Introduction
Distant metastasis is the major cause of death in
patients with colorectal cancer (CRC). Obtaining a better
understanding of the molecular mechanisms underlying distant
metastasis is required in order to facilitate the development of
effective therapeutic strategies for patients with CRC. In many
cases of CRC, invasion is associated with dedifferentiation of
neoplastic tumor cells in invasive regions. At the invasive front
of primary tumors, cancer cells can acquire a mesenchymal-like
phenotype and express mesenchymal markers such as α-SMA and
vimentin, thus resembling epithelial-mesenchymal transition
(EMT).
EMT plays a crucial role in gastrulation in early
embryonic development and organ formation (1) and is considered to coordinate
molecular steps in the process of distant metastasis. It has been
reported that EMT permits both invasion and emigration in various
solid tumors (2) and is associated
with a poor prognosis in patients with CRC (3). EMT has been classified as a unique
process in which epithelial cells undergo marked morphologic
changes characterized by the transition from an epithelial
cobblestone phenotype to a motile, elongated fibroblastic phenotype
(4,5). The hallmark of EMT is the loss of
epithelial homotypic adhesion molecules such as E-cadherin and
γ-catenin and the gain of mesenchymal markers such as vimentin and
fibronectin. EMT is triggered by environmental stresses such as
inflammation, reactive oxygen species and hypoxia (6) and is mediated intracellularly through
different transcription factors, including the Snail family and
Twist (7).
It is known that hypoxic or anoxic areas are
heterogeneously distributed within solid tumors (8,9) and
tumor hypoxia can be associated with clinical responses to both
chemotherapy and radiotherapy or development of metastases
(10). Cancer tissues develop
pathophysiological microenvironments during growth that are
characterized by irregular microvascular networks and regions of
chronically and transiently ischemic cells. Ischemic cancer cells
are known to be reperfused by neovascularization or decreases in
tissue pressure (11). Many cancer
cells are exposed to anoxia/reoxygenation.
Anoxia/reoxygenation (A/R) generates reactive oxygen
species (ROS) that activate NF-κB. A/R is also involved in cancer
initiation, promotion and progression (12). A/R induces point mutations,
deletions and gene amplification by creating oxidative damage,
thereby allowing tumors to acquire malignant potential and
increasing the metastatic potential (13). Moreover, it has been reported that
A/R increases the expression of VEGF and induces matrix
metalloproteinase (MMP) activation, both of which are involved in
tumor growth, invasion and metastasis (14–16).
A/R-induced ROS may cause EMT in cancer cells; however, to date, it
remains to be elucidated whether A/R induces EMT in cancer cells.
Hence, the objective of this study was to clarify whether A/R
induces EMT in human colon cancer cells.
Materials and methods
Cell line and cell culture
The human colon adenocarcinoma cell line HT-29 was
obtained from the American Type Culture Collection (Rockville, MD).
The HT-29 cells were grown in RPMI-1640 medium (Gibco BRL,
Gaithersburg, MD) supplemented with 10% fetal calf serum (FCS) and
penicillin (100 U/ml)/streptomycin (100 μg/ml). The cell cultures
were maintained in a humidified atmosphere of 95% air and 5%
CO2 at 37°C.
Reagents and antibodies
All chemicals were prepared immediately before use.
Lactacystin was purchased from Biomol International L.P.
(Farmingdale, NY), MG-132 was purchased from Cayman Chemical Co.
(Ann Arbor, MI) and anti-E-cadherin antibodies were purchased from
Wako Pure Chemical (Osaka, Japan).
Anoxia/reoxygenation protocol
The in vitro model of anoxia/reoxygenation
used in this study is similar to a previously described model
(5,17). Briefly, the medium was changed to
RPMI only just before the experiment was conducted, and then
confluent HT-29 colon cancer cells were exposed to anoxia by
incubation in a Plexiglass chamber that was continuously purged (1
l/min) with an anoxic gas mixture (95% N2–5%
CO2). The chamber pO2 level was monitored
during the entire experiment using an oxygen electrode (model OM-1,
Microelectrodes, Londonderry, NH). Reoxygenation was performed by
exposing the cancer cells to normoxia (21% O2-5%
CO2-74% N2) in a CO2 incubator.
The control cells were exposed to normoxia.
Morphologic analysis
Phase-contrast microscopy was undertaken using CKX41
(Olympus, Tokyo, Japan) at ×100 magnification. The images were
captured using the Flovel Filing System (Flovel, Tokyo, Japan).
Immunocytochemistry
The HT-29 cells cultured on 35-mm μ-dishes (iBidi,
Munich, Germany) were incubated with the primary antibodies
(anti-E-cadherin antibodies, Wako Pure Chemical) in
phosphate-buffered saline (PBS) containing 5% FCS and 2 mM
CaCl2 for 2 h at room temperature followed by 30 min of
incubation with fluorescence-labeled secondary antibodies (Alexa
Fluor 594, Life Technologies, Tokyo, Japan) visualized using
epi-illumination on a laser scanning confocal microscope
(Olympus).
Western blotting
Whole-cell extracts were prepared as follows. The
HT-29 cells were retrieved with a cell scraper and collected by
centrifugation at 2,500 rpm/min at 4°C for 5 min. After removing
the upper PBS, the cell pellets were lysed in lysis buffer
(CelLytic M; Sigma-Aldrich Co., St. Louis, MO), stirred and
incubated on ice for 15 min. The supernatants were collected and
stored at −80°C.
The total proteins were mixed with an SDS sample
buffer. The samples were then subjected to 10% SDS-PAGE and blotted
onto a polyvinylidene fluoride membrane (Atto Corporation, Tokyo,
Japan). The membrane was then incubated with 10% EzBlock (Atto
Corporation) in TBS-T (10 mM Tris-Cl, pH 8.0; 150 mM NaCl, 0.1%
Tween-20 v/v) for 30 min at room temperature and washed with TBS-T
three times. The membrane was incubated for 1 h at room temperature
with the primary antibodies (anti-human E-cadherin IgG, mouse
monoclonal, Wako Pure Chemical) in TBS-T (diluted 1:500) and then
incubated with the secondary anti-mouse IgG antibodies (GE
Healthcare, Tokyo, Japan) in TBS-T (diluted 1:1000) for 1 h at room
temperature. Immunocomplexes were detected using western blotting
(ECL Plus; GE Healthcare Bio-Sciences K.K., Tokyo, Japan).
