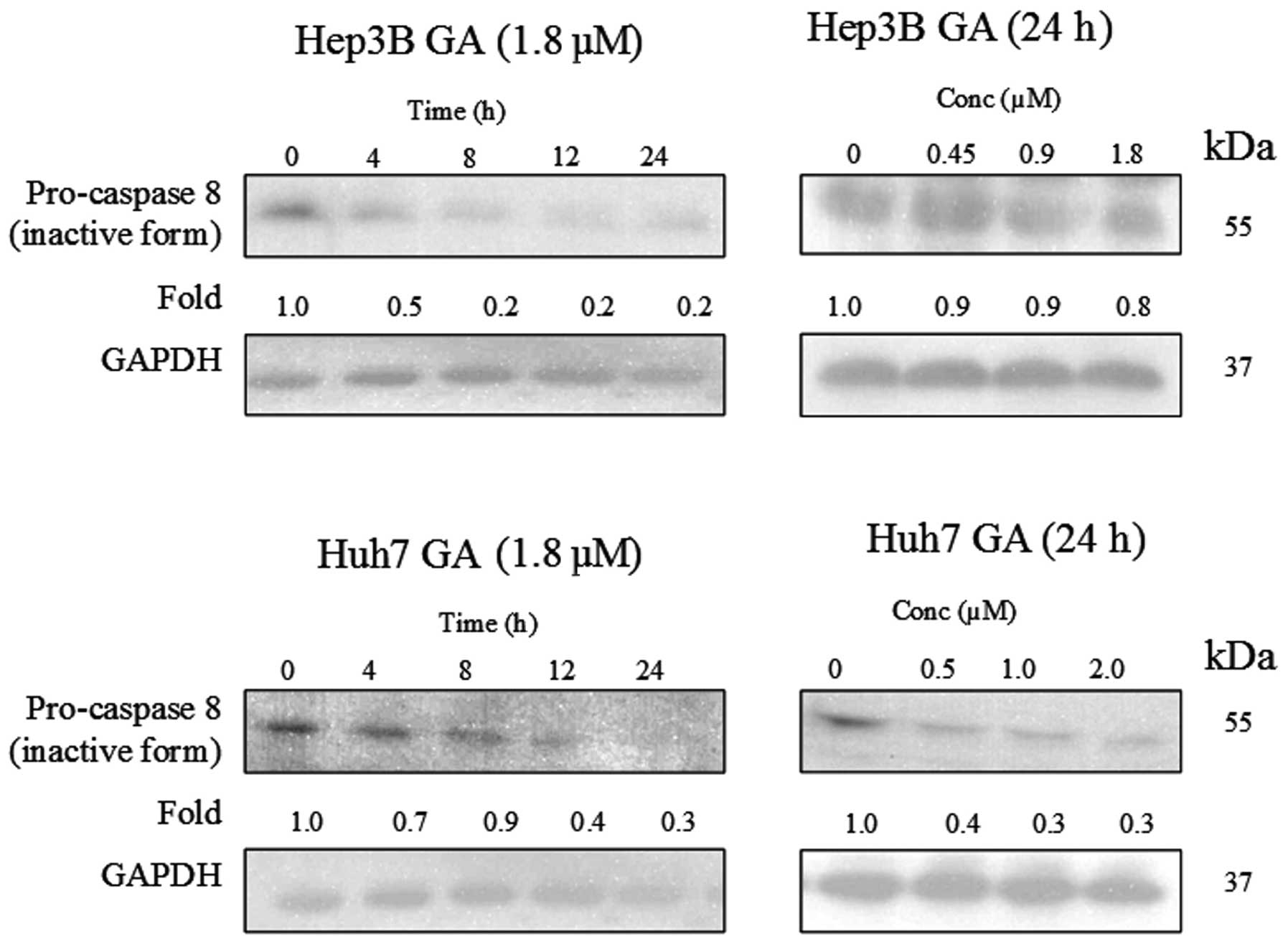
of caspase-8 in the death receptor pathway and caspase-9 in the
mitochondrial pathway were determined using western blot analysis
