Introduction
The Philadelphia chromosome, a hallmark of chronic
myeloid leukaemia (CML), generates the fusion oncoprotein, Bcr-Abl,
and a constitutively active tyrosine kinase (1,2).
Chronic phase CML is well managed in most patients with tyrosine
kinase inhibitors (TKIs), most notably imatinib, whereas the
advanced stage of CML known as blast crisis is a highly aggressive
drug-resistant disease and is analogous to acute leukaemia
(3).
Despite the success of imatinib for the treatment of
CML, treatment failures due to resistance and relapse are still
common. Bcr-Abl amplification, mutations or the acquisition of
Bcr-Abl-independent mechanisms have been shown to play a role in
the development of resistance (4).
In addition, relapse is common when imatinib is withdrawn, even in
patients who achieve a complete molecular response and this can
re-emerge as the same imatinib susceptible disease (5). This highlights the need for
potentially life-long treatment and the possibility of resistant
disease emerging later on.
We and others have shown that the induction of
autophagy in response to treatment with imatinib is associated with
persistence and recovery of a subpopulation of leukaemia cells
(6,7). Autophagy is a survival mechanism
initiated in response to cellular stress or starvation. It removes
damaged organelles and aggregated proteins and can recycle
cytoplasmic content (8). Inhibition
of autophagy can significantly reduce the recovery of CML cells
following imatinib treatment (7).
This is one of the most promising strategies for improving the
curative ability of imatinib.
Currently, there are no specific autophagy
inhibitors. There are, however, a limited number of pharmacological
agents that have been reported to disrupt processes or organelles
required for effective autophagy. Combining such agents with
imatinib may therefore compromise the autophagic recovery employed
by leukaemic cells, potentially improving treatment regimens for
CML. We evaluated three agents in combination with imatinib:
chloroquine, vincristine and brefeldin A. Chloroquine, a
lysosomotropic agent, damages lysosomal integrity leading to the
inhibition of autophagosome turnover (9) and is currently in clinical trials in
combination with imatinib for treatment of CML (10). The second agent in the present
study, vincristine, is a microtubulin de-polymerising agent which
arrests cells in the G2/M phase. Evidence suggests that
cells undergoing mitosis are resistant to autophagic induction
(11) and therefore,
G2/M inhibitors may have anti-autophagic activity in
combination regimes. Vincristine has also been reported to impede
autophagy by disrupting the microtubulin network and inhibiting the
trafficking required for autophagosome and lysosomal fusion
(12). The third agent in this
study, brefeldin A disrupts protein transport in the Golgi
compartment (13). While this may
lead to endoplasmic reticulum (ER) stress and potentially increase
autophagy, brefeldin A has been reported to block an alternative
autophagic pathway (14). This may
be due to involvement of the Golgi region in the initiation of
certain types of autophagy.
In the present study, we investigated the autophagic
response of K562 CML cells following imatinib treatment alone and
combination treatment with potential autophagy-disrupting agents.
Our data indicate that the early implementation of imatinib
combined with autophagy-disrupting agents, reduces cell recovery
and may potentially improve the curative ability of imatinib in CML
patients.
Materials and methods
Cell culture/reagents
K562 cells (blast crisis CML) were obtained from
Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ)
and were maintained in RPMI-1640 medium, HEPES modification,
supplemented with 10% foetal calf serum, 1% penicillin and
streptomycin (Sigma-Aldrich, Ireland Ltd., Dublin, Ireland) and 2
mmol/l L-glutamine (Gibco-BRL, Paisley, UK) cultured at 37°C in 5%
CO2. Chloroquine, vincristine and brefeldin A were all
from Sigma-Aldrich Ireland Ltd. Imatinib was obtained from Novartis
Pharma AG (Basel, Switzerland).
Cell viability and cell cycle
analysis
Cells were resuspended in PBS with 50 μg/μl
propidium iodide (PI; Sigma-Aldrich Ireland). FL-2 fluorescence was
measured using a FACSCalibur® flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA). Statistical
significance was determined using the Student’s t-test. P-values of
<0.05 were considered statistically significant. For the
prolonged viablity analysis, following the initial 24-h treatment,
the drug was removed and cells were resuspended in fresh media.
Viability was assessed over the subsequent 5 days. For cell cycle
analysis, cells were fixed in 70% ethanol prior to PI staining.
Cell morphology and
immunofluorescence
For cell morphology, cytospun cells were stained
using Pro-Diff (Braidwood Laboratories, Ireland). For
immunofluorescence staining, the cytospun cells were fixed in 4%
paraformaldelyde for 20 min and washed in PBS. Permeabilisation and
blocking were carried out using 0.005% Saponin/PBS and 0.2%
BSA/PBS, respectively. For antibody staining, LC3 (Abgent, San
Diego, CA, USA) and Beclin 1 (Cell Signaling Technology, UK) were
incubated (1:200) for 1 h at room temperature. After incubation
with the appropriate secondary Alexa Fluor antibody, samples were
mounted with Gold Plus with DAPI antifade reagent (Invitrogen,
Dublin, Ireland). Images were captured using a DP70 digital
microscope camera and Olympus DP-Soft823 version 3.2 software. All
images are representative of at least three separate
experiments.