RNA isolation and quantitative
RT-PCR
The expression levels of E-cadherin, vimentin, Snail
and ZEB1 mRNA were determined using real-time PCR. The samples used
for mRNA isolation were removed from the colon cancer cells (HT29).
Total RNA was isolated using the acid guanidinium phenol chloroform
method with Isogen (Nippon Gene Co. Ltd., Tokyo, Japan). The
isolated RNA was stored at −70°C until use in real-time PCR. In the
latter, 1 μg of extracted RNA was reverse-transcribed into
first-strand complementary DNA (cDNA) using the High Capacity cDNA
Reverse Transcription kit (Applied Biosystems, Foster City, CA).
Real-time PCR for E-cadherin, vimentin, Snail, ZEB-1 and GAPDH was
performed using the 7300 Real-time PCR system (Applied Biosystems)
using the DNA-binding dye SYBR® Green to detect the PCR
products. The primers had the following sequences: E-cadherin,
sense 5′-GTCAGTTCAGACTCCAG CCC-3′ and antisense
5′-AAATTCACTCTGCCCAGG ACG-3′; vimentin, sense 5′-TCTACGAGGAGGAGATGC
GG-3′ and antisense 5′-GGTCAAGACGTGCCAGAGAC-3′; Snail, sense
5′-ACCACTATGCCGCGCTCTT-3′ and antisense 5′-GGTCGTAGGGCTGCTGGAA-3′;
ZEB-1, sense 5′-TGGG ATCAACCACCAATGG-3′ and antisense 5′-AAGTAACCC
TGTGTATTTCTGGATGA-3′; GAPDH, sense 5′-ACCACA GTCCATGCCATCACT-3′ and
antisense 5′-CCATCACGC CACAGTTTCC-3′.
ELISA assay for NF-κB activation
Nuclear extracts were prepared using the Nuclear
Extract kit (Active Motif, Tokyo, Japan). The DNA-binding activity
of the p65 and p50 NF-κB subunits in the nuclear extracts was
measured using the TransAM NF-κB p50, p52, p65 and Family kit
(Active Motif) at 490 nm according to the manufacturer’s
instructions. The results were expressed as the optical density
(OD).
Statistical analysis
All analyses were performed using the GraphPad Prism
4 program (GraphPad Software Inc., San Diego, CA). The results are
presented as the mean ± SEM. An analysis of variance (ANOVA) and
Student’s t-test were used to compare the mean values. The
criterion for statistical significance was taken as P<0.05.
Results
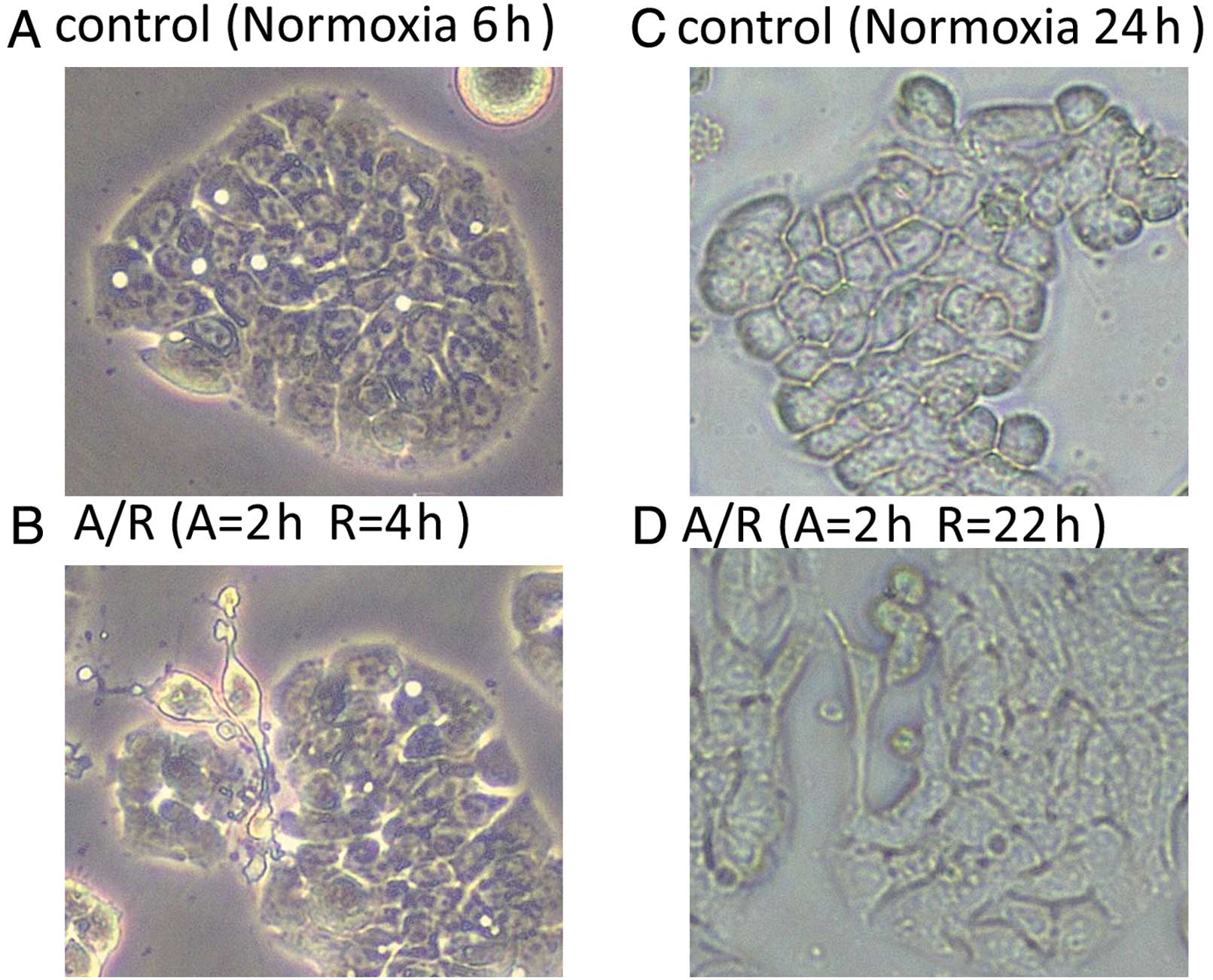
Anoxia/reoxygenation induces morphologic
changes consistent with EMT in HT-29 cells
HT-29 cells were exposed to 6 or 24 h of normoxia
and 2 h of anoxia followed by 4 or 22 h of reoxygenation,
respectively. A phase contrast analysis (original magnification:
×100) was used to detect morphological changes in HT29 cells under
normoxic or A/R conditions. The cells exposed to A/R began to lose
cell contact and acquired an elongated, fusiform morphology with
dendritic processes, consistent with mesenchymal transition
(Fig. 1).
