Introduction
The t(8;21) is the second most common chromosomal
abnormality in AML, accounting for 10–15% of cases with discernible
translocations, and characteristically induces a leukemia with the
French-American-British (FAB) M2 phenotype. As previously
documented (1–3), AML1-ETO exerts the dominant negative
effect on AML1-dependent transcriptional activation, mostly through
interaction of its ETO moiety with nuclear co-repressors N-CoR and
Sin3A that recruit the histone deacetylases (HDACs), resulting in
transcriptional repression by deacetylating histones and creating
repressive chromatic structures. A simple model of AML1-ETO
function in leukemogenesis reflects its dominant negative effects
on AML1 target genes, to a large extent via the aberrant
recruitment of epigenetic modifiers such as HDACs and DNA
methyltransferases (DNMTs).
A direct transcriptional regulation by AML1-ETO
through the AML1 DNA-binding activity has been demonstrated for a
few genes, notably the anti-apoptotic gene Bcl-2 (4), the hematopoietic lineage regulator
gene CCAAT/enhancer-binding protein α (CEBPA) (5) and the cell cycle regulator
p14ARF(6).
The Bcl-2 gene is a highly conserved member of the
Bcl-2 family and constitutes an important regulator of apoptosis.
Bcl-2 can prevent or delay apoptosis in several cell types
(7). Klampfer et al(4) identified a consensus DNA binding
sequence for AML1 (TGT/cGGT) in the 5′ regulatory region of the
Bcl-2 gene and demonstrated that both AML1 and AML1-ETO proteins
can bind to this site. Regulation of the Bcl-2 promoter by
AML1-ETO, but not by the normal AML1 proteins, indicates a unique
biological activity of the fusion protein (4).
The C/EBP genes are believed to be critically
involved in hematopoietic differentiation and leukemogenesis
(8). Approximately 10–15% of AML
samples have inactivating mutations of CEBPA, and the forced
expression of C/EBPα in AML cells can induce terminal
differentiation, emphasizing the important contribution of C/EBPα
to AML leukemogenesis (5,9). AML1-ETO may contribute to
leukemogenesis by specifically inhibiting AML1 and CEBPA-dependent
activation of myeloid promoters and blocking differentiation
(10).
The p53 tumor suppressor pathway is arguably the
most important checkpoint control pathway in human cancer. A third
component of the p53 pathway is the p14ARF tumor
suppressor, which regulates the p53-dependent oncogene checkpoint
(11). Loss of p14ARF
impairs p53-mediated growth arrest and/or apoptosis in response to
activated oncogenes. In addition, cells lacking either p53 or
p14ARF fail to undergo replicative crisis and are
immortal (12). The p53 promoter
does not contain any perfect AML1 DNA-binding sites (TGT/cGGT), but
the human p14ARF promoter contains eight such sites
(6).
In this study, using AML1-ETO-expressing cell line
U937-A/E as an in vitro model, we performed chromatin
immunoprecipitation (ChIP) assays to investigate how the binding of
AML1-ETO affected the chromatin structure of its target genes
(Bcl-2, CEBPA and p14ARF) and thus caused deregulated
gene expression associated with growth arrest and differentiation
block. Our study identified Bcl-2, CEBPA and p14ARF as
additional pathogenic targets for a leukemia fusion protein and
provided evidence that linked the epigenetic silencing of Bcl-2,
CEBPA and p14ARF loci to the growth arrest and
differentiation block of myeloid precursors. Thus, suppression of
these gene expressions correlated with significant alterations in
the chromatin structure at the promoters may play a key role in the
proliferation and differentiation underlying leukemogenesis.
Materials and methods
Clinical samples
Leukemic cells of nine non-t(8;21) AML patients and
nine t(8;21) AML patients who were diagnosed as the M2 subtype
according to the FAB classification, were prepared from bone marrow
cells or peripheral blood mononuclear cells, following approval by
the Hospital Ethics Committee with signed consent provided by the
patients.
Cell culture
Human myeloid U937 cells were maintained in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at
37°C in a humidified incubator with an atmosphere of 5%
CO2. U937-Mock and U937-A/E cells were maintained in
RPMI-1640 medium supplemented with 10% FBS and 0.5 mg of
G418/ml.
ChIP assay
ChIP assays were performed by using a ChIP Express
kit (Millipore Biotechnology), according to the manufacturer’s
instructions.
Statistical analysis
All values in the figures are expressed as the means
± SD. To determine statistical significance, the values were
compared using two-group t-tests, and P-values <0.05 were
considered to indicate statistically significant differences.