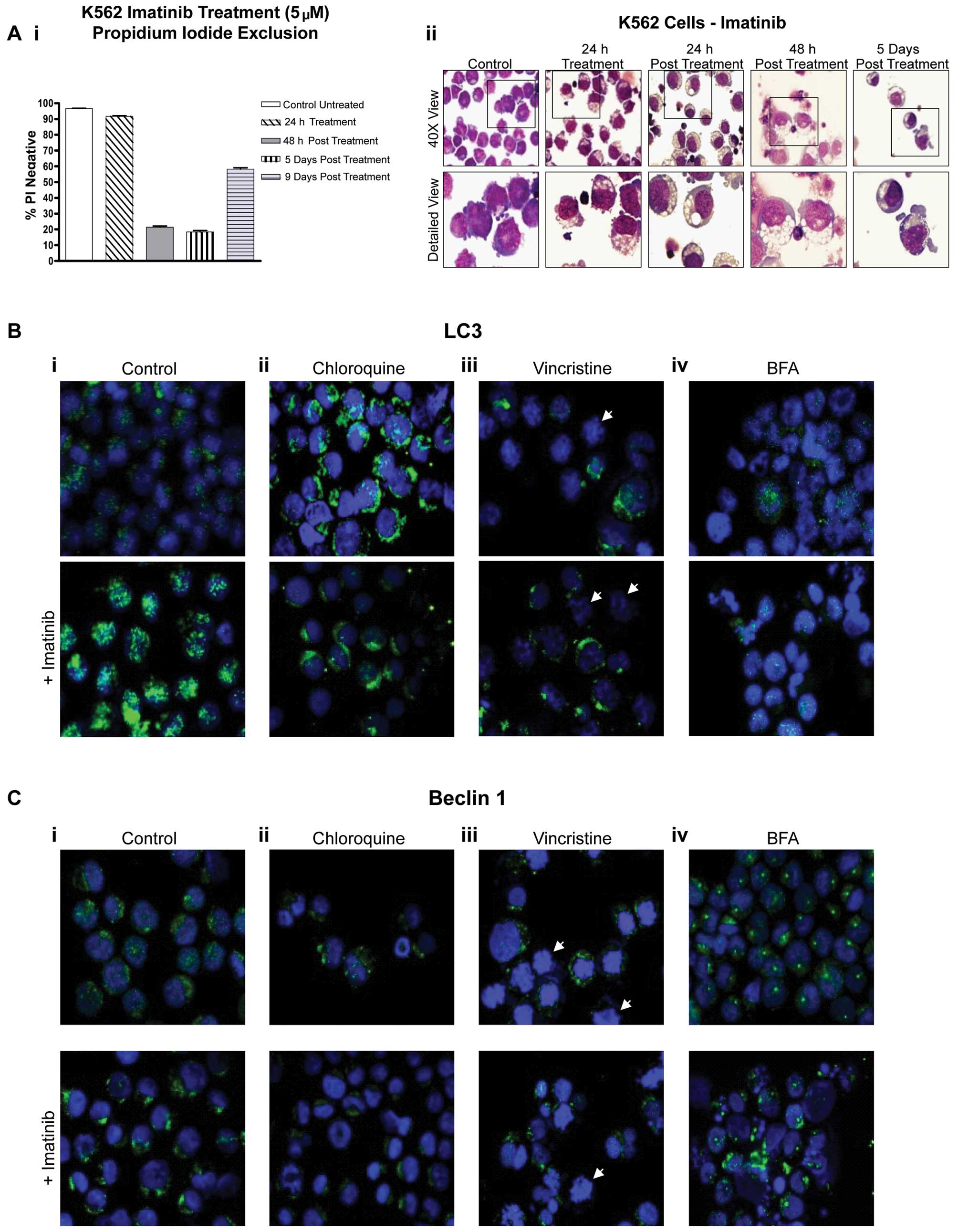
Results
Distribution of LC3 and Beclin 1
following treatment with chloroquine, vincristine and brefeldin A
with and without imatinib
We previously demonstrated that induction of
autophagy in K562 CML cells limits the cytotoxic effect of imatinib
and facilitates the recovery of treated cells (6). Viability and morphologic data for K562
cells are shown in Fig. 1A to
enable comparison with the subsequent combination treatments. In
the present study, we examined whether combinations of imatinib
with potential autophagy-disrupting agents, chloroquine,
vincristine and brefeldin A, alter expression of LC3 and Beclin 1
(autophagy markers).
Chloroquine
K562 cells were treated with 5 μM imatinib, 25 μM
chloroquine or a combination of both for 24 h. Cells treated with
chloroquine alone displayed increased LC3 staining when compared to
that in the control K562 cells (Fig.
1B-ii, upper panel) which was likely due to the inhibition of
autophagosome turnover. K562 cells treated with chloroquine and
imatinib together also showed increased LC3 expression in some
cells when compared to the controls. This expression, however, was
significantly reduced when compared to that in cells treated with
either agent alone and was localised to a peri-nuclear region
(Fig. 1B-ii, lower panel),
suggesting impairment at either initiation or trafficking in
addition to compromised turnover. Beclin 1 expression was also
increased in cells treated with imatinib, exhibiting a more
peri-nuclear localised staining pattern (Fig. 1C-i, lower panel). Cells treated with
chloroquine alone or the combination with imatinib displayed
diminished staining of Beclin 1, again suggesting that initiation
of autophagy may be impaired in addition to turnover (Fig. 1C-ii). The distribution of both
markers suggests that autophagy was impeded in the
chloroquine-treated cells.