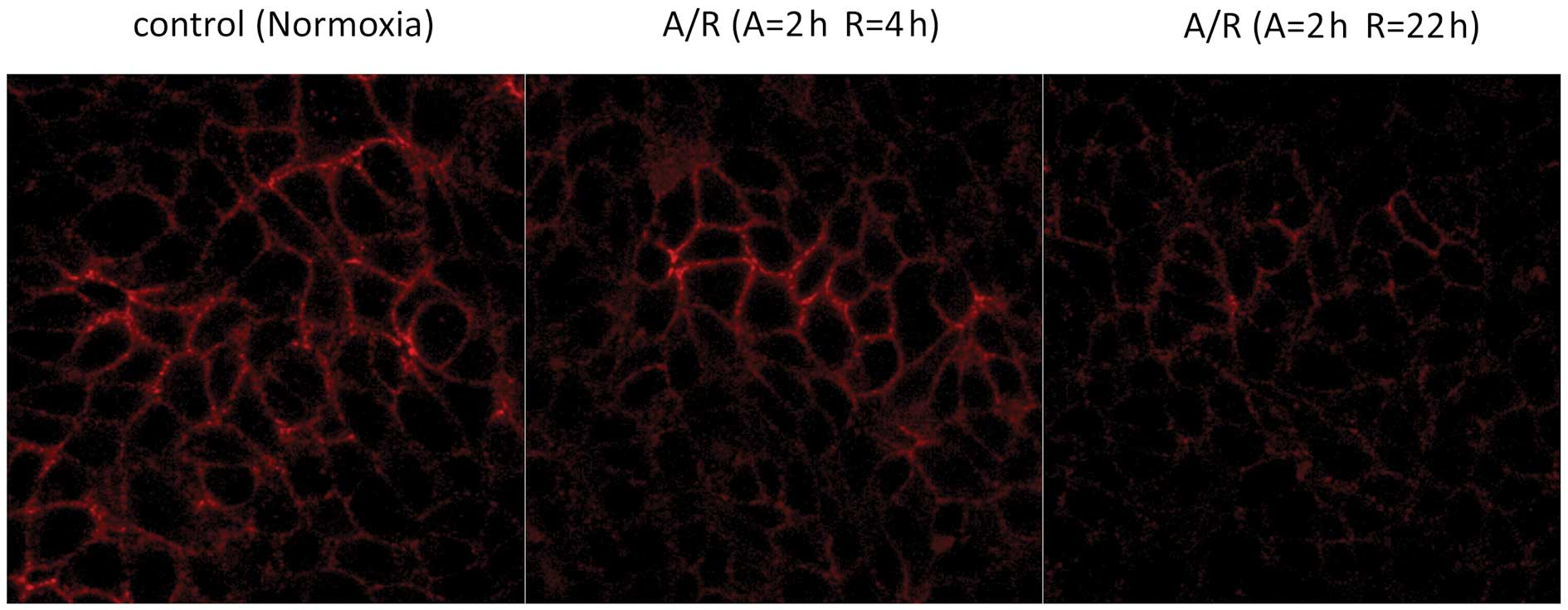
Expression of E-cadherin on the cell
surface after anoxia/reoxygenation (immunofluorescence)
In the control cells exposed to normoxia,
immunofluorescent staining showed that the expression of E-cadherin
was localized to the lateral aspects of the plasma membranes in a
continuous, linear staining pattern. In the cells exposed to 2 h of
anoxia followed by 4 or 22 h of reoxygenation, the linear staining
pattern appeared to be weaker (Fig.
2).
Effects of anoxia/reoxygenation on
molecular markers of EMT
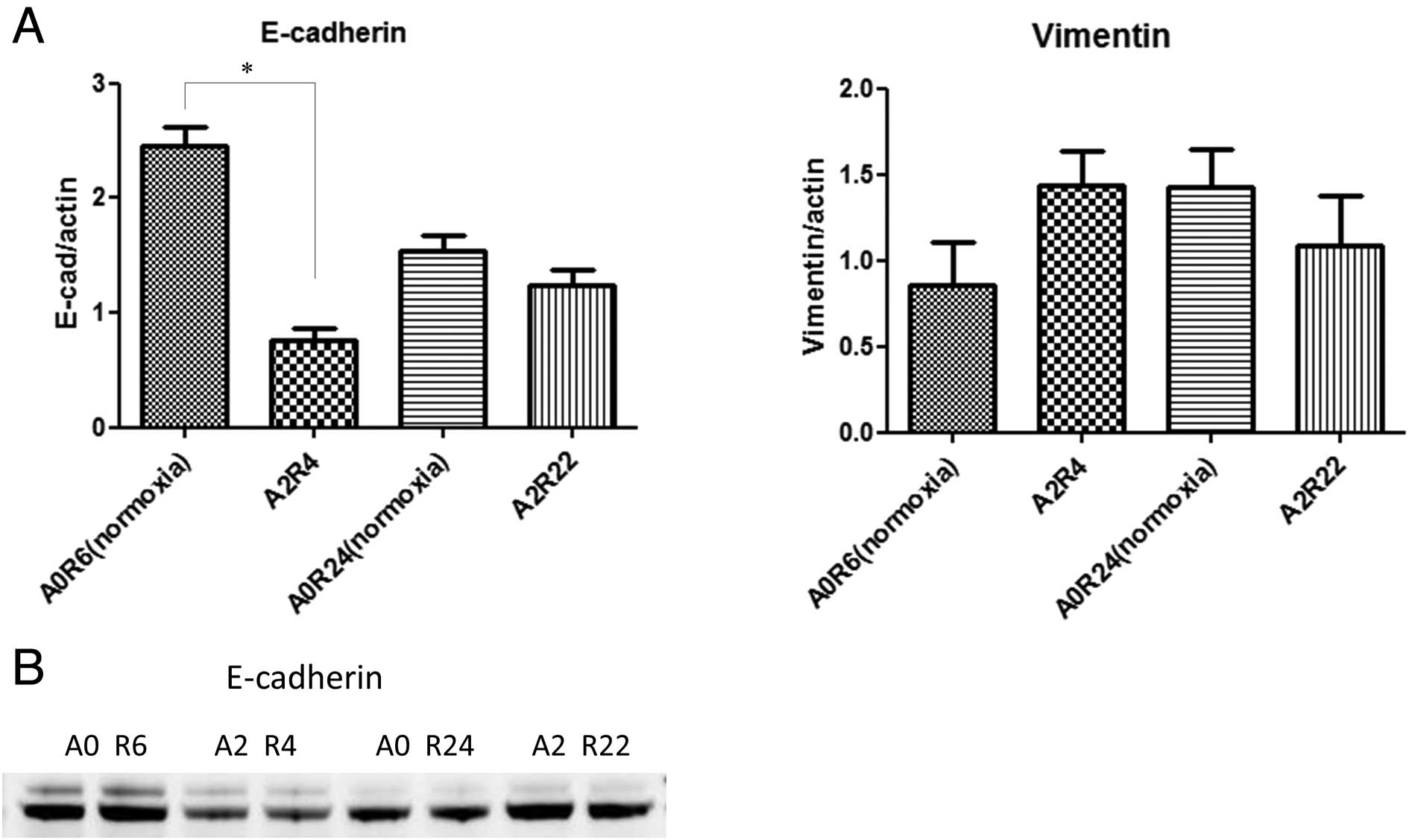
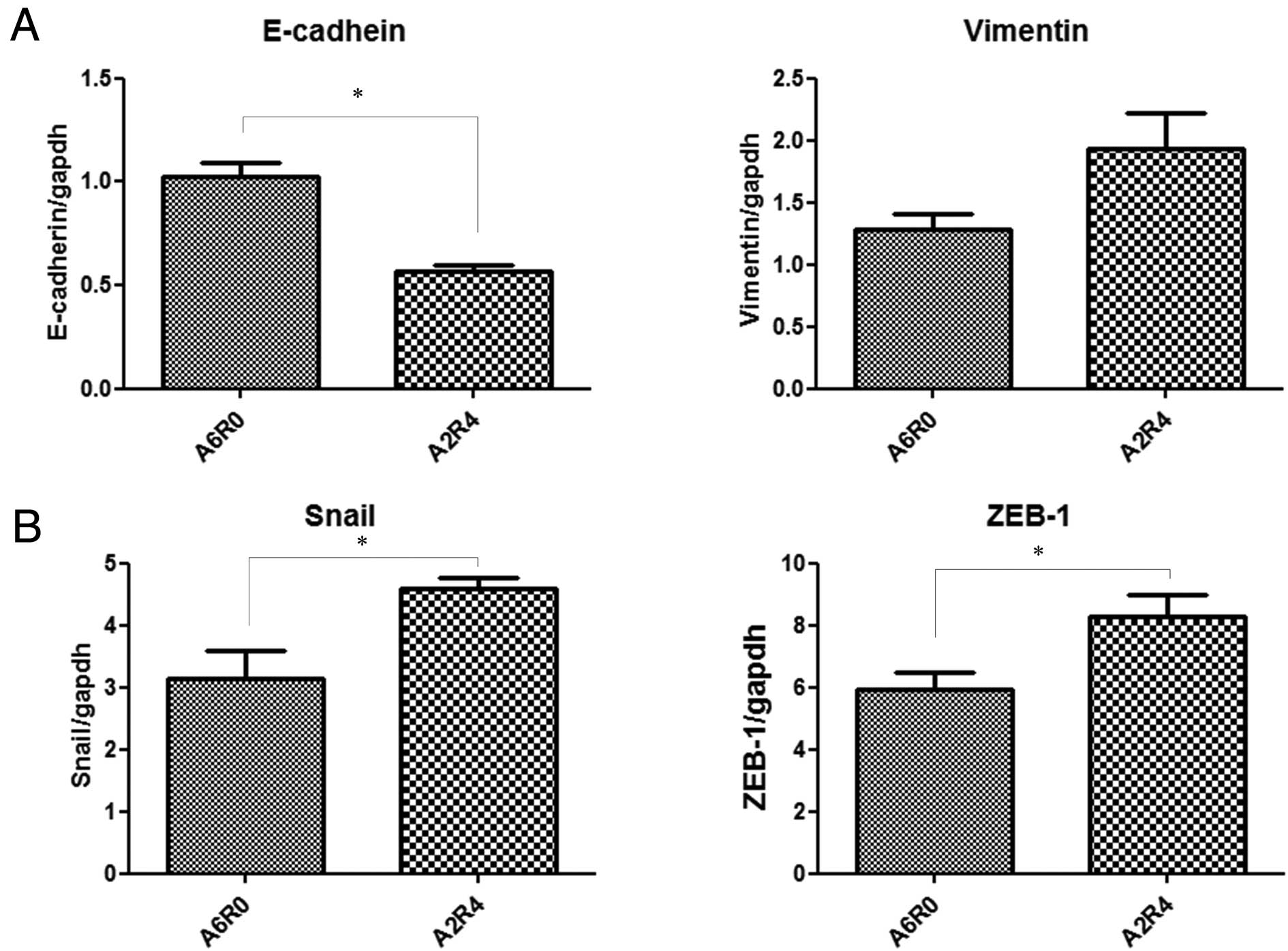
Quantitative RT-PCR of the E-cadherin and vimentin
mRNA expression in HT29 cells was performed after 6 or 24 h of
normoxia and 2 h of anoxia followed by 4 or 22 h of reoxygenation.
Two hours of anoxia followed by 4 h of reoxygenation significantly
downregulated the expression of E-cadherin mRNA and tended to
upregulate the expression of vimentin mRNA (Fig. 3A).
Western blotting was performed on the cell lysates
obtained from the anoxia/reoxygenation-exposed HT-29 cells. No
significant losses of E-cadherin were detectable after 2 h of
anoxia followed by 22 h of reoxygenation relative to that observed
after 6 h of normoxia. In contrast, a clear and consistent
reduction in the total amount of E-cadherin was seen after 2 h of
anoxia followed by 4 h of reoxygenation relative to that observed
after 6 h of normoxia (Fig.