Results
Expression of AML1-ETO reduces
proliferation, induces apoptosis and blocks myeloid
differentiation
To investigate the potential direct role of AML1-ETO
in the growth, survival, and differentiation of myeloid leukemic
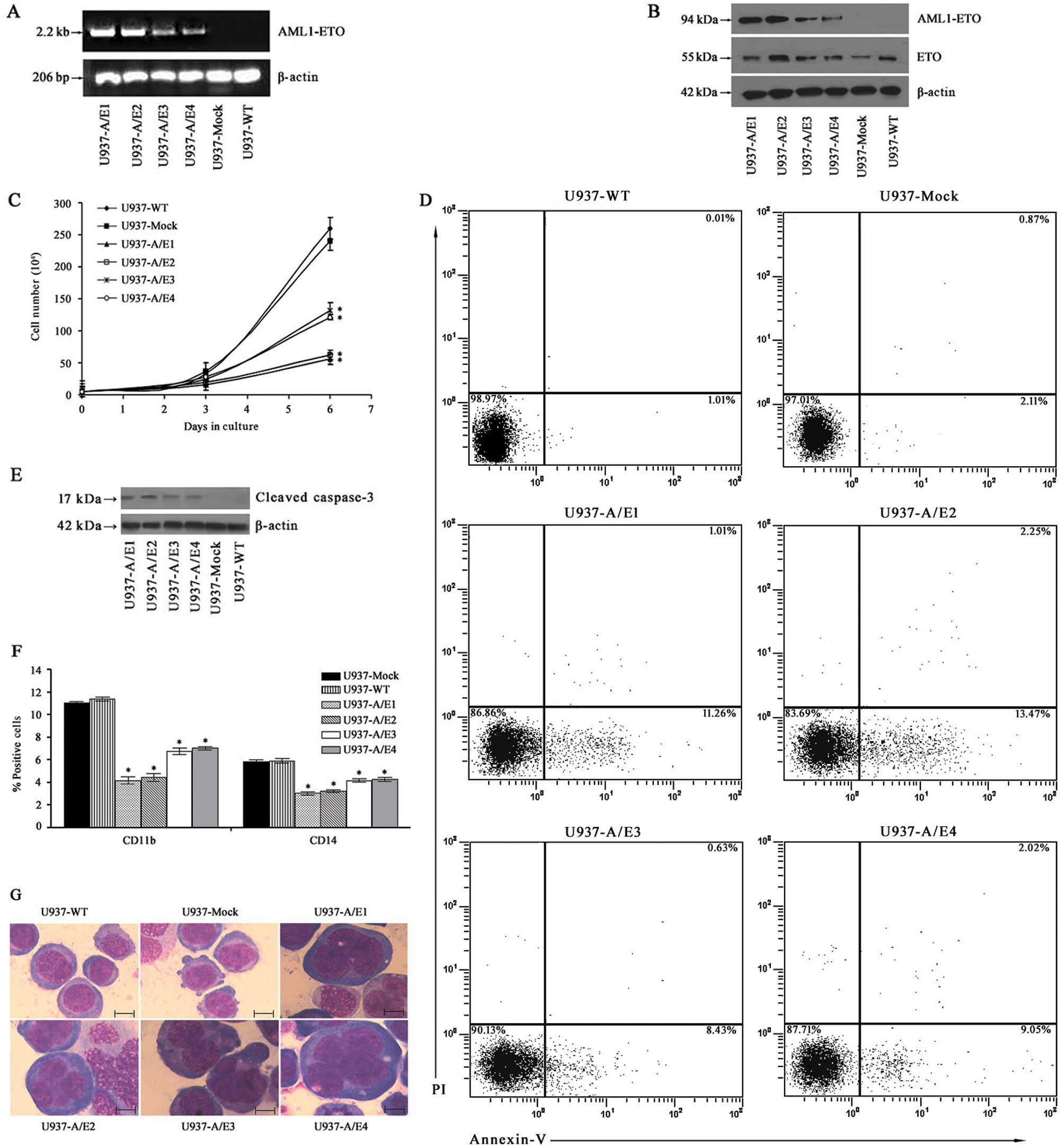
cells, we made AML1-ETO-expressing U937 cell lines. As indicated in
Fig. 1A and B, relatively high
levels of AML1-ETO expression were clearly observed in U937-A/E1
and 2 at both mRNA and protein levels. The effects of AML1-ETO
expression on cell growth were evaluated by comparing the growth
curves of U937-A/E, U937-Mock and U937-WT cells. Analyses of the
proliferative ability indicated that cell growth in
AML1-ETO-transfected cells was significantly decreased in
comparison to empty vector-transfected cells and non-transfected
cells (P<0.01) (Fig. 1C). There
was no significant difference between the proliferation rates of
U937-Mock and U937-WT cells, indicating that the effect was due
solely to the expression of AML1-ETO.
We performed experiments to determine how the
expression of AML1-ETO affected the apoptosis. The results
demonstrated that in U937-A/E cells but not in U937-WT and
U937-Mock cells, apoptotic cells were statistically significantly
increased, although to a lower degree, as evidenced by the Annexin
V assay (Fig. 1D). In order to
further confirm the effects of AML1-ETO expression on enhancing
apoptosis, changes in caspase-3 protein were analyzed in U937-A/E
cells. As shown in Fig. 1E,
AML1-ETO expression also significantly enhanced activation of
caspase-3, an indicator of cell apoptosis, as indicated by the
appearance of active fragment 17 kDa of cleaved caspase-3 on the
blot.
Then, we examined whether the expression of AML1-ETO
fusion protein had an influence on the differentiation capacity of
U937 cells. As markers for myeloid differentiation, the expression
of CD11b and CD14 was monitored via FACS analysis. CD11b + cell %
was 4.1–7.0% in U937-A/E1–4 cells, which was significantly lower
than that in U937-Mock cells (11.4%) and U937-WT cells (11.0%)
(P<0.01) (Fig. 1F). Moreover,
the expression of CD14 antigen was decreased by 1.5–2-fold as
compared with the control cells (P<0.01) (Fig. 1F). These data correspond to the
lower differentiation morphological changes of AML1-ETO-transfected
cells such as expanded cell size and increased nuclei/cytoplasm
ratio with larger nuclei observed in the morphological examination
of Wright-Giemsa-stained cytospins (Fig. 1G).
Therefore, it appears that AML1-ETO expression
induces growth arrest in leukemic U937 transformants, as
demonstrated by the reduced growth rate. Furthermore, the
expression of AML1-ETO significantly inhibited the differentiation
of U937-A/E cells. These cells lost their original lymphoblast-like
morphology without displaying granulocytic morphology and exhibited
a block of differentiation at an early stage, as previously
reported (16).