Vincristine
We investigated whether vincristine disrupts
imatinib-induced autophagy. Cells were treated for 24 h with 10 nM
vincristine, 5 μM imatinib or a combination of both. K562 cells
treated with vincristine alone demonstrated a mixed LC3 staining
pattern. Cells displaying mitotic disturbances [NB irregular
DAPI-stained nuclei (white arrows)] did not express LC3. In
contrast, cells with normal nuclei exhibited LC3 staining analogous
to that in the control cells or was slightly elevated (Fig. 1B-iii, upper panel). A similar
pattern was observed in the combination-treated cells (Fig. 1B-iii, lower panel). Beclin 1 was
also absent or significantly reduced in the majority of
vincristine-treated cells displaying nuclear mitotic abnormalities
(Fig. 1C-iii, upper panel). Cells
treated with the combination of imatinib and vincristine
demonstrated reduced overall Beclin 1 expression when compared to
expression in the cells treated with imatinib alone (Fig. 1C-iii, lower panel). This
reduced/absent Beclin 1 staining, particularly in the
G2/M-arrested cells, indicated a reduced capacity to
initiate autophagy.
Brefeldin A
K562 cells were treated with 1 μg/ml brefeldin A
(BFA), 5 μM imatinib or a combination of both for 24 h. Cells
treated with BFA alone showed reduced expression of LC3 when
compared to that in the control cells (Fig. 1B-iv, upper panel). In cells treated
with the combination of imatinib and BFA, LC3 staining was
dramatically reduced compared to staining in cells treated with
imatinib alone (Fig. 1B-iv, lower
panel). Therefore, BFA impeded autophagy initiation in cells
treated with imatinib. Notably, BFA treatment alone induced
dramatic condensation of Beclin 1 to single peri-nuclear foci
(Fig. 1C-iv, upper panel). Cell
treated with the combination treatment exhibited a mixture of
single discrete Beclin 1 foci and less condensed peri-nuclear
localisation (Fig. 1C-iv, lower
panel) suggesting that autophagy initiation was impaired.
Effect of chloroquine and imatinib
treatment on cell viability and recovery
K562 cells were treated with 5 μM imatinib, 25 μM
chloroquine or a combination of both for 24 h and cell viability
was assessed over 5 days (Fig. 2A).
K562 viability was unaffected by treatment with chloroquine alone
at any time point. Forty-eight hours post treatment, the viability
of K562 cultures treated with imatinib alone was 23.7±0.3%.
However, following the combination therapy, cell viability was much
lower at 4.7±0.1%, indicating a marked improvement when combining
chloroquine and imatinib, which was not observed following the 24-h
treatment. This combination effect was sustained as indicated by
the absence of any recovery at day 5 (Fig. 2A).