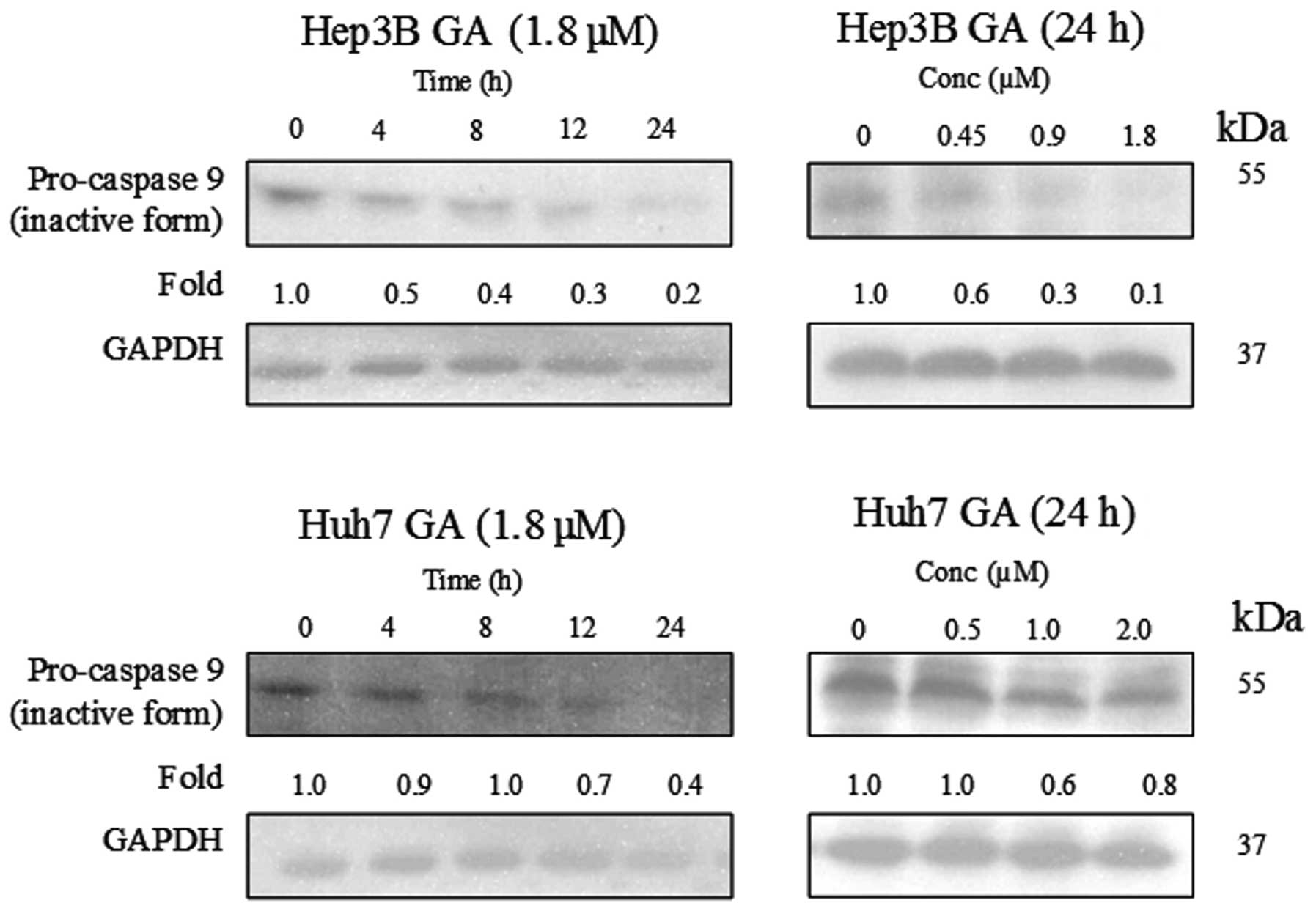
(Figs. 8–10). The results indicate that the
cleavage of the active form of caspases-8 and -9 after GA-treatment
occurred in a time- and dose-dependent manner (Fig. 11).