Bcl-2, CEBPA and p14ARF
expression are downregulated by the AML1-ETO fusion protein
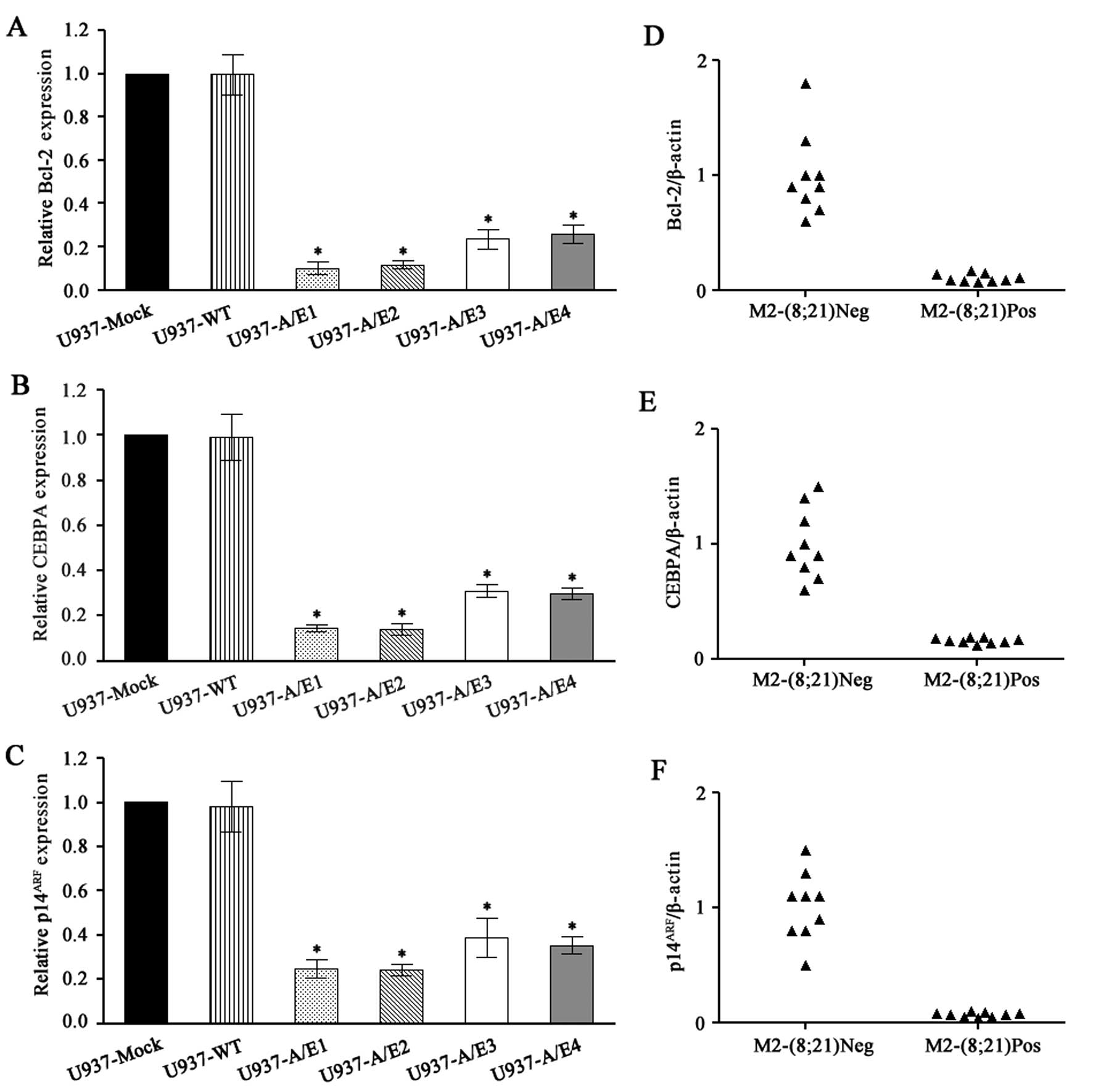
A quantitative reverse transcription PCR (qRT-PCR)
assay was used to assess the mRNA expression of Bcl-2, CEBPA,
p14ARF and GAPDH. This assay was tested on U937
AML1-ETO-expressing cells and U937 non-expressing cells. We
observed that AML1-ETO-expressing cells contained markedly reduced
levels of Bcl-2, CEBPA and p14ARF as compared with
control-transfected cells or wild-type cells (P<0.001) (Fig. 2A–C).
The same assay was applied to assess Bcl-2, CEBPA
and p14ARF mRNA levels in primary leukemia cells of AML
patients with or without t(8;21). Two cell lines derived from
t(8;21) leukemia patient cells showed higher expression of Bcl-2
(4,13). However, studies using 29 (14) and 17 (15) primary t(8;21) leukemia patient
samples indicated that Bcl-2 expression was generally downregulated
compared to that for non-leukemic or non-t(8;21) AML samples. We
also confirmed a reduced Bcl-2 mRNA level in patients with
t(8;21)-containing AML (P<0.001) (Fig. 2D), consistent with a previous report
(16).
This was also the case with sorted cells from
patients suffering from a leukemia with or without a t(8;21),
indicating that the presence of AML1-ETO led to a significant
downregulation of CEBPA expression (P<0.001) (Fig. 2E).
