Introduction
Hepatocellular carcinoma (HCC) is one of the most
lethal malignancies (1) ranking as
the third leading-cause of cancer-related mortality worldwide
(2). The disease is highly lethal
due to its aggressive metastasis, and is usually diagnosed at an
advanced stage. Increasing evidence indicates that epithelial to
mesenchymal transition (EMT) (3)
plays a pivotal role in tumorgenesis and tumor progression.
E-cadherin downregulation is a hallmark molecular event in EMT, and
contributes to the detachment of cancer cells as single cells,
which acquire a mesenchymal phenotype and undergo dissemination and
invasion. Loss of E-cadherin expression has been shown in
undifferentiated HCC, to proceed to intrahepatic metastasis of HCC
(4). The underlying mechanisms of
E-cadherin loss in cancer include epigenetic methylation of the
E-cadherin coding CDH1 gene (5–7),
transcriptional repression by inhibitory transcription factors
(8,9), and deletion of chromosome 16q24 locus
(10,11), and loss of heterozygosity (LOH) of
the CDH1 gene (5,9,12). In
HCC, the loss of E-cadherin expression is closely associated with
LOH at the E-cadherin locus and methylation of CpG islands in the
promoter region (5). C-terminal
binding protein 1 (CtBP1) is a transcriptional corepressor which
mediates E-cadherin repression, and plays a key role in EMT
(13). Transcription factors such
as Zeb1, Zeb2 (SIP1), Snail, Twist and Slug play a key role in EMT
in various cellular contexts (14).
Among these factors, the activity of Zeb1 and Zeb2 (SIP1) depends
on CtBP1 (15). We previously
detected CtBP1 expression in multiple types of cancers using tissue
array, and showed that CtBP1 was upregulated in HCC tissues in
comparison with paired normal tissues (unpublished data). Thus, the
present study was conducted to further investigate the expression
of CtBP1 and its target protein E-cadherin in HCC patient
specimens, and the impact of the knockdown of CtBP1 on the
biological behavior of HepG2 human HCC cells, with the aim to
evaluate the role of CtBP1 in HCC.
Materials and methods
Tissue microarray and tissue samples
The liver cancer tissue microarray (LVC1722) was
purchased from Patomics (Richmond, CA, USA), which included 52
cases of liver tumor tissues (39 HCC, 5 clear cell HCC, 4
intrahepatic cholangiocarcinomas, 3 mixed hepatocellular
carcinoma-cholangiocarcinomas, 1 sarcoma) and 17 cases of non-tumor
liver tissues. Each tumor case included double tumor tissue cores
and one paired adjacent normal tissue core.
Tumor tissues and adjacent non-tumor tissues of 10
HCC patients, who underwent curative hepatic resection between
March 2012 and June 2012, were provided by the Department of
Hepatobiliary Surgery, Affiliated Hospital of Guilin Medical
College, Guilin, China, and used for preparation of 10 pairs of
protein extracts. The study was approved by the Ethics Committee of
the Affiliated Hospital of Guilin Medical College. Curative
resection was defined as removal of all recognizable tumors with
clear microscopic margins. Specimens were obtained immediately
after surgical resection. None of the patients were treated by any
preoperative therapy. The patients included 8 men and 2 women,
ranging from 31 to 78 years, with an average age of 54 years. Tumor
stage was defined according to the Tumor, Node, Metastasis (TNM)
classification system of the American Joint Committee on
Cancer/International Union Against Cancer (16).
Cell line
HepG2 cells were purchased from the American Type
Culture Collection (ATCC; Rockville, MD, USA). Cells were cultured
in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
heat-inactived fetal bovine (FBS) serum and 0.01% penicillin and
streptomycin at 37°C in a humidified incubator with 5%
CO2.