Caspase-8 activation in GA-treated cells
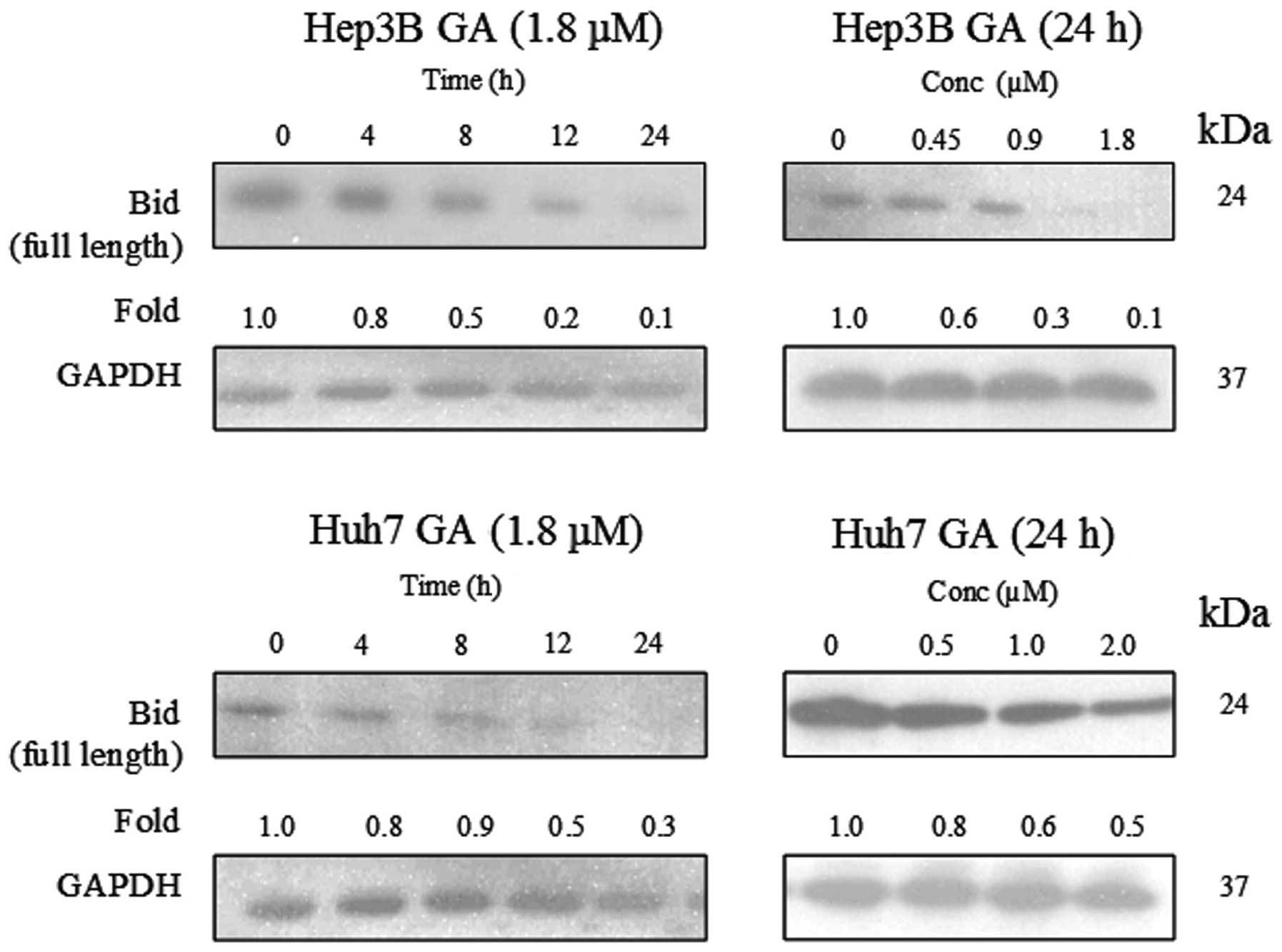
leads to Bid cleavage
The involvement of Bid and its relationship with
caspase-8 activation in GA-induced apoptosis were investigated.