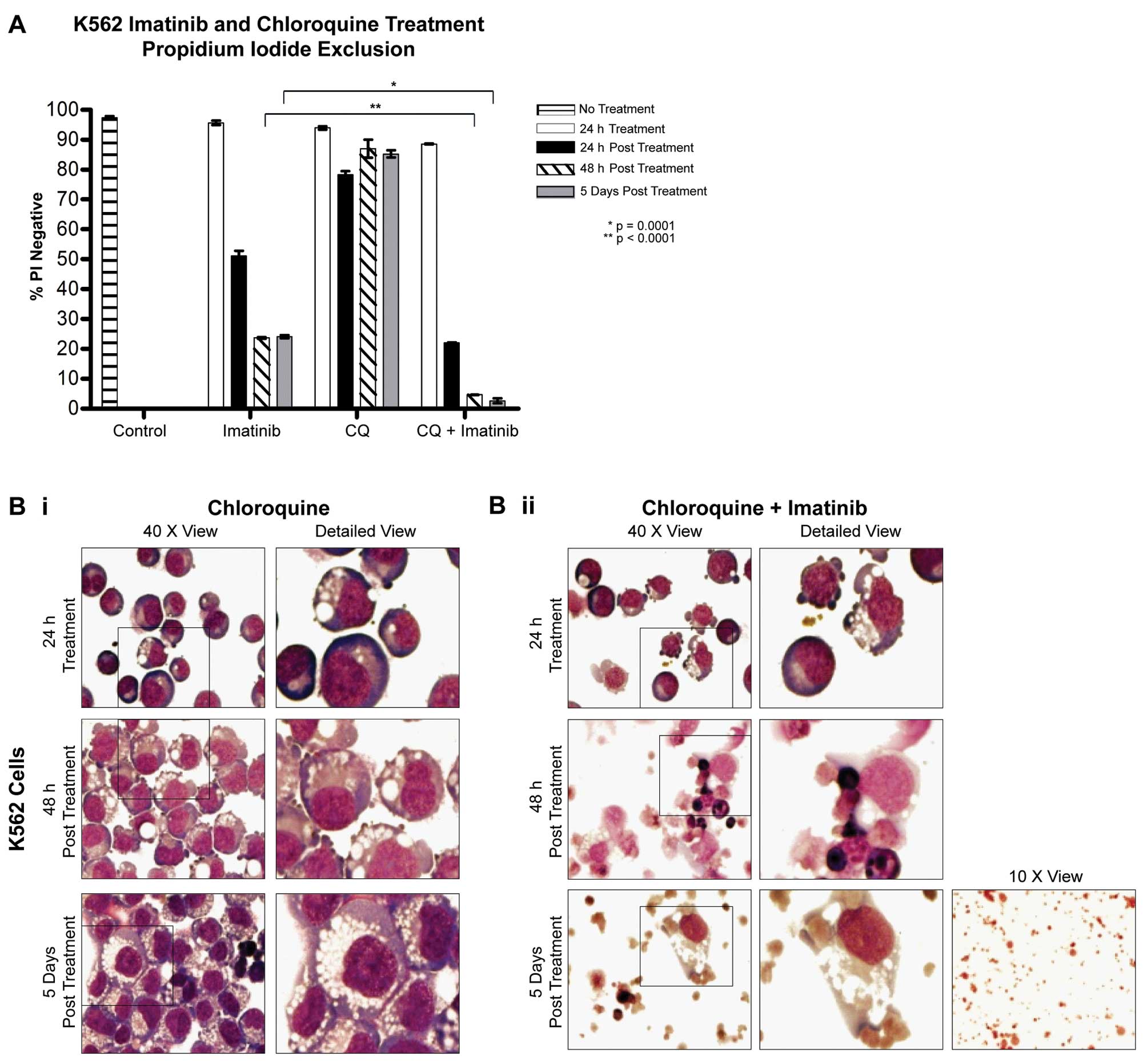 | Figure 2Effects of the combination of
chloroquine and imatinib on cell viability and morphology. (A)
Viability of K562 cells following treatment with 5 μM imatinib, 25
μM chloroquine or a combination of both treatments for 24 h and
following drug withdrawal. The viability of cells treated with the
combination was significantly reduced compared to the viability of
cells treated with imatinib alone following drug withdrawal. (B)
Morphology of K562 cells treated with (i) 25 μM chloroquine or (ii)
5 μM imatinib and 25 μM chloroquine. Significant vesicular
accumulation was noted in cells treated with chloroquine alone, or
imatinib alone (Fig. 1A-ii),
whereas this was significantly reduced in cells treated with the
combination, and cells were fragmented or necrotic. Upper panels,
24-h treatment; middle panel, 48 h post treatment; lower panel, 5
days post treatment. Left hand column: magnification, ×40; right
hand column: enlarged section of left hand column indicated by a
box. Third column of lower panel: magnification, ×10 (5 days post
treatment). CQ, chloroquine. |
Cells treated with chloroquine alone began to
display vesicle accumulation in a small percentage of cells at 24 h
(Fig. 2B-i, upper panel). At 48 h
and 5 days post chloroquine treatment alone, cells showed an
extensive build-up of vacuoles in their cytoplasm (Fig. 2B-i, middle and lower panel),
consistent with previous reports (9). Notably, this did not significantly
affect the viability of the culture.
Cells treated with the combination of imatinib and
chloroquine for 24 h showed limited cytoplasmic vacuoles (Fig. 2B-ii, upper panel) in contrast to the
high level of vacuoles in cultures treated with imatinib alone
(Fig. 1A-ii). At 48 h post imatinib
and chloroquine combination treatment, cells exhibited a mixed
morphology, with fragmented or intact nuclei, small dark shrunken
cells and/or necrotic cells. At 5 days post treatment, only cell
fragments remained (Fig. 2B-ii,
middle and lower panel). The combination of chloroquine and
imatinib was clearly a superior treatment when compared to either
therapy alone, improving cytotoxicity and reducing the potential
for cells to recover following treatment.
Vincristine and imatinib treatment
As vincristine limited the autophagy induced by
imatinib (Fig. 1B and C), we
investigated whether this agent improves cytotoxicity. Cells were
treated for 24 h with 10 nM vincristine, 5 μM imatinib or a
combination of both. Five days post drug removal, the viability of
the cells treated with the combination of vincristine and imatinib
was reduced to 3.5±0.1%. This was statistically significant