Immunohistochemistry
The CtBP (C1) and E-cadherin (6F9) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Slides of the tissue microarray underwent heat antigen
retrieval in citrate buffer (0.01 M, pH 6.0), and were incubated
with the primary antibody for 1 h at room temperature. Secondary
antibody incubation and DAB coloring (MaxVision™ HRP-Polymer
anti-mouse IHC kit and DAB kit from Maxim, Fuzhou, China) were
conducted following the manufacturer’s instructions. Scoring of the
immunohistochemical staining was carried out by 3 pathologists at
Guilin Medical College. Briefly, each sample was examined under a
light microscope, and the number of positive cells and staining
intensity were scored (17). The
proportion of positive cells was scored according to 5 graded
scales (0, none; 1, <25%; 2, ≥25% and <50%; 3, ≥50% and
<75%; and 4, ≥75%), and the average staining intensity of the
positive cells according to 4 scales (0, none; 1, weak; 2,
intermediate; and 3, strong). The proportion and intensity scores
were then summed to provide a total score, which ranged from 0 to
7, and the specimens were categorized into 2 groups according to
the overall scores: i) low expression, ≤4 points; and ii) high
expression, 5–7 points.
Western blotting
Proteins from clinical specimens and HepG2 cells
were extracted with lysis buffer (Beyotime Biotechnology, Shanghai,
China), separated by sodium dodecyl sulfate-10% polyacrylamide gel
electrophoresis and transferred onto a polyvinylidene difluoride
membrane. The membrane was blocked with 5% skim milk in TBST (20 mM
Tris-HCl, 150 mM NaCl and 0.1% Tween-20, pH 7.5) for 1 h and
incubated overnight with the primary antibodies at a proper
dilution at 4°C. The dilution of CtBP (C1), E-cadherin (6F9) and
β-actin (ZsBio, Beijing, China) were 1:1,500. After being washed
with TBST buffer, the membranes were incubated with horseradish
peroxidase-conjugated secondary antibody (ZsBio) for 1 h at room
temperature and detection was carried out by enhanced
chemiluminescence detection system (MultiScience Biotech, Shanghai,
China). Intensity of the bands was quantified by densitometry and
normalized to that of β-actin.
CtBP1 knockdown by RNAi in HepG2
cells
The day before transfection, HepG2 cells in the
logarithmic growth phase were trypsinized, counted, and seeded in
6-well and 96-well plates at an appropriate density. When the cells
achieved 80% confluency, they were transfected with CtBP1 siRNA
(Santa Cruz Biotechnology, Inc.) using X-tremeGENE transfection
reagents (Roche, USA). Control cells were those franked with
control siRNA-A (Santa Cruz Biotechnology, Inc.), X-tremeGENE
transfection reagents alone or medium only (Opti-MEM). After
overnight incubation, the medium was replaced with complete DMEM
for further incubation for 72 h in 6-well plates for western blot
assay and for 24, 48, 72, 96 h in 96-well plates for cell viability
assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT).
MTT assay
HepG2 cells were resuspended into single-cell
suspensions, and 100 μl (each portion containing 2,000 cells) was
seeded into the wells of a 96-well culture plate. The cells were
incubated overnight to adherent monolayer of cells, and transfected
with siRNA as above. After further incubation for 24, 48, 72 and 96
h, 20 μl of MTT solution (5 mg/ml) was added to each well and
incubated for an additional 4 h. Finally, the medium was aspirated,
and 150 μl of DMSO was added and mixed thoroughly to dissolve the
dye crystals. Optical absorbance was read on a microplate reader
(BioTek Instruments, Inc., Winooski, VT, USA) at a wavelength of
490 nm. Each group was plated in 6-wells, and the experiment was
repeated 3 times.
Determination of cell cycle distribution
and apoptosis by flow cytometry
HepG2 cells at 80% confluency were cultured first in
serum-free medium for 24 h to synchronize and then in complete
medium for 24 h. The cells were trypsinized and washed with PBS and
fixed overnight with cold 70% ethanol at −20°C. The fixed cells
were washed first with citrate phosphate buffer and then with PBS.
The cells were then incubated in RNase solution (100 μg/ml) at 37°C
for 30 min, and stained in propidium iodide solution (100 μg/ml in
PBS) at room temperature for 30 min for analysis by flow cytometry
(BD Biosciences, Franklin Lakes, NJ, USA) for cell cycle
distribution and the proportion of apoptotic cells. The data are
shown as the fraction of cells in the different cell cycle phases.
Apoptotic cells were defined as those with a DNA volume of
hypodiplomar chromatins. Each group was examined in triplicate.