Full length Bid (24 kDa) was found to be cleaved in a time- and
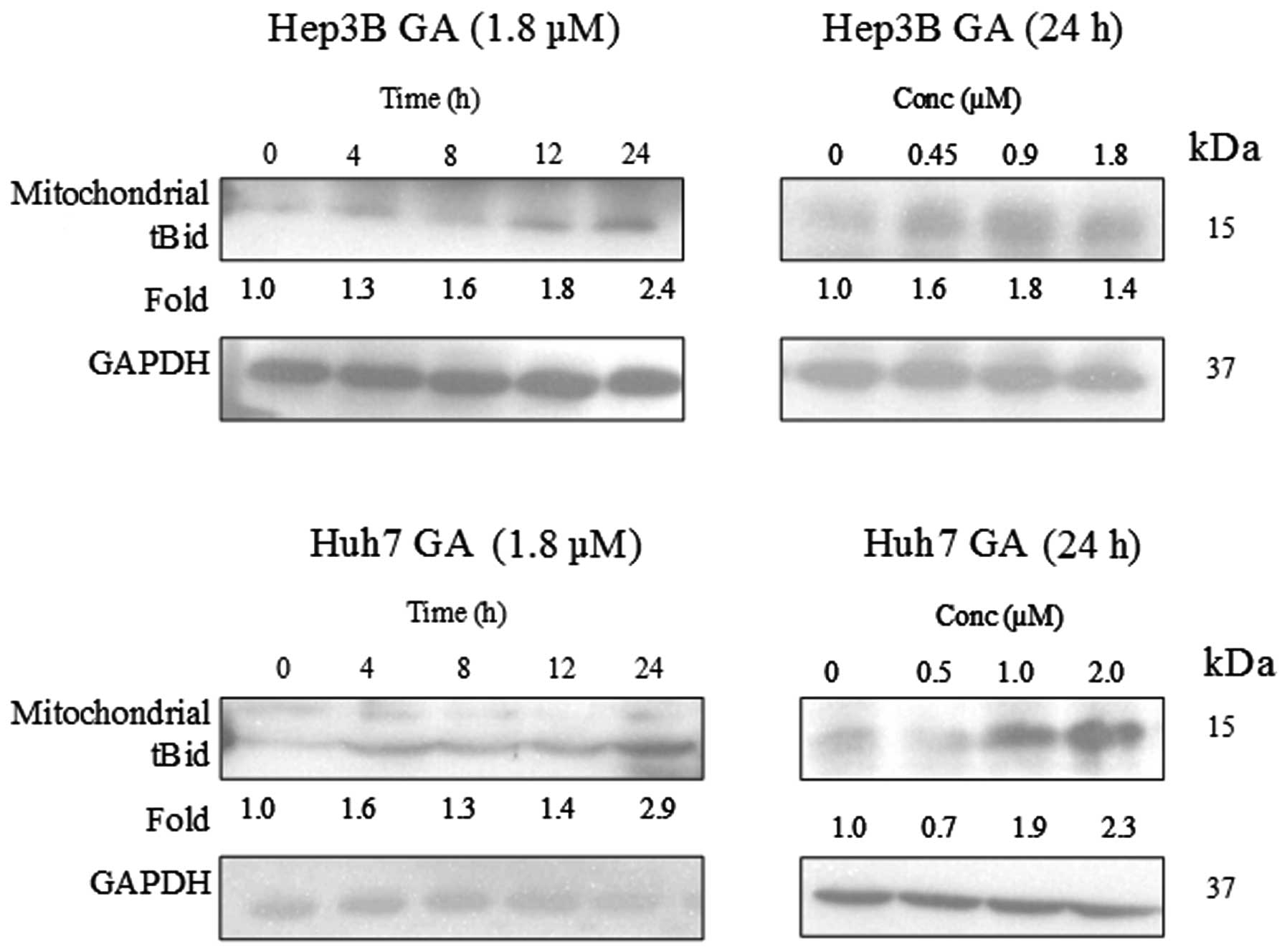
dose-dependent manner upon GA treatment (Fig. 12). A cell fractionation experiment
was performed to investigate the level of tBid in the mitochondria.
Fig. 13 indicates an accumulation
of tBid in the mitochondria in the HCC cell line.