3B).
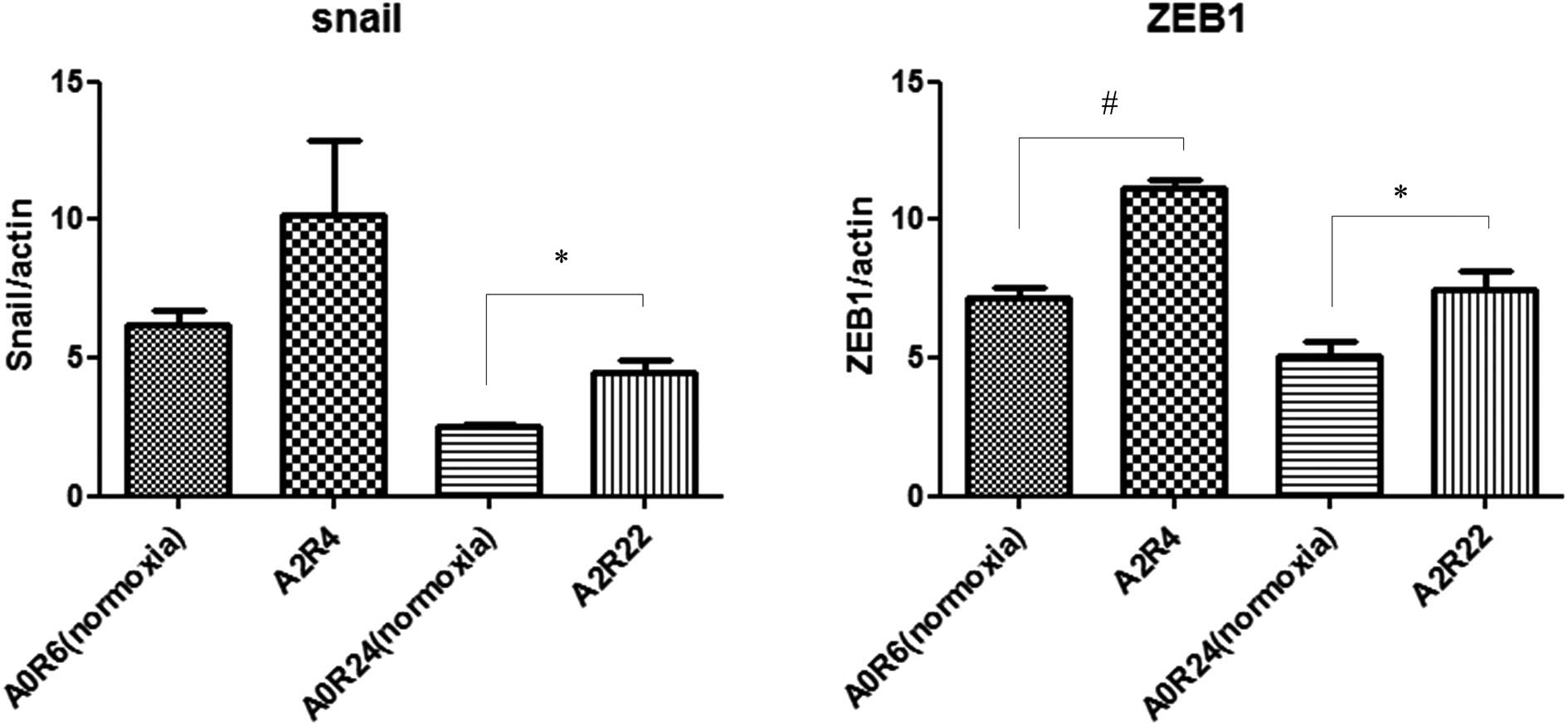
Quantitative RT-PCR of the Snail and ZEB-1 mRNA
expression in HT29 cells was performed after 6 or 24 h of normoxia
and 2 h of anoxia followed by 4 or 22 h of reoxygenation. Two hours
of anoxia followed by 4 or 22 h of reoxygenation upregulated both
Snail and ZEB-1 mRNA (Fig. 4).
These results indicate that stimulation with 2 h of anoxia followed
by 4 h of reoxygenation induces EMT to a greater extent than
stimulation with 2 h of anoxia followed by 22 h of reoxygenation.
Therefore, subsequent experiments were performed under A/R
conditions with 2 h of anoxia followed by 4 h of reoxygenation.
Comparison of the effects of anoxia and
anoxia/reoxygenation on molecular markers of EMT
Quantitative RT-PCR of the E-cadherin and vimentin
mRNA expression in HT29 cells was performed after 6 h of anoxia and
2 h of anoxia followed by 4 h of reoxygenation. Two hours of anoxia
followed by 4 h of reoxygenation significantly downregulated
E-cadherin mRNA and tended to upregulate vimentin mRNA (Fig. 5A).
Quantitative RT-PCR of the Snail and ZEB-1 mRNA
expression in HT29 cells was performed after 6 h of anoxia and 2 h
of anoxia followed by 4 h of reoxygenation. Two hours of anoxia
followed by 4 h of reoxygenation upregulated both Snail and ZEB-1
mRNA (Fig. 5B). These results
indicate that stimulation with 2 h of anoxia followed by 4 h of
reoxygenation induces EMT a greater extent than stimulation with 6
h of anoxia.