(P=0.0015) when compared to imatinib treatment alone (Fig. 3A).
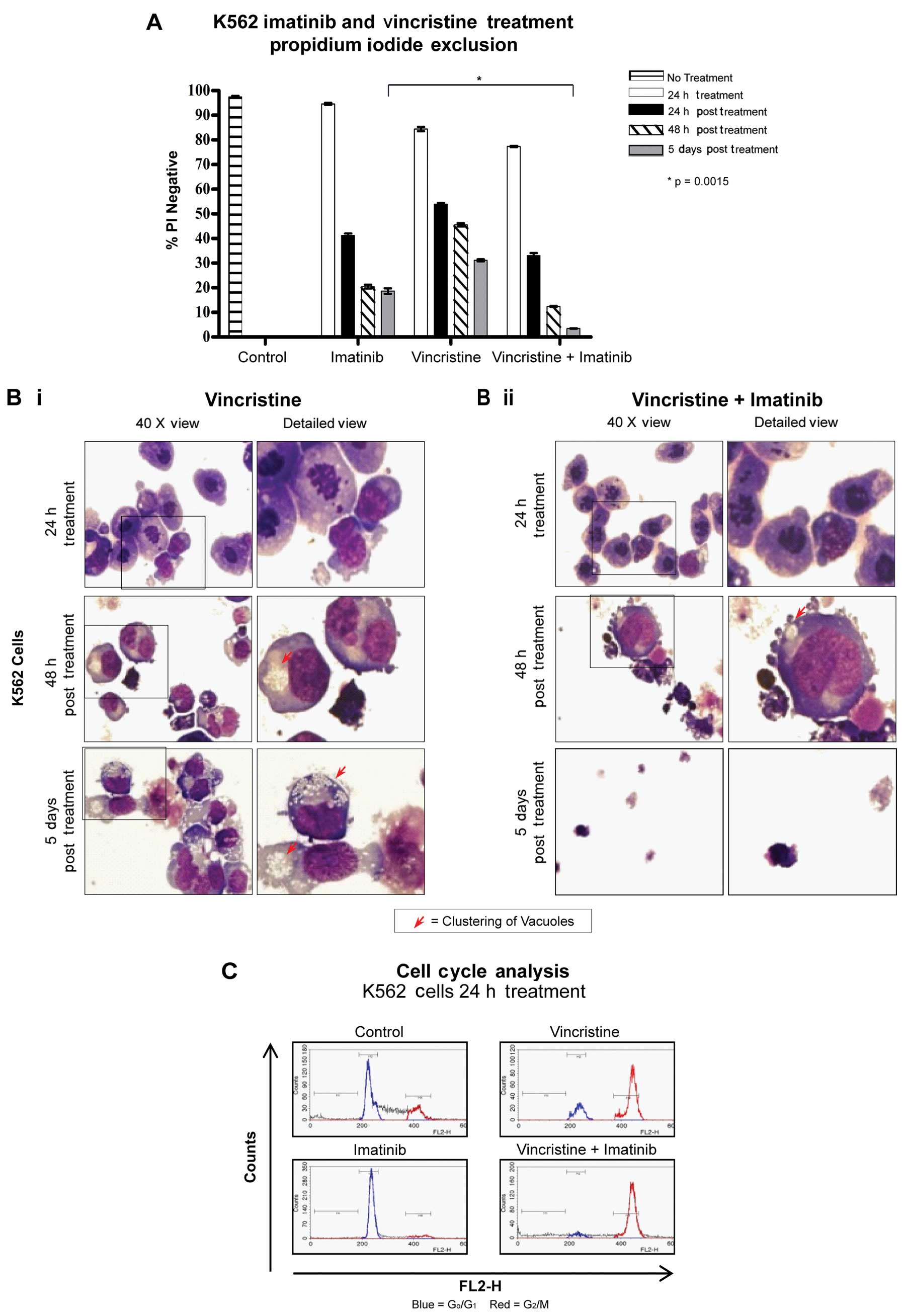 | Figure 3Effects of vincristine alone and in
combination with imatinib on cell viability, morphology and cell
cycle distribution. (A) Viability of K562 cells following treatment
with 5 μM imatinib, 10 nM vincristine or a combination of both for
24 h and up to 5 days post drug withdrawal. The viability of the
cells treated with the combination was significantly reduced
compared to the viability of cells treated with imatinib alone
following drug withdrawal. (B) Morphology of K562 cells following
treatment with (i) 10 nM vincristine, or (ii) combination of 5 μM
imatinib and 10 nM vincristine. Cells treated with the combination
treatment showed minor vesicle clustering in some cells and
predominantly cell fragments at later time points. Upper panels,
24-h treatment; middle panel, 48 h post treatment; lower panel, 5
days post treatment. Left hand column: magnification, ×40; right
hand column: enlarged section of left hand columns indicated by a
box. Red arrows, clustering of vacuoles. (C) Cell cycle analysis of
K562 cells treated with imatinib, vincristine or both. All
vincristine treated cells were arrested in G2/M phase.
x-axis, FL2-H; y-axis, cell number. Blue,
G0/G1; red, G2/M. |
Morphological analysis of vincristine-treated cells
indicated that the majority of cells were arrested in mitosis at 24
h (Fig. 3B-i, left upper panel). In
the cell treated with the combination of vincristine and imatinib,
this morphology dominated and there was an absence of cytoplasmic
vesicularisation (Fig. 3B-ii, right
upper panel). At 48 h post vincristine treatment some cells
exhibited tight clustering of cytoplasmic vacuoles (red arrows) and
numerous multi-nucleated cells were evident (Fig. 3B-i, middle panel). This was also
consistent with earlier LC3 staining in a limited number of cells,
which appeared confined to a peri-nuclear area (Fig. 1B). The combination treatment at 48 h
showed fewer cells with vesicles and many fragmented cells
(Fig. 3B-ii, middle panel). Five
days post treatment, cells treated with vincristine alone showed
accumulation of cytoplasmic vesicles in the remaining intact cells,
similar to cells treated with imatinib, indicative of cells
attempting recovery; some vesicular clustering was still evident
(Fig. 3B-i, lower panel). The
combination treatment showed only cell fragments and demonstrated a
clear advantage over the single agents in reducing culture
viability (Fig. 3B-ii, lower
panel). Cell cycle analysis confirmed that a 24-h treatment with
vincristine with or without imatinib, arrested cells in the
G2/M phase of the cell cycle (Fig. 3C).