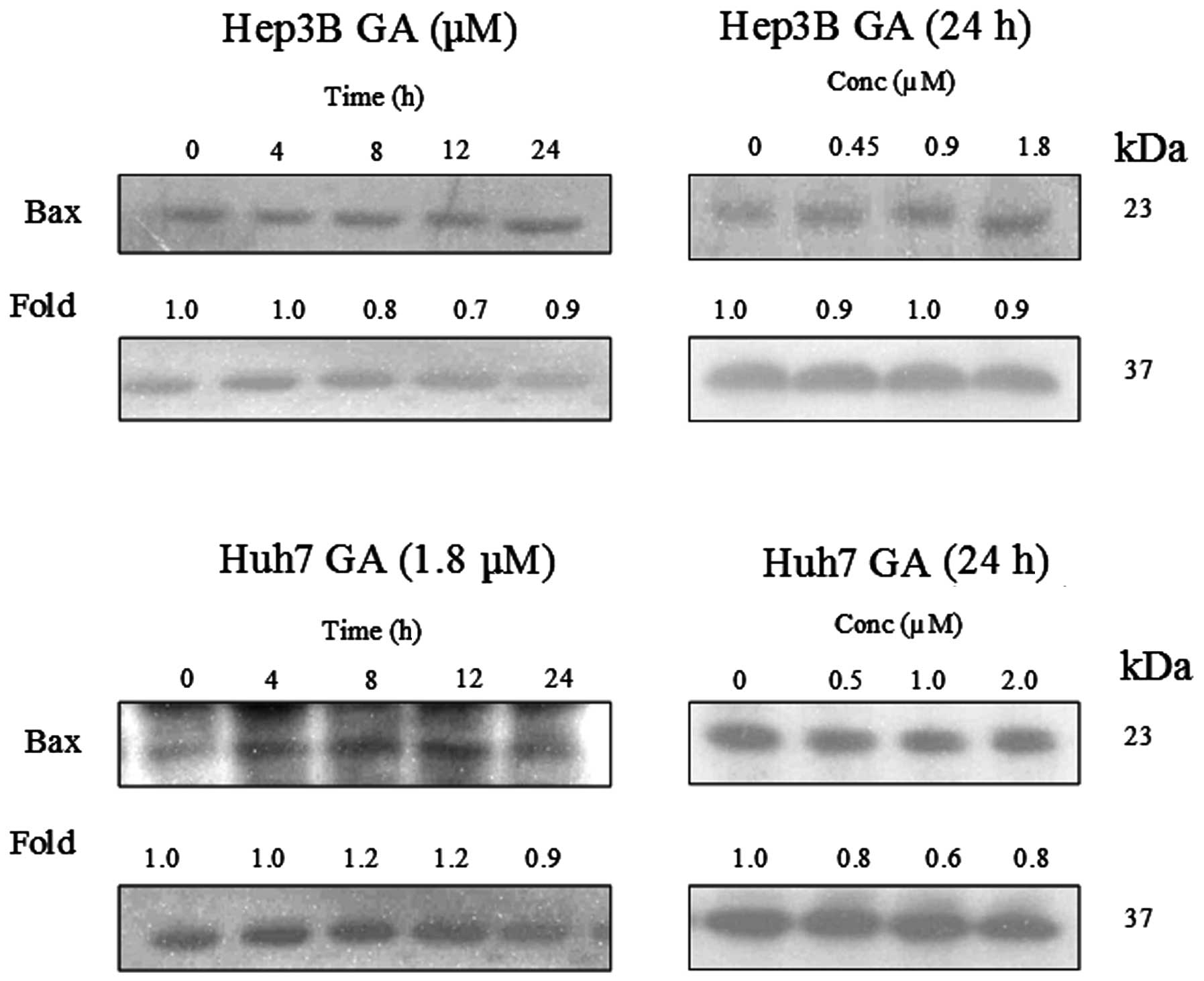
GA induces Bax conformational changes and
cytochrome c release
It has been reported that Bax plays an important
role in Bid-mediated apoptosis through conformational change and
mediation of signaling process (11,12).
Fig. 14 shows that the expression
level of Bax was constant in GA-induced apoptosis. In order to
further examine the role of Bax in GA-induced apoptosis,
immunoprecipitation (IP) of Bax using Bax (6A7) monoclonal antibody
was performed. This antibody recognizes the Bax protein with
conformational change but not the native form (11). It was reported that the
conformational change of Bax was triggered by the truncated form of
Bid (tBid) (13). This tBid is
subsequently translocated to the mitochondria with Bax. The
translocation of tBid to mitochondria was shown in western blot
analysis (Fig. 15). Cytochrome
c, which is originally present in the mitochondria, was
released.