The p14ARF locus is rarely deleted in AML
(17). Whereas p14ARF
mRNA levels are low in normal peripheral blood cells and bone
marrow, the levels of p14ARF were increased in most AML
samples studied, suggesting that this checkpoint was activated
(18). We tested whether AML1-ETO
prevents the increase of p14ARF in patients with
t(8;21)-containing AML. Analysis of p14ARF mRNA levels
in 18 AML samples indicated that p14ARF mRNA levels were
lower in t(8;21)-containing AML samples. The p14ARF
expression values were normalized to β-actin expression. The
t(8;21)-negative samples expressed a range of p14ARF,
with a mean ratio of p14ARF: β-actin of 1.0. By
contrast, the t(8;21)-containing samples on average expressed
markedly reduced levels of p14ARF (P<0.001) with a
mean p14ARF: β-actin ratio of only 0.07 (Fig. 2F).
We therefore concluded that the expressions of
Bcl-2, CEBPA and p14ARF mRNA are specifically inhibited
in leukemia cells that have the AML1-ETO fusion gene. These results
are similar to previous findings (5,16,19,20),
validating the quantitative accuracy of the RT-PCR assay.
AML1-ETO triggers the heterochromatic
silencing of Bcl-2, CEBPA and p14ARF promoter
regions
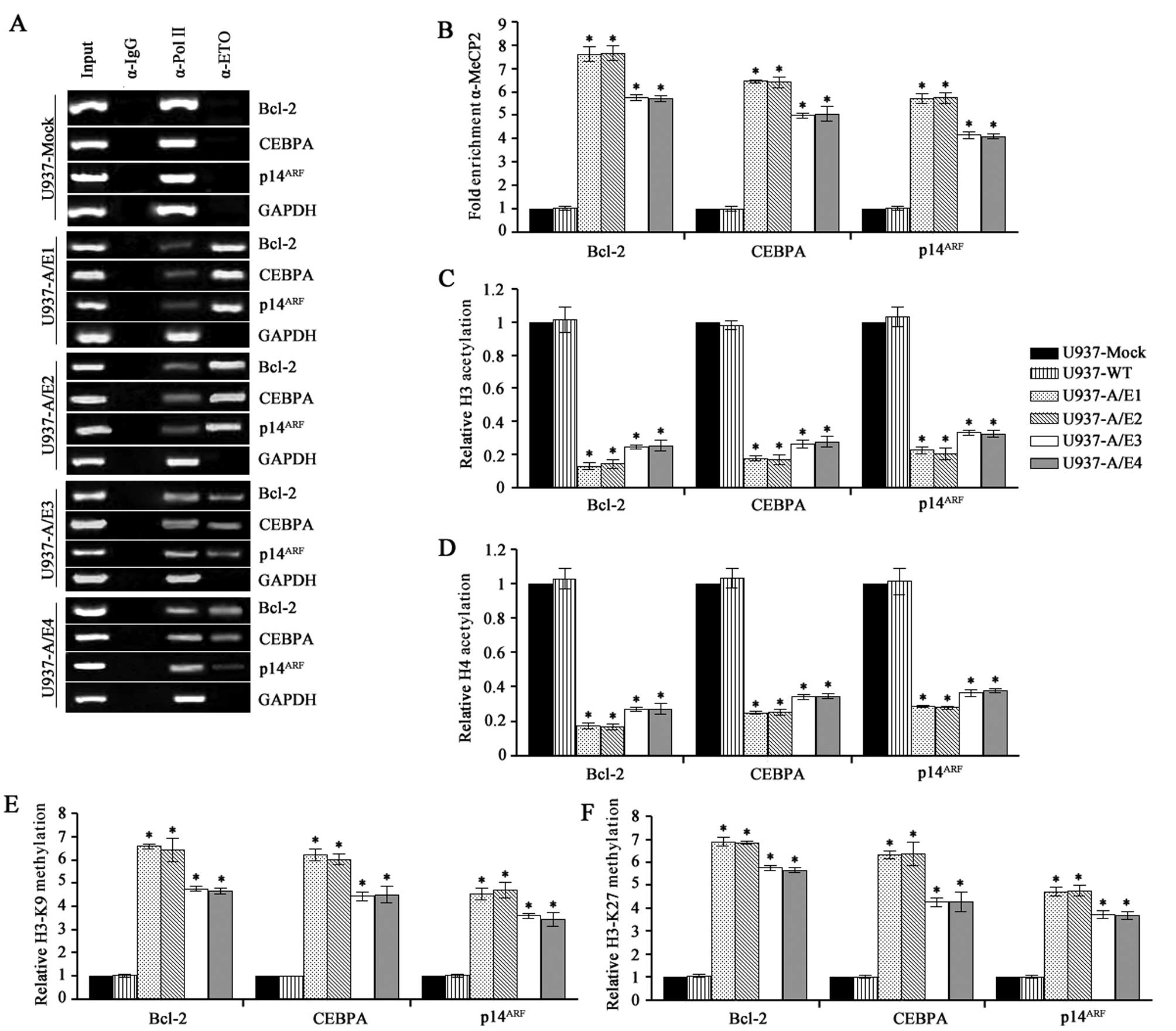
AML1-ETO maintains the ability of AML1 to bind the
consensus sequence TGT/cGGT on target gene promoters and acts as a
dominant-negative repressor of AML1 targeting genes, including
Bcl-2, CEBPA and p14ARF(5,16,19,20).