Effects of anoxia/reoxygenation on NF-κB
in HT29 cells (ELISA)
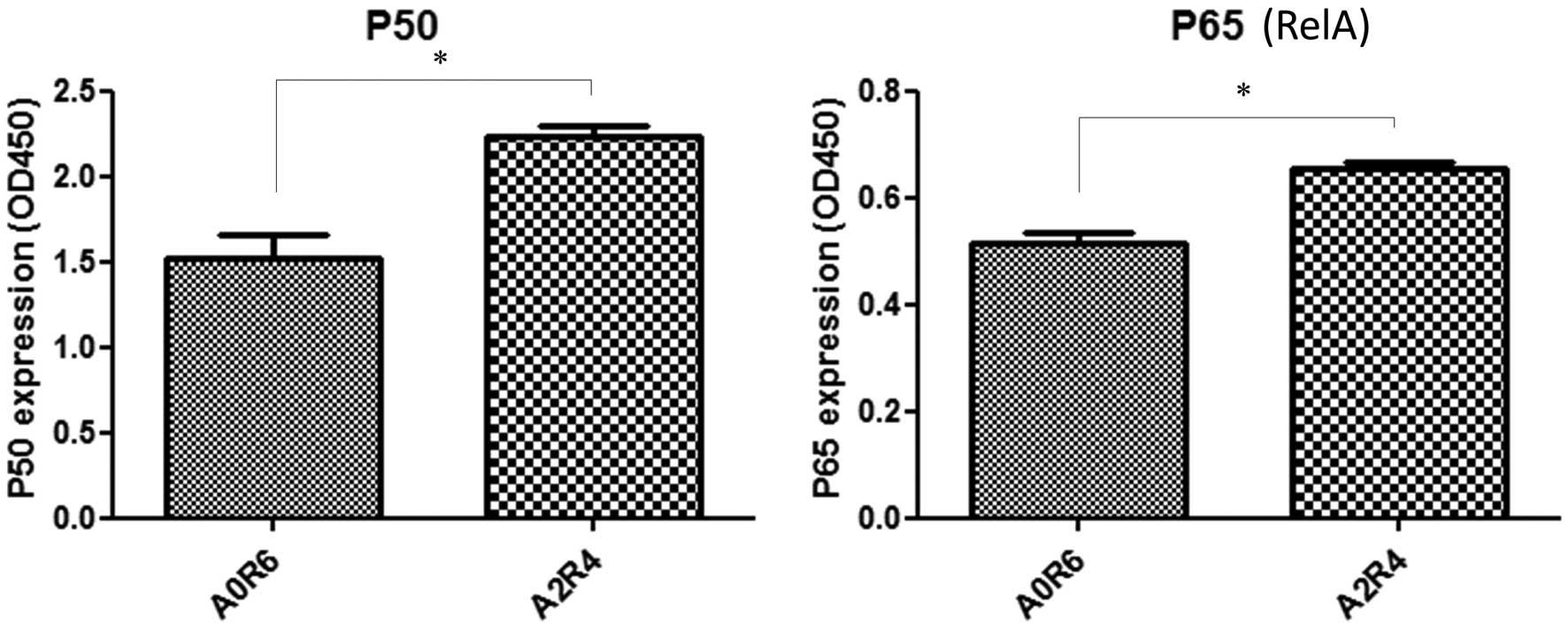
Nuclear proteins were extracted from HT29 cells
after 2 h of anoxia followed by 4 h of reoxygenation and 6 h of
normoxia. The optical density (OD) exhibited by the active forms of
p50 and p65 proteins in the nuclear extracts of the HT29 cells was
significantly increased after 2 h of anoxia followed by 4 h of
reoxygenation compared to that observed after 6 h of normoxia
(Fig. 6).
Effects of inhibition of NF-κB activation
(proteasome inhibitor) on the transcription of E-cadherin after
anoxia/reoxygenation (RT-PCR)
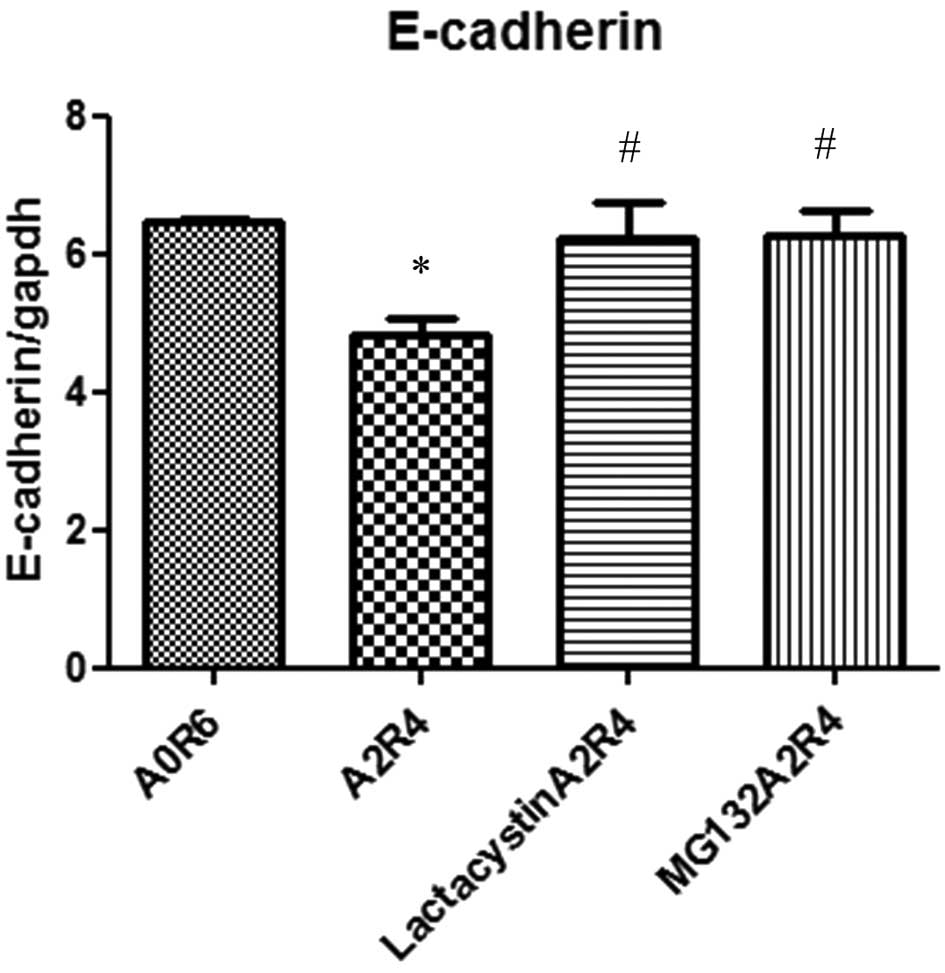
To evaluate the contribution of the transcription
factor NF-κB on anoxia/reoxygenation-induced downregulation of
E-cadherin, HT-29 cells were treated with proteasome inhibitors
(lactacystin or MG132). Treatment with the proteasome inhibitors
alone had no significant effects on the expression of E-cadherin
(data not shown). HT-29 cells were pretreated with lactacystin (10
μM) or MG132 (10 μM) for 1 h. Quantitative RT-PCR of the E-cadherin
mRNA expression in HT29 cells was performed after 6 h of normoxia
and 2 h of anoxia followed by 4 h of reoxygenation. Decreases in
the E-cadherin mRNA expression were attenuated when the cells were
pretreated with lactacystin or MG132 (Fig. 7).