Discussion
In the present study we provided insights into the
effects of GA on the inhibition of two different types of liver
cancer cells. Present chemotherapy based on the treatment of liver
cancer with drugs is not yet satisfactory. Specific anticancer
agents against different types of liver cancer need to be
developed. We demonstrated that GA induced apoptosis via the
mitochondrial pathway in two types of liver cancer cells. p53
protein is a crucial factor in cellular stress responses and acts
as an essential tumor suppressor (14). Upon activation, p53 controls the
signaling process associated with the cell cycle based on the
severity of the DNA damage. Thus, it can inhibit cell cycle
progression or induce apoptosis. More than 50% of human tumors have
been reported to have p53 mutations which affect p53 function. We
found that GA induced caspase-associated apoptotic pathway which is
important for induction of caspase-regulated apoptosis. We detected
caspase protein in both cancer cells. The two HCC cell lines differ
in the expression of one tumor suppressor protein, p53. Hep3B is
with deleted p53 while Huh7 has mutated p53. Both cells are p53
deficient. p53, also known as tumor protein 53, is a tumor
suppressor protein that in humans is encoded by the TP53
gene. p53 plays a crucial role in regulation of the cell cycle. The
functions of the tumor suppressor gene have become an important
target to prevent cancer. As such, p53 has been described as ‘the
guardian of the genome’ due to its pivotal role in conserving the
stability of genome and preventing mutation (14). p53 tumor suppressor is one of our
defenses against damage due to radiation, carcinogens and viruses.
When DNA damage occurs, p53 levels rise and initiate protective
measures. p53 binds to several regulatory sites in the genome that
consequently halts cell division through the process of programmed
cell death, or apoptosis. The mediation of p53 provides a common
drug target for cancer development. However, the regulation of cell
cycle in p53-deficient cells may be through different apoptotic
pathways. A recent study showed that p53 can mediate apoptosis by
repression of HPV oncogenes and upregulation of tumor suppressor
proteins in human cancer cells (15). Hep3B contains an integrated
hepatitis B virus (HBV) genome in the cell which is highly
associated with HCC development. However, the present findings
showed that GA induces apoptosis in Hep3B and Huh7 cell death
through caspases and is independent of p53-associated pathway. The
results suggest the operative role of GA may be involved in killing
HBV genome integrated in HCC through regulation of other apoptotic
processes. A previous study indicated that GA has no influence on
the viability of primary cultured rat hepatocytes (16), suggesting that GA causes less harm
to normal liver cells than to its cancer counterparts. These
findings also reveal that the apoptosis of liver cancer cells is
regulated by different complex systems involving large numbers of
molecules and that GA-mediated signaling is only part of the
regulatory system. The existence of the p53-independent regulatory
signals in apoptosis in liver cancer cells may represent an
alternate approach for inhibition of cancer cell growth to avoid
complete liver damage. To investigate the possible mechanism that
led to the cell death of Hep3B and Huh7 cells, different parameters
related to apoptosis including cell shrinkage and budding, DNA
fragmentation and condensation, as well as cytosolic DNA content
were measured. Both the death receptor pathway and the
mitochondrial pathway were found to be involved in GA-induced
apoptosis. Several studies showed that Bid is an important
pro-apoptotic protein in cross-linking the extrinsic cell death
receptor signaling pathway to mitochondria upon caspase-8-mediated
cleavage (17). These findings are
consistent with the previous report that integration of Bax and
tBid to the outer membrane followed by the release of cytochrome
c is a possible prerequisite for the mitochondrial apoptotic
pathway (13,18). GA induces apoptosis in p53-deficient
cancer cells. GA exerts inhibitory effects on Hep3B and Huh7 cell
lines through similar mode of action. Based on these results, it
seems unlikely that GA-induced apoptosis is due to the p53 status.
This study showed the anticancer activity of GA is mediated via
both the caspases in the extrinsic death receptor pathway and the
mitochondria-dependent pathway. The present study demonstrated that
deletion or deficiency of p53 does not affect cell cycle. The
present results indicate that GA positively regulates the cancer
cell apoptosis. Therefore, GA activity in liver cancer cells may
represent one of the molecular mechanisms involved in anticancer
agent-induced apoptosis. The low concentration of GA towards cancer
cells is one of the fundamental criteria for efficient drug
development and targeting. The results suggest that GA can be
developed as an anticancer drug for p53-deficient cancer cells. The
present study on GA may provide a promising therapeutic strategy
for different types of liver cancer.
Acknowledgements
This study was in part supported by grant no.
6903292 kindly provided by Luck Tissue MFY Ltd. We thank Mr. Matt
Cheung for his technical assistance.
References
|
1
|
Asano J, Chiba K, Tada M and Yoshii T:
Cytotoxic xanthones from Garcinia hanburyi. Phytochemistry.