ChIP using primers that encompassed the Bcl-2, CEBPA and
p14ARF promoter regions was performed with anti-ETO
antibody to verify these AML1-ETO targets enriched within the
AML1-ETO bound genomic sequences in U937-AML1-ETO cells and as a
negative control in U937-empty vector. All comparisons in the cell
lines were made between AML1-ETO-expressing and non-expressing
cells. DNA sequences specifically precipitated by anti-ETO antibody
in AML1-ETO-expressing cells (but not in AML1-ETO-negative cells)
most likely represent the AML1-ETO-specific targets. We detected
the Bcl-2, CEBPA and p14ARF promoter sequences in
anti-ETO immune complexes, but not in control immune complexes
(Fig. 3A), indicating that Bcl-2,
CEBPA and p14ARF are direct and specific targets of the
t(8;21) fusion protein.
The oncogenic properties of AML1-ETO are linked to
its ability to form oligomeric complexes with increased affinity
for HDAC and DNMTs rendering AML1-ETO a potent transcriptional
repressor of AML1-target genes (21). ChIP analysis also revealed the
presence of methyl-CpG binding protein 2 (MeCP2) at the Bcl-2,
CEBPA and p14ARF promoter regions occupied by AML1-ETO
in U937-A/E1–4 (P<0.001) (Fig.
3B). We therefore investigated whether the aberrant recruitment
of MeCP2 activities by AML1-ETO modifies nucleosomal histone tails
on the Bcl-2, CEBPA and p14ARF promoters. Using ChIP
analysis, we focused on several modifications of histone H3 (AcH3,
tri-mK27 and tri-mK9) and the acetylated forms of histone H4 in the
same cell lines that were used for AML1-ETO target identification.
These modifications are mutually exclusive, whereby H3-K9
trimethylation or H3-K27 trimethylation is a hallmark of inactive
chromatin and acetylation of H3 or H4 is found at active loci
(22,23). As illustrated in Fig. 3C and D, H3 and H4 histones are
hyperacetylated at the Bcl-2, CEBPA and p14ARF promoter
regions in U937-Mock and U937-WT cells, while decreased acetylation
levels are measurable in U937-A/E1–4 cells (P<0.001). The
reduced histone acetylation in AML1-ETO-expressing cells suggested
a hindered transcription at these chromatin sites on the Bcl-2,
CEBPA and p14ARF genes. ChIP assay performed using
antibodies against H3-K9 trimethylation and H3-K27 trimethylation
demonstrated that AML1-ETO-expressing cells had a marked
trimethylation level of H3-K9 and H3-K27 at the Bcl-2, CEBPA and
p14ARF promoters. By contrast, few or no promoters with
trimethylation of H3-K9 and H3-K27 were observed in
AML1-ETO-non-expressing cells (P<0.001) (Fig. 3E and F). The higher level of histone
methylation in AML1-ETO-expressing cells paralleled with
significantly lower levels of H3 and H4 acetylation. These changes
are consistent with the induction of a repressive chromatin
configuration by AML1-ETO in its direct target genes.
Treatment of demethylating agent or HDAC
inhibitor partially reverses Bcl-2, CEBPA and p14ARF
suppression
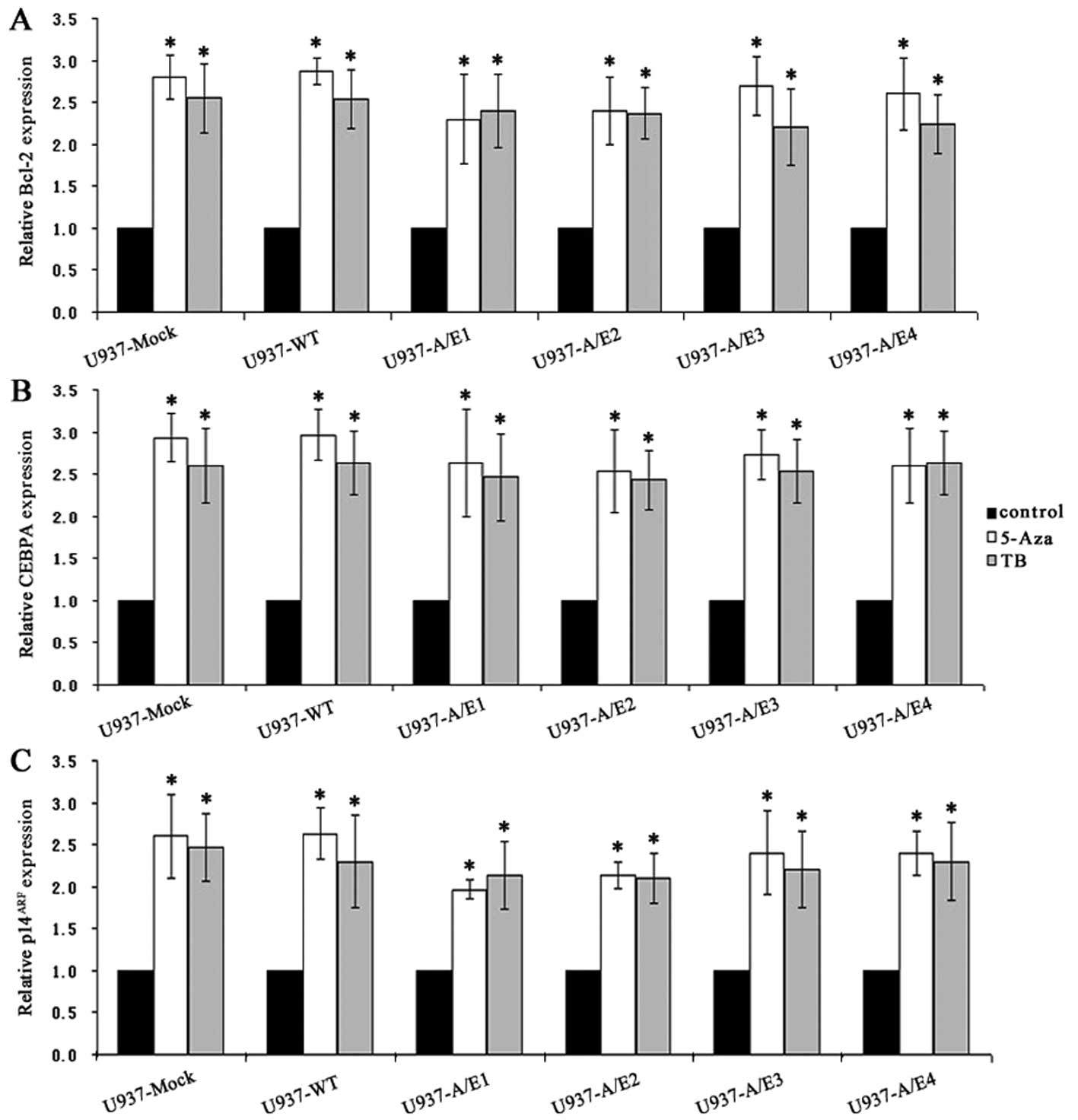
We next treated the AML1-ETO-positive and -negative
U937 cells with either the DNMT inhibitor 5-Aza or the HDAC
inhibitor TB, respectively. Both 5-Aza and TB increased the
expression of Bcl-2, CEBPA and p14ARF by ~2- to 3-fold
(P<0.001) (Fig. 4A–C). In
addition, 5-Aza impaired the ability of anti-MeCP2 antibody to
immunoprecipitate naked DNA surrounding the region of AML1 binding
sites on Bcl-2, CEBPA and p14ARF gene promoters
(P<0.001) (Fig. 4D). On the
other hand, a significant increase (P<0.001) in chromatin H3 and
H4 acetylation of Bcl-2, CEBPA and p14ARF was observed
in cells treated with TB compared with untreated cells (Fig. 4E and F).
Consistent with the increased Bcl-2, CEBPA and
p14ARF mRNA levels, 5-Aza treatment showed demethylation
of CpGs and TB treatment resulted in enhanced accumulation of
acetylated histone H3 or H4 at the Bcl-2, CEBPA and
p14ARF promoters. These results indicated that DNA
methylation and HDAC were simultaneously and independently
operative in this model, and both contributed to gene regulation in
U937 cells.
Discussion
Cancer is a genetic and epigenetic disease (24,25).
The contribution of epigenetic mechanisms for a correct cell