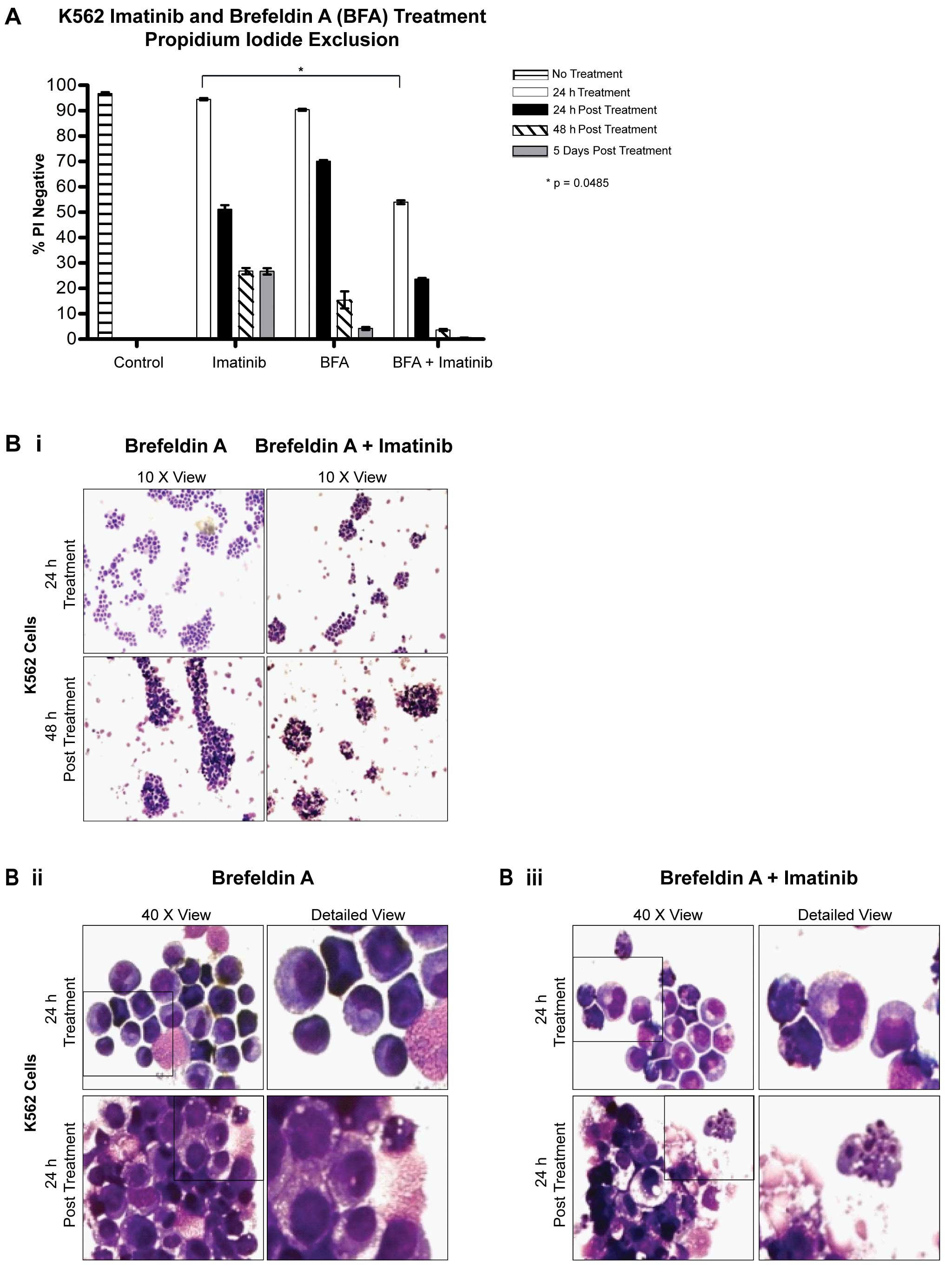
Brefeldin A and imatinib treatment
Immunofluorescence of LC3 and Beclin 1 indicated
that BFA inhibited autophagy (Fig. 1B
and C). K562 cells were treated with 1 μg/ml BFA, 5 μM imatinib
or a combination of both for 24 h and viability was assessed
thereafter. When cells were treated with a combination of BFA and
imatinib for 24 h, viability decreased to 59.9±2.8% (Fig. 4A) (P=0.0485; combination treatment
compared to imatinib treatment alone at 24 h). At 48 h post drug
withdrawal, viable cells were barely detectable following the
combined BFA and imatinib treatment.
Analysis of cell morphology, following treatment
with BFA as a single treatment or in combination with imatinib,
showed K562 cells forming clusters (Fig. 4B-i). These clusters consisted of
cells displaying a mixture of morphologies, the majority of which
had intact (but condensed) nuclei and an absence of cytoplasmic
vacuoles. A small number of cells (<10%) exhibited nuclear
fragmentation. At 24 h post treatment there was also evidence of
necrosis (Fig. 4B-ii and -iii,
lower panel). While the effect of BFA as a single agent was
impressive, the response of cells to the combination therapy was
more rapid.
Discussion
Following imatinib treatment, just under half of all
patients achieve a complete molecular response (CMR) (15). However, after discontinuation of
imatinib treatment, the majority of patients suffer relapse and
recurrence of disease (5,16). We and others have previously shown
that the recovery of leukaemic cells is aided by autophagy and by
inhibiting the autophagic response we can improve the cytotoxic
impact of imatinib (6,7). We, therefore, assessed whether
disruption of other processes important for autophagy, reduces the
recovery following imatinib withdrawal. The viability and recovery
of CML-derived K562 cells were significantly reduced following the
combination therapies.
The long-term viability of K562 cells treated with
chloroquine and imatinib was significantly reduced when compared to
long-term viability in cells treated with imatinib alone. Our
analysis is consistent with another study of CML cell lines and CML
patient samples which showed an improvement in imatinib-induced
cytotoxicity with chloroquine (7).
It is currently unclear, however, whether inhibition of late
vesicular/lysosomal fusion is the sole reason for this enhancement.