41:815–820. 1996. View Article : Google Scholar
|
|
2
|
Li S, Song JZ, Han QB, Qiao CF and Xu HX:
Improved high-performance liquid chromatographic method for
simultaneous determination of 12 cytotoxic caged xanthones in
gamboges, a potential anticancer resin from Garcinia
hanburyi. Biomed Chromatogr. 22:637–644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu
W, Lu N, Wu Z, You Q and Guo Q: Reactive oxygen species
accumulation contributes to gambogic acid-induced apoptosis in
human hepatoma SMMC-7721 cells. Toxicology. 260:60–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Palempalli UD, Gandhi U, Kalantari P,
Vunta H, Arner RJ, Narayan V, Ravindran A and Prabhu KS: Gambogic
acid covalently modifies IkappaB kinase-beta subunit to mediate
suppression of lipopolysaccharide-induced activation of NF-kappaB
in macrophages. Biochem J. 419:401–409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shu W, Chen Y, Li R, Wu Q, Cui G, Ke W and
Chen Z: Involvement of regulations of nucleophosmin and
nucleoporins in gambogic acid-induced apoptosis in Jurkat cells.
Basic Clin Pharmacol Toxicol. 103:530–537. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen J, Gu H, Lu N, Yang Y, Liu W, Qi Q,
Rong J, Wang XT, You QD and Guo QL: Microtubule depolymerization
and phosphorylation of c-Jun N-terminal kinase-1 and p38 were
involved in gambogic acid induced cell cycle arrest and apoptosis
in human breast carcinoma MCF-7 cells. Life Sci. 83:103–109. 2008.
View Article : Google Scholar
|
|
7
|
Bortner CD and Cidlowski JA: Cell
shrinkage and monovalent cation fluxes: role in apoptosis. Arch
Biochem Biophys. 462:176–188. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oberhammer FA, Hochegger K, Fröschl G,
Tiefenbacher R and Pavelka M: Chromatin condensation during
apoptosis is accompanied by degradation of lamin A+B, without
enhanced activation of cdc2 kinase. J Cell Biol. 126:827–837.
1994.
|
|
9
|
Bonfoco E, Krainc D, Ankarcrona M,
Nicotera P and Lipton SA: Apoptosis and necrosis: two distinct
events induced, respectively, by mild and intense insults with
N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell
cultures. Proc Natl Acad Sci USA. 92:7162–7166. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duke RC and Cohen JJ: IL-2 addiction:
withdrawal of growth factor activates a suicide program in
dependent T cells. Lymphokine Res. 5:289–299. 1986.PubMed/NCBI
|
|
11
|
Nechushtan A, Smith CL, Hsu YT and Youle
RJ: Conformation of the Bax C-terminus regulates subcellular
location and cell death. EMBO J. 18:2330–2341. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang S, Zhao S, Bai L, Guan M, Mo J and
Lan L: Melatonin restores normal Bax and Bcl-2 protein expression
in the subgranular zone of the dentate gyrus in pinealectomized
rats. Neural Regen Res. 6:2129–2133. 2011.
|
|
13
|
Gross A, Jockel J, Wei MC and Korsmeyer
SJ: Enforced dimerization of BAX results in its translocation,
mitochondrial dysfunction and apoptosis. EMBO J. 17:3878–3885.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Latonen L and Laiho M: Cellular UV damage
response - functions of tumor suppressor p53. Biochim Biophys Acta.
1755:71–89. 2005.PubMed/NCBI
|
|
15
|
Munagala R, Kausar H, Munjal C and Gupta
RC: Withaferin A induces p53-dependent apoptosis by repression of
HPV oncogenes and upregulation of tumor suppressor proteins in
human cervical cancer cells. Carcinogenesis. 32:1697–1705. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Y, Yang L, You QD, Nie FF, Gu HY,
Zhao L, Wang XT and Guo QL: Differential apoptotic induction of
gambogic acid, a novel anticancer natural product, on hepatoma
cells and normal hepatocytes. Cancer Lett. 256:259–266. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Zhu H, Xu C and Yuan J: Cleavage of
BID by caspase mediates the mitochondrial damage in the Fas pathway
of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Lu N, Dai Q, Wei L, Zhao Q, Li Z, He
Q, Dai Y and Guo Q: GL-V9, a newly synthetic flavonoid derivative,
induces mitochondrial-mediated apoptosis and G2/M cell cycle arrest
in human hepatocellular carcinoma HepG2 cells. Eur J Pharmacol.
670:13–21. 2011. View Article : Google Scholar
|