function is highlighted by the effects of their deregulation in
cooperation with genetic alterations leading to the establishment
and progression of tumors. Heterochromatic gene silencing
represents an alternative oncogenic mechanism to gene mutation or
deletion for the transcriptional repression of tumor suppressor
genes (24).
Reduced expression or loss of function in
hematopoietic malignancies has been studied extensively, and loss
of C/EBPα function is thought to contribute as an early event to
leukemogenesis by inhibiting myeloid differentiation (9). Hypermethylation in the upstream region
of the promoter-associated CpG island of CEBPA has previously been
detected in lung cancer as well as in head and neck squamous cell
carcinoma (26,27). In hematopoietic tumor cell lines,
CpG island hypermethylation of the proximal CEBPA promoter region
was associated with transcriptional silencing, and treatment with
the demethylating agent 5-aza-2′-deoxycytidine resulted in C/EBPα
reexpression and promoter demethylation (28). Wouters et al provided first
evidence for the importance of C/EBPα methylation in a small
subgroup of AML (29). The
epigenetic contribution to C/EBPα deregulation has been
investigated and the aberrant DNA methylation in the upstream
promoter of C/EBPα has been shown to be a frequent event in AML
(28).
Here, we showed that the myeloid transcription
factor C/EBPα was specifically downregulated in AML patients with
the AML1-ETO of the FAB-M2 subtype or U937
AML1-ETO-expressing cells. U937-A/E clones exhibited lower
differentiation morphological changes such as expanded cell size
and increased nuclei/cytoplasm ratio with larger nuclei associated
with a decreased expression of cell surface markers CD11b and CD14.
This altered differentiation potential is correlated with the
downregulation of C/EBPα upon expression of AML1-ETO. Therefore,
the epigenetic dysregulation including MeCP2 binding, H3 and H4
hypoacetylation as well as hypertrimethylation of H3-K9 or K27 may
be a common alternative or complementary mechanism of interfering
with C/EBPα function.
It has previously been reported that the AML1-ETO
fusion protein was able to induce anti-apoptotic Bcl-2 expression
in vitro(4), while Burel
et al(16) and Lu et
al(20) as well as the present
study showed an AML1-ETO-induced decrease in Bcl-2 expression. On
the contrary, AML1-ETO increased the expression of Bak protein, a
pro-apoptotic member of the Bcl-2 family that plays an important
role in regulating mitochondrial membrane permeability during
apoptosis (30). The induction of
AML1-ETO in U937T-A/E cells causes a progressive cell cycle arrest
in G0/G1 phase. Moreover, ectopic expression of Bcl-2 delays
apoptosis without preventing AML1-ETO-induced G1/G0 arrest
(16). Our results are in agreement
with this and showed that AML1-ETO could markedly downregulate the
expression of Bcl-2 by inducing repressive chromatin structure at
its promoter. It has been suggested that the overexpression of the
anti-apoptotic protein Bcl-2 in chronic lymphocytic leukemia (CLL)
is caused by hypomethylation of the promoter region of the Bcl-2
gene (31). However, methylation of
the 5′ region of apoptosis-associated genes is a common finding in
patients with bladder carcinoma (32). This finding is noteworthy as DNA
hypermethylation is often associated with decreased gene
expression, and in the case of Bcl-2, this would be expected to
promote apoptosis rather than tumor growth. Inhibition of
proliferation or apoptosis would not be favorable to the
propagation of clonal cells harboring the t(8;21) translocation. If
growth arrest and apoptosis are general features associated with
the expression of AML1-ETO, we hypothesize that AML1-ETO-modulated
apoptosis-regulating genes and/or proteins may become the targets
for secondary ‘hit’ that contributes to the pathogenesis of
AML1-ETO-associated leukemia. It may be inferred that some genetic
or/and epigenetic alterations of apoptosis-related genes have
appeared in these AML1-ETO-positive AML cells, which may overcome
the apoptosis-enhancing effect of AML1-ETO.