Our analysis of LC3 and Beclin 1 distribution in treated cells (at
24 h prior to cell death) showed reduced LC3 accumulation and
substantially decreased Beclin 1 expression in the
combination-treated cells. Reduced vesicular content was also
evident. It is possible that this was a consequence of a lack of
autophagosome turnover providing negative feedback to reduce
autophagy initiation. Chloroquine derivatives have been prescribed
for many years for other conditions such as malaria or for
rheumatoid arthritis where hydroxyl-chloroquine is considered to be
less toxic. Treatment regimens including hydroxyl-chloroquine have
now entered CML clinical trials (10). It has also been incorporated in at
least 12 clinical trials for solid tumours treated with various
agents (reviewed in ref. 17).
The combination of imatinib and the microtubule
inhibitor vincristine was more effective than either therapy alone
and reduced the recovery of K562 cells following drug withdrawal.
The arrested cells in the vincristine-treated populations also
showed the greatest reduction in levels of both LC3 and Beclin 1.
Mitotic inhibitors, which include vinca alkaloids as well as
taxanes, are well established in the clinical setting and have
formed the basis for a number of chemotherapeutic regimens.
Vincristine has been incorporated in treatment regimens for acute
leukaemias and blast crisis CMLs. It has also been combined with
other non-targeted therapies such as cisplatin or etoposide for
treatment of small-cell lung cancer (18). It is possible that these types of
agents have dual activity: disruption of cell division and
impairment of autophagy.
Treatment of K562 cells with BFA had the most
dramatic effects on distribution of autophagy markers. Cytotoxicity
was accelerated in cells treated with a combination of BFA and
imatinib; however, BFA was also highly effective alone 5 days after
drug withdrawal. BFA has been reported to be effective against
pancreatic, gliobastoma and prostate cancer (19–21). A
generation of new derivatives such as breflate (22–24) is
overcoming problems associated with the low water solubility of
BFA, making it an interesting candidate for future clinical
trials.
We previously showed that specific autophagy
knockdown (siRNA approach) reduced the recovery of imatinib-treated
cells (6). However, in the
combinations presented here we cannot conclusively attribute the
enhanced cytotoxicity of the combined treatments with imatinib to
autophagy inhibition. These agents have other activities. The aim
of the present study was to indicate that certain pharmacological
agents can also impede autophagy as part of their activity and this
may be an important factor in the design of combination treatment
regimes where the efficacy of one of the agents is limited by
autophagy.
Newer TKIs such as nilotinib (Bcr-Abl inhibitor) or
dasatinib and INNO-406 (src family kinase inhibitors) developed to
improve treatment of imatinib-intolerant or -resistant leukaemia
have also been reported to be limited by autophagy (7,25). We
and others have also shown that the induction of autophagy is not
limited to the treatment with TKIs but also with other non-targeted
therapies such as VP16 or SAHA (6,26).
Autophagy has also been reported to be a barrier in achieving
successful treatment of other cancer types, including breast,
prostate and oesophageal cancer treated with tamoxifen, sorafenib
or 5-fluorouracil (27–29).
An important part of the rationale for using
autophagy inhibitors in cancer is the potential for the eradication
of the more resistant residual cells and importantly the
transformed stem cells which can lead to re-population of disease.
Hydroxyl-chloroquine has been reported to be effective at targeting
primitive CML cells including colony forming cells and long-term
culture-initiating cells (7).
Notably, autophagy has been reported to be important for normal
stem cell longevity (30). This
suggests that normal cells may also be affected by prolonged
treatment with autophagy inhibitors. These studies highlight the
need for the development of more selective inhibitors and a better
understanding of their effects on normal cells for effective
clinical management.
The present study suggests that the early
implementation of regimes which incorporate an
autophagy-deregulating agent in combination with TKIs may reduce
the residual disease present in CML. Importantly, this will remain
largely targeted if the autophagy inhibitor has limited toxicity on
its own. Regimes designed to reduce autophagy may limit the
development of TKI resistance and the toxicity associated with dose
escalation or less targeted treatments.
Acknowledgements
We are eternally grateful to the late Professor
Gerald C. O’Sullivan, Cork Cancer Research Centre, for helpful
discussions and support throughout this study. We also thank Dr
Baukje Elzinga, Cork Cancer Research Centre, for her input and
advice. This study was funded by the Children’s Leukaemia Research
Project, Cancer Research Ireland, the Higher Education Authority of
Ireland and the Cork Cancer Research Centre.
References
|
1
|
Nowell PC and Hungerford DA: A minute
chromosome in human chronic granulocyte leukeamia. Science.
132:1497–1501. 1960.
|
|
2
|
Melo JV: The diversity of BCR-ABL fusion
proteins and their relationship to leukemia phenotype. Blood.
88:2375–2384. 1996.PubMed/NCBI
|
|
3
|
Calabretta B and Perrotti D: The biology
of CML blast crisis. Blood. 103:4010–4022. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Quentmeier H, Eberth S, Romani J, Zaborski
M and Drexler HG: BCR-ABL1-independent PI3Kinase activation causing
imatinib-resistance. J Hematol Oncol. 4:62011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cortes J, O’Brien S and Kantarjian H:
Discontinuation of imatinib therapy after achieving a molecular
response. Blood. 104:2204–2205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crowley LC, Elzinga BM, O’Sullivan GC and
McKenna SL: Autophagy induction by Bcr-Abl-expressing cells
facilitates their recovery from a targeted or nontargeted
treatment. Am J Hematol. 86:38–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bellodi C, Lidonnici MR, Hamilton A, et
al: Targeting autophagy potentiates tyrosine kinase
inhibitor-induced cell death in Philadelphia chromosome-positive
cells, including primary CML stem cells. J Clin Invest.