Discussion
Transient cycles of A/R are known to occur within
solid tumors and can be associated with metastatic development
(11). Although it has been
reported that various environmental stressors such as inflammation
and hypoxia can induce EMT, the effects of A/R stress on EMT have
not yet been clarified. In this study, we showed that A/R stress
induces morphologic and molecular alterations consistent with EMT
in human colon cancer cells. Furthermore, we showed that activation
of nuclear factor NF-κB is a critical event in A/R-induced EMT
since A/R-induced EMT was abolished almost completely by
pretreatment with proteasome inhibitors. This study is the first
report demonstrating that A/R stress can strongly induce EMT in
colon cancer cells.
Hypoxia is a common condition found in various solid
tumors and can be associated with resistance to chemotherapy and
radiation therapy. Ischemic regions can exhibit intermittent reflow
and associated redox stress (8,18). It
has been shown that cycling through periods of acute hypoxia
followed by reoxygenation leads to the promotion of tumor cell
survival (19) and increased
metastatic development (20). Maqat
et al, using the 19F MRI technique, recently
demonstrated that spontaneous fluctuations in tumor pO2
occur regardless of the basal oxygenation state (i.e., in both
oxygenated and hypoxic regions) (11). In addition, it has been reported
that hypoxia (anoxia)/reoxygenation occurs in tumors (8,9,13).
Hypoxia (anoxia)/reoxygenation stress leads to the generation of a
large amount of ROS from the mitochondria, and redox alterations
modulate various mitogenic and survival signaling pathways,
including NF-κB activation, and contribute to cancer cell
proliferation and invasion (12).
Although A/R-induced ROS generation and/or NF-κB activation may
cause EMT in cancer cells, this issue has not been addressed to
date.
EMT not only plays crucial roles in the formation of
the body plan and physiological responses to injury, but also is an
important element in cancer progression (1,21–23).
The process of EMT involves the loss of epithelial cell-cell
junction and upregulation of mesenchymal markers such as vimentin
and fibronectin. Epithelial cancer cells acquire migratory and
invasive properties during the EMT process. It has been reported
that EMT-inducing signals emanating from the tumor-associated
stroma, notably HGF (hepatocyte growth factor), EGF, PDGF and
TGF-β, are responsible for the induction of a series of
EMT-inducing transcription factors such as Snail, Slug, zinc finger
E-box binding homeobox 1 (ZEB1) and Twist (22,24,25).
Multiple lines of evidence indicate that these EMT-inducing
transcription factors are regulated either directly or indirectly
by NF-κB (5,26–28).
We and others have indicated that A/R stress generates a large
amount of intracellular ROS and rapidly and strongly activates
NF-κB (12,29). In agreement with the results of
these previous studies, there were greater amounts of intracellular
ROS generation under the A/R conditions than under normoxia or
anoxia (data not shown), and NF-κB activity was significantly
enhanced under the A/R conditions compared to that observed under
the normoxic conditions.
Recent studies have demonstrated that hypoxia and
the overexpression of hypoxia-inducible factor-1a (HIF-1α) promotes
EMT in tumor cells (30,31). In the present study, A/R promoted
EMT more strongly than hypoxia alone in a short period of stress
time (6 h). We found that EMT changes (fibroblastoid phenotype,
Snail transcription and changes in E-cadherin) appeared within 6 h
after A/R treatment, whereas other studies demonstrated that EMT
changes induced by hypoxia are detected 24 to 72 h from the
beginning of hypoxia (32). Cannito
et al demonstrated that hypoxia-dependent EMT changes occur
through a biphasic mechanism. Early EMT-related events induced by
hypoxia (GSK-3β inhibition and Snail translocation) are dependent
on the transient intracellular increased generation of ROS. Later
EMT-related events (migration and invasiveness) are sustained by
HIF-1α- and vascular endothelial growth factor (VEGF)-dependent
mechanisms (32). It has been
demonstrated that acutely and chronically hypoxic cells coexist in
solid tumors, and the fraction of acutely hypoxic cells is larger
than the fraction of chronically hypoxic cells (33). Therefore, in solid tumors, A/R
stress may play a more important role than hypoxic stress in
inducing EMT. A/R occurs repeatedly in solid tumors (11), and A/R cycles can induce EMT and
have been implicated in invasion and metastasis. Moreover, it is
possible that a large amount of ROS induced by A/R is a strong
trigger for EMT, and VEGF and HIF-1α expressed under hypoxic
conditions may contribute to maintaining the EMT phenotype.
Conducting in vivo animal studies is required to clarify the
mechanisms underlying the effects of fluctuations in tumor blood
flow on EMT induction.
In conclusion, A/R rapidly and strongly induces EMT
in human colon cancer cells (HT29) via the NF-κB-dependent
transcriptional pathway. These observations strongly suggest that
A/R stress plays a pathogenic role in tumor progression by inducing
EMT. A/R-induced molecules, such as NF-κB and ROS, may therefore be
good targets for inhibiting A/R-induced EMT and invasiveness in
colonic cancer therapy.
Acknowledgements
This study was partially supported by JSPS KAKENHI
Grant no. 23590891. S.K., T.I. and T.O. are affiliated with a
donation-funded department from Takara Bio Inc. Y.N. received
scholarship funds from Otsuka Pharmaceutical Co., Ltd. and Takeda
Pharmaceutical Co., Ltd.
References
|
1
|
Greenburg G and Hay ED: Epithelia
suspended in collagen gels can lose polarity and express
characteristics of migrating mesenchymal cells. J Cell Biol.