Methylation at the p14ARF promoter to
suppress gene expression has been observed in some tumor cell
lines, particularly in colorectal cancer (33,34).
In our study, we found that the recruitment of MeCP2 to
p14ARF chromatin in AML1-ETO-expressing cell lines
correlates with lower levels of H3 and H4 acetylation and higher
levels of H3 (Lys9 and Lys27) trimethylation resulting in the
silence of the p14ARF gene. AML1-ETO suppressed the
p14ARF promoter and reduced endogenous levels of
p14ARF expression in multiple cell types (6). Our results support this and provide an
explanation for the observed reduced p14ARF expression.
Thus, AML1-ETO-mediated suppression of p14ARF may
disrupt both p53-dependent and p53-independent growth suppression
pathways to extend the lifespan of myeloid progenitor cells,
allowing more opportunities to acquire additional mutations,
ultimately leading to leukemia.
Epigenetic alterations are increasingly recognized
as important contributors to human cancer pathogenesis and DNMT and
HDAC inhibitors have recently been incorporated into the treatment
of AML1-ETO leukemias (35–37). Their ability to reverse the
inhibition of myeloid-specific genes helps to re-establish a normal
differentiation program. Our findings indicate that demethylating
agents or HDAC inhibitors can relieve Bcl-2, CEBPA and
p14ARF suppression in AML1-ETO-expressing cells through
a mechanism that involves inversion of epigenetic alterations.
Despite the changes in expression pattern of Bcl-2,
CEBPA and p14ARF in primary bone marrow cells of
AML1-ETO-positive AML-M2 patients were similar to those in U937-A/E
cells when compared to the AML1-ETO-negative cells, to date we have
not found the significant and consistent alterations of DNA/histone
modifications at these genes in primary cells of AML1-ETO-positive
AML patients (data not shown). This may be due to the heterogeneity
of the primary marrow cells of the patients and the limitation of
techniques. However, one would expect to apply epigenetic markers
for diagnosis, stratification and especially as an indicator in
epigenetic regulatory treatment in leukemia patients in the
future.
Collectively, we provided the first evidence for
modifications of the chromatin structure at the Bcl-2, CEBPA and
p14ARF promoters occupied by the AML1-ETO fusion
protein. Our data are therefore consistent with a model by which
the binding of AML1-ETO leads to alterations in the chromatin
structure of its target genes. These findings underscore the
importance of epigenetic alteration mediated silencing of these
genes in leukemogenesis. It is noteworthy to compare the chromatin
structure of different AML1-ETO target genes in order to better
understand the molecular details of the deregulation of gene
expression by this oncoprotein. If so, these mechanisms may be
potential targets for therapeutic strategies based on the reversal
of epigenetic silencing in t(8;21)-positive leukemias.
Acknowledgements
The authors thank Dr Qing-Yu Wu for providing
pcDNA3.1/V5-His-TOPO TA vector. This study was supported by grants
from the National Natural Scientific Foundation of China (no.
81070402), the National Key Scientific Projects of China (no.
2011CB933501) and the Priority Academic Program Development of
Jiangsu Higher Education Institutions (PAPD).
References
|
1
|
Chevallier N, Corcoran CM, Lennon C, et
al: ETO protein of t(8;21) AML is a corepressor for Bcl-6 B-cell
lymphoma oncoprotein. Blood. 103:1454–1463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rowley JD: Molecular genetics in acute
leukemia. Leukemia. 14:513–517. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maiques-Diaz A, Chou FS, Wunderlich M, et
al: Chromatin modifications induced by the AML1-ETO fusion protein
reversibly silence its genomic targets through AML1 and Sp1 binding
motifs. Leukemia. 26:1329–1337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klampfer L, Zhang J, Zelenetz AO, Uchida H
and Nimer SD: The AML1/ETO fusion protein activates transcription
of BCL-2. Proc Natl Acad Sci USA. 93:14059–14064. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pabst T, Mueller BU, Harakawa N, et al:
AML1-ETO downregulates the granulocytic differentiation factor
C/EBPalpha in t(8;21) myeloid leukemia. Nat Med. 7:444–451. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Linggi B, Muller-Tidow C, van de Locht L,
et al: The t(8;21) fusion protein, AML1 ETO, specifically represses
the transcription of the p14(ARF) tumor suppressor in acute myeloid
leukemia. Nat Med. 8:743–750. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krug U, Ganser A and Koeffler HP: Tumor
suppressor genes in normal and malignant hematopoiesis. Oncogene.