119:1109–1123. 2009. View
Article : Google Scholar
|
|
8
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
9
|
Degtyarev M, De Maziere A, Orr C, et al:
Akt inhibition promotes autophagy and sensitizes PTEN-null tumors
to lysosomotropic agents. J Cell Biol. 183:101–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holyoakes T; Medical Research Council.
Research Portofolio: A randomised phase II trial of IM versus
HCQ/IM for patients with CML in MCyR with residual disease by
Q-PCR. http://www.mrc.ac.uk/ResearchPortfolio/Grant/Record.htm?GrantRef=G0900882&CaseId=15301.
|
|
11
|
Eskelinen EL, Prescott AR, Cooper J, et
al: Inhibition of autophagy in mitotic animal cells. Traffic.
3:878–893. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Groth-Pedersen L, Ostenfeld MS,
Hoyer-Hansen M, Nylandsted J and Jaattela M: Vincristine induces
dramatic lysosomal changes and sensitizes cancer cells to
lysosome-destabilizing siramesine. Cancer Res. 67:2217–2225. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vetterlein M, Niapir M, Ellinger A,
Neumüller J and Pavelka M: Brefeldin A-regulated retrograde
transport into the endoplasmic reticulum of internalised wheat germ
agglutinin. Histochem Cell Biol. 120:121–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nishida Y, Arakawa S, Fujitani K, et al:
Discovery of Atg5/Atg7-independent alternative macroautophagy.
Nature. 461:654–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aguayo A and Couban S: State-of-the-art in
the management of chronic myelogenous leukemia in the era of the
tyrosine kinase inhibitors: evolutionary trends in diagnosis,
monitoring and treatment. Leuk Lymphoma. 50(Suppl 2): 1–8. 2009.
View Article : Google Scholar
|
|
16
|
O’Brien SG, Guilhot F, Goldman JM, et al:
International randomized study of interferon versus STI571 (IRIS)
7-year follow-up: sustained survival, low rate of transformation
and increased rate of major molecular response (MMR) in patients
(pts) with newly diagnosed chronic myeloid leukemia in chronic
phase (CMLCP) treated with imatinib (IM). (ASH Annual Meeting
Abstracts). Blood. 112:1862008.
|
|
17
|
Amaravadi RK, Lippincott-Schwartz J, Yin
X-M, et al: Principles and current strategies for targeting
autophagy for cancer treatment. Clin Cancer Res. 17:654–666. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lassen U, Kristjansen PE, Osterlind K, et
al: Superiority of cisplatin or carboplatin in combination with
teniposide and vincristine in the induction chemotherapy of
small-cell lung cancer. A randomized trial with 5 years follow up.
Ann Oncol. 7:365–371. 1996.PubMed/NCBI
|
|
19
|
Pommepuy I, Terro F, Petit B, et al:
Brefeldin A induces apoptosis and cell cycle blockade in
glioblastoma cell lines. Oncology. 64:459–467. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chapman JR, Tazaki H, Mallouh C and Konno
S: Brefeldin A-induced apoptosis in prostatic cancer DU-145 cells:
a possible p53-independent death pathway. BJU Int. 83:703–708.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Larsson DE, Wickström M, Hassan S, OBerg K
and Granberg DAN: The cytotoxic agents NSC-95397, brefeldin A,
bortezomib and sanguinarine induce apoptosis in neuroendocrine
tumors in vitro. Anticancer Res. 30:149–156. 2010.PubMed/NCBI
|
|
22
|
Fox BM, Vroman JA, Fanwick PE and Cushman
M: Preparation and evaluation of sulfide derivatives of the
antibiotic brefeldin A as potential prodrug candidates with
enhanced aqueous solubilities. J Med Chem. 44:3915–3924. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Phillips LR, Wolfe TL, Malspeis L and
Supko JG: Analysis of brefeldin A and the prodrug breflate in
plasma by gas chromatography with mass selective detection. J Pharm
Biomed Anal. 16:1301–1309. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Anadu NO, Davisson VJ and Cushman M:
Synthesis and anticancer activity of brefeldin A ester derivatives.
J Med Chem. 49:3897–3905. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kamitsuji Y, Kuroda J, Kimura S, et al:
The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and
different modes of cell death execution in Bcr-Abl-positive
leukemias. Cell Death Differ. 15:1712–1722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carew JS, Nawrocki ST, Kahue CN, et al:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qadir MA, Kwok B, Dragowska WH, et al:
Macroautophagy inhibition sensitizes tamoxifen-resistant breast
cancer cells and enhances mitochondrial depolarization. Breast
Cancer Res Treat. 112:389–403. 2008. View Article : Google Scholar
|
|
28
|
Ullen A, Farnebo M, Thyrell L, et al:
Sorafenib induces apoptosis and autophagy in prostate cancer cells
in vitro. Int J Oncol. 37:15–20. 2010. View Article : Google Scholar
|
|
29
|
O’Donovan TR, O’Sullivan GC and McKenna
SL: Induction of autophagy by drug-resistant esophageal cancer
cells promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011.PubMed/NCBI
|
|
30
|
Mortensen M, Soilleux EJ, Djordjevic G, et
al: The autophagy protein Atg7 is essential for hematopoietic stem
cell maintenance. J Exp Med. 208:455–467. 2011. View Article : Google Scholar : PubMed/NCBI
|