95:333–339. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spaderna S, Schmalhofer O, Hlubek F, Berx
G, Eqer A, Merkel S, et al: A transient, EMT-linked loss of
basement membranes indicates metastasis and poor survival in
colorectal cancer. Gastroenterology. 131:830–840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED, et al: Epithelial-mesenchymal and
mesenchymal-epithelial transitions in carcinoma progression. J Cell
Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Min C, Eddy SF, Sherr DH and Sonenshein
GE: NF-κB and epithelial to mesenchymal transition of cancer. J
Cell Biochem. 104:733–744. 2008.
|
|
6
|
Wang Y and Zhou BP: Epithelial-mesenchymal
transition in breast cancer progression and metastasis. Chin J
Cancer. 30:603–611. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jouppila-Mättö A, Närkiö-Mäkelä M, Soini
Y, Pukkila M, Sironen R, Tuhkanen H, et al: Twist and snai1
expression in pharyngeal squamous cell carcinoma stroma is related
to cancer progression. BMC Cancer. 11:3502011.PubMed/NCBI
|
|
8
|
Okunieff P, Fenton B and Chen Y: Past,
present, and future of oxygen in cancer research. Adv Exp Med Biol.
566:213–222. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vaupel P, Schlenqer K, Knoop C and Hockel
M: Oxygenation of human tumors: evaluation of tissue oxygen
distribution in breast cancers by computerized O2
tension measurements. Cancer Res. 51:3316–3322. 1991.PubMed/NCBI
|
|
10
|
Fokas E, McKenna WG and Muschel RJ: The
impact of tumor microenvironment on cancer treatment and its
modulation by direct and indirect antivascular strategies. Cancer
Metastasis Rev. 31:823–842. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maqat J, Jordan BF, Cron GO and Gallez B:
Noninvasive mapping of spontaneous fluctuations in tumor
oxygenation using 19F MRI. Med Phys. 37:5434–5441. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morgan MJ and Liu ZG: Crosstalk of
reactive oxygen species and NF-κB signaling. Cell Res. 21:103–115.
2011.
|
|
13
|
Rofstad EK: Microenvironment-induced
cancer metastasis. Int J Radiat Biol. 76:589–605. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Binker MG, Binker-Cosen AA, Richards D,
Gaisano HY, de Cosen RH and Cosen-Binker LI: Hypoxia-reoxygenation
increase invasiveness of PANC-1 cells through Rac1/MMP-2. Biochem
Biophys Res Commun. 393:371–376. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nicould IB, Jones CM, Pierce JM, Earl TM,
Matrisian LM, Chari RS, et al: Warm hepatic ischemia-reperfusion
promotes growth of colorectal carcinoma micrometastases in mouse
liver via matrix metalloproteinase-9 induction. Cancer Res.
67:2720–2728. 2007. View Article : Google Scholar
|
|
16
|
Man K, Nq KT, Lo CM, Ho JW, Sun BS, Sun
CK, et al: Ischemia-reperfusion of small liver remnant promotes
liver tumor growth and metastases - activation of cell invasion and
migration pathways. Liver Transpl. 13:1669–1677. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kokura S, Yoshida N, Imamoto E, Ueda M,
Ishikawa T, Uchiyama K, Kuchide M, Naito Y, Okanoue T and Yoshikawa
T: Anoxia reoxygenation down-regulates the expression of E-cadherin
in human colon cancer cell lines. Cancer Lett. 211:79–87. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teicher BA: Hypoxia and drug resistance.
Cancer Metastasis Rev. 13:139–168. 1994. View Article : Google Scholar
|
|
19
|
Martinive P, Defresne F, Bouzin C, Saliez
J, Lair F, Grégoire V, et al: Preconditioning of the tumor
vasculature and tumor cells by intermittent hypoxia: implications
for anticancer therapies. Cancer Res. 66:11736–11744. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cairns RA, Kalliomaki T and Hill RP: Acute
(cyclic) hypoxia enhances spontaneous metastasis of KHT murine
tumors. Cancer Res. 61:8903–8908. 2001.PubMed/NCBI
|
|
21
|
Jing Y, Han Z, Zhang S, Liu Y and Wei L:
Epithelial-mesenchymal transition in tumor microenvironment. Cell
Biosci. 1:292011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thiery JP: Epithelial-mesenchymal
transitions in tumor progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee MY and Shen MR: Epithelial-mesenchymal
transition in cervical carcinoma. Am J Transl Res. 4:1–13.
2012.PubMed/NCBI
|
|
24
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Medici D, Hay ED and Olsen BR: Snail and
Slug promote epithelial-mesenchymal transition through
beta-catenin-T-cell factor-4-dependent expression of transforming
growth factor-beta3. Mol Biol Cell. 19:4875–4887. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bachelder RE, Yoon SO, Franci C, de
Herreros AG and Mercurio AM: Glycogen synthase kinase-3 is an
endogenous inhibitor of Snail transcription: implications for the
epithelial-mesenchymal transition. J Cell Biol. 168:29–33. 2005.
View Article : Google Scholar
|
|
27
|
Chua HL, Bhat-Nakshatri P, Clare SE,
Morimiya A, Badve S and Nakashatri H: NF-kappaB represses
E-cadherin expression and enhances epithelial to mesenchymal
transition of mammary epithelial cells: potential involvement of
ZEB-1 and ZEB-2. Oncogene. 26:711–724. 2007. View Article : Google Scholar
|
|
28
|
Takeda K, Takeuchi O, Tsujimura T, Itami
S, Adachi O, Kawai T, et al: Limb and skin abnormalities in mice
lacking IKKalpha. Science. 284:313–316. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rupec RA and Baeuerle PA: The genomic
response of tumor cells to hypoxia and reoxygenation. Differential
activation of transcription factors AP-1 and NF-kappa B. Eur J
Biochem. 234:632–640. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng ZX, Sun B, Wang SJ, Gao Y, Zhang YM,
Zhou HX, et al: Nuclear factor-κB-dependent epithelial to
mesenchymal transition induced by HIF-1α activation in pancreatic
cancer cells under hypoxic conditions. PLoS One. 6:e237522011.
|
|
31
|
Shi J, Wan Y and Di W: Effect of hypoxia
and re-oxygenation on cell invasion and adhesion in human ovarian
carcinoma cells. Oncol Rep. 20:803–807. 2008.PubMed/NCBI
|
|
32
|
Cannito S, Novo E and Compaqnone A: Redox
mechanisms switch on hypoxia-dependent epithelial-mesenchymal
transition in cancer cells. Carcinogenesis. 29:2267–2278. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rofstad EK, Galappathi K, Mathiesen B and
Ruud EB: Fluctuating and diffusion-limited hypoxia-induced
metastasis. Clin Cancer Res. 13:1971–1978. 2007. View Article : Google Scholar : PubMed/NCBI
|