21:3475–3495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ho PA, Alonzo TA, Gerbing RB, et al:
Prevalence and prognostic implications of CEBPA mutations in
pediatric acute myeloid leukemia (AML): a report from the
Children’s Oncology Group. Blood. 113:6558–6566. 2009.PubMed/NCBI
|
|
10
|
Zhang DE, Hetherington CJ, Meyers S, et
al: CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2)
synergistically activate the macrophage colony-stimulating factor
receptor promoter. Mol Cell Biol. 16:1231–1240. 1996.
|
|
11
|
Sherr CJ and Weber JD: The ARF/p53
pathway. Curr Opin Genet Dev. 10:94–99. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamijo T, Zindy F, Roussel MF, et al:
Tumor suppression at the mouse INK4a locus mediated by the
alternative reading frame product p19ARF. Cell. 91:649–659. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kohzaki H, Ito K, Huang G, Wee HJ,
Murakami Y and Ito Y: Block of granulocytic differentiation of
32Dcl3 cells by AML1/ETO(MTG8) but not by highly expressed Bcl-2.
Oncogene. 18:4055–4062. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shikami M, Miwa H, Nishii K, et al: Low
BCL-2 expression in acute leukemia with t(8;21) chromosomal
abnormality. Leukemia. 13:358–368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Banker DE, Radich J, Becker A, et al: The
t(8;21) translocation is not consistently associated with high
Bcl-2 expression in de novo acute myeloid leukemias of adults. Clin
Cancer Res. 4:3051–3062. 1998.PubMed/NCBI
|
|
16
|
Burel SA, Harakawa N, Zhou L, Pabst T,
Tenen DG and Zhang DE: Dichotomy of AML1-ETO functions: growth
arrest versus block of differentiation. Mol Cell Biol.
21:5577–5590. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ruas M and Peters G: The p16INK4a/CDKN2A
tumor suppressor and its relatives. Biochim Biophys Acta.
1378:F115–F177. 1998.PubMed/NCBI
|
|
18
|
Taniguchi T, Chikatsu N, Takahashi S, et
al: Expression of p16INK4A and p14ARF in hematological
malignancies. Leukemia. 13:1760–1769. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hiebert SW, Reed-Inderbitzin EF, Amann J,
Irvin B, Durst K and Linggi B: The t(8;21) fusion protein contacts
co-repressors and histone deacetylases to repress the transcription
of the p14ARF tumor suppressor. Blood Cells Mol Dis.
30:177–183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu Y, Xu YB, Yuan TT, et al: Inducible
expression of AML1-ETO fusion protein endows leukemic cells with
susceptibility to extrinsic and intrinsic apoptosis. Leukemia.
20:987–993. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fazi F, Zardo G, Gelmetti V, et al:
Heterochromatic gene repression of the retinoic acid pathway in
acute myeloid leukemia. Blood. 109:4432–4440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eberharter A and Becker PB: Histone
acetylation: a switch between repressive and permissive chromatin.
Second in review series on chromatin dynamics. EMBO Rep. 3:224–229.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kouzarides T: Histone methylation in
transcriptional control. Curr Opin Genet Dev. 12:198–209. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar
|
|
26
|
Tada Y, Brena RM, Hackanson B, Morrison C,
Otterson GA and Plass C: Epigenetic modulation of tumor suppressor
CCAAT/enhancer binding protein alpha activity in lung cancer. J
Natl Cancer Inst. 98:396–406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bennett KL, Hackanson B, Smith LT, et al:
Tumor suppressor activity of CCAAT/enhancer binding protein alpha
is epigenetically down-regulated in head and neck squamous cell
carcinoma. Cancer Res. 67:4657–4664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hackanson B, Bennett KL, Brena RM, et al:
Epigenetic modification of CCAAT/enhancer binding protein alpha
expression in acute myeloid leukemia. Cancer Res. 68:3142–3151.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wouters BJ, Jordà MA, Keeshan K, et al:
Distinct gene expression profiles of acute myeloid/T-lymphoid
leukemia with silenced CEBPA and mutations in NOTCH1. Blood.
110:3706–3714. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kiefer MC, Brauer MJ, Powers VC, et al:
Modulation of apoptosis by the widely distributed Bcl-2 homologue
Bak. Nature. 374:736–739. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanada M, Delia D, Aiello A, Stadtmauer E
and Reed JC: bcl-2 gene hypomethylation and high-level expression
in B-cell chronic lymphocytic leukemia. Blood. 82:1820–1828.
1993.PubMed/NCBI
|
|
32
|
Friedrich MG, Weisenberger DJ, Cheng JC,
et al: Detection of methylated apoptosis-associated genes in urine
sediments of bladder cancer patients. Clin Cancer Res.
10:7457–7465. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Esteller M, Tortola S, Toyota M, et al:
Hypermethylation-associated inactivation of p14(ARF) is independent
of p16(INK4a) methylation and p53 mutational status. Cancer Res.
60:129–133. 2000.PubMed/NCBI
|
|
34
|
Benanti JA, Wang ML, Myers HE, Robinson
KL, Grandori C and Galloway DA: Epigenetic down-regulation of ARF
expression is a selection step in immortalization of human
fibroblasts by c-Myc. Mol Cancer Res. 5:1181–1189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hollenbach PW, Nguyen AN, Brady H, et al:
A comparison of azacitidine and decitabine activities in acute
myeloid leukemia cell lines. PLoS One. 5:e90012010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Buchi F, Spinelli E, Masala E, et al:
Proteomic analysis identifies differentially expressed proteins in
AML1/ETO acute myeloid leukemia cells treated with DNMT inhibitors
azacitidine and decitabine. Leuk Res. 36:607–618. 2012. View Article : Google Scholar
|
|
37
|
Zapotocky M, Mejstrikova E, Smetana K,
Stary J, Trka J and Starkova J: Valproic acid triggers
differentiation and apoptosis in AML1/ETO-positive leukemic cells
specifically. Cancer Lett. 319:144–153. 2012. View Article : Google Scholar : PubMed/NCBI
|