Determination of the invasive capability
of HepG2 cells
Quantitative analysis of the invasive capability of
HepG2 cells were performed. Transwell chambers (Corning Life
Sciences) were washed with serum-free medium. Matrigel at an
appropriate dilution was added to the polycarbonate membrane of the
Transwell to make an artificial basement membrane by which the
chamber was divided into upper and lower chambers. A total of
2×105 cells in 200 μl of serum-free DMEM were inoculated
into the upper chamber of the Transwell invasion system, and 500 μl
DMEM containing 10% FBS was added to the lower chamber. After
incubation for 24 h, the cells on the upper side of the basement
membrane were removed with a sterile cotton swab, and the cells
that invaded to the lower side of the basement membrane were
stained with crystal violet. The cells passing through the
Transwell polycarbonate membrane were counted under a Leica
microscope. The number of cells represent the cell invasive
capability. Six random high power fields were selected for each
sample, and the experiment was repeated 3 times.
Results
CtBP1 is upregulated in HCC and is
inversely associated with E-cadherin expression
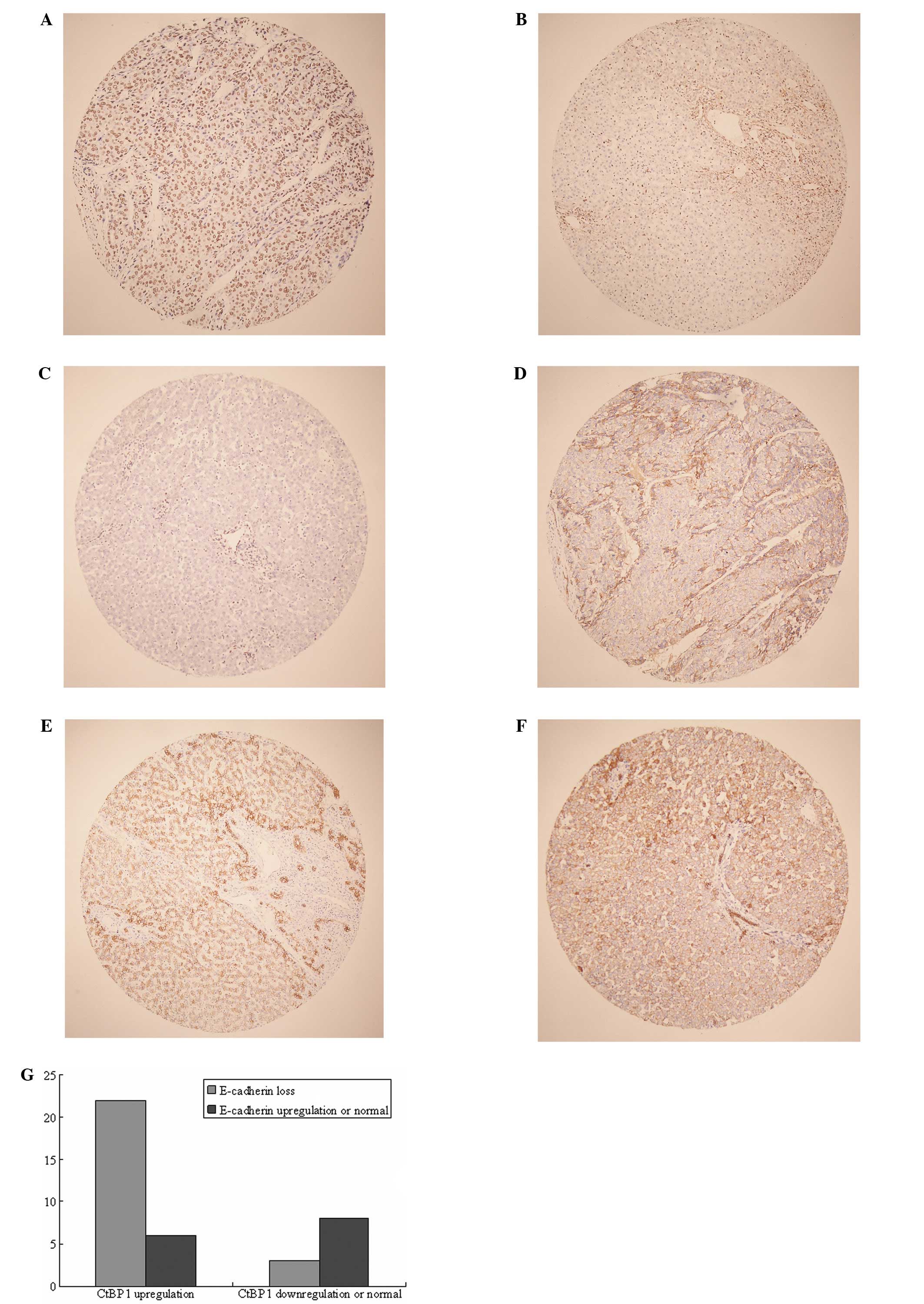
CtBP1 was overexpressed in the HCC tumors when
compared with that in the non-malignant paired adjacent tissues and
normal liver tissues of the non-malignant patients (Fig. 1A-C). In contrast to CtBP1, E-caherin
was inversely downregulated in the HCC tumor tissues (Fig. 1D-F). Among the 28 cases exhibiting
CtBP1 upregulation, 22 showed E-cadherin loss or decreased
expression, and 3 exhibited upregulated and 3 normal expression
(Fig. 1G). Notably, in the
specimens with low CtBP1 expression, 8 out of 11 cases showed
reciprocal upregulation or normal expression of E-cadherin
(Fig. 1G, right).
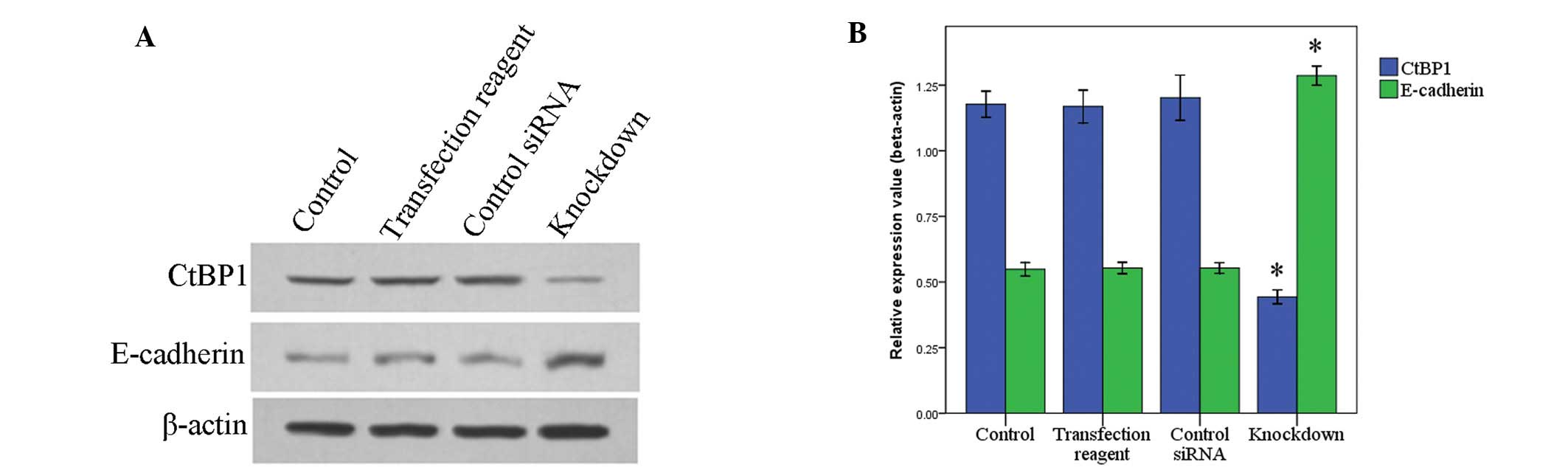
Western blotting showed that CtBP1 was overexpressed
in 8 cases among the 10 cases of HCC tissues as compared with the
paired non-tumor tissues (Fig. 2).
Among these 8 cases, 6 cases showed E-cadherin downregulation
(Fig. 2).
CtBP1 knockdown by siRNA results in
increased E-cadherin expression in HepG2 cells and decreased
invasive ability
CtBP1 was knocked down by siRNA transfection. In
accord with the reduction in CtBP1, E-cadherin was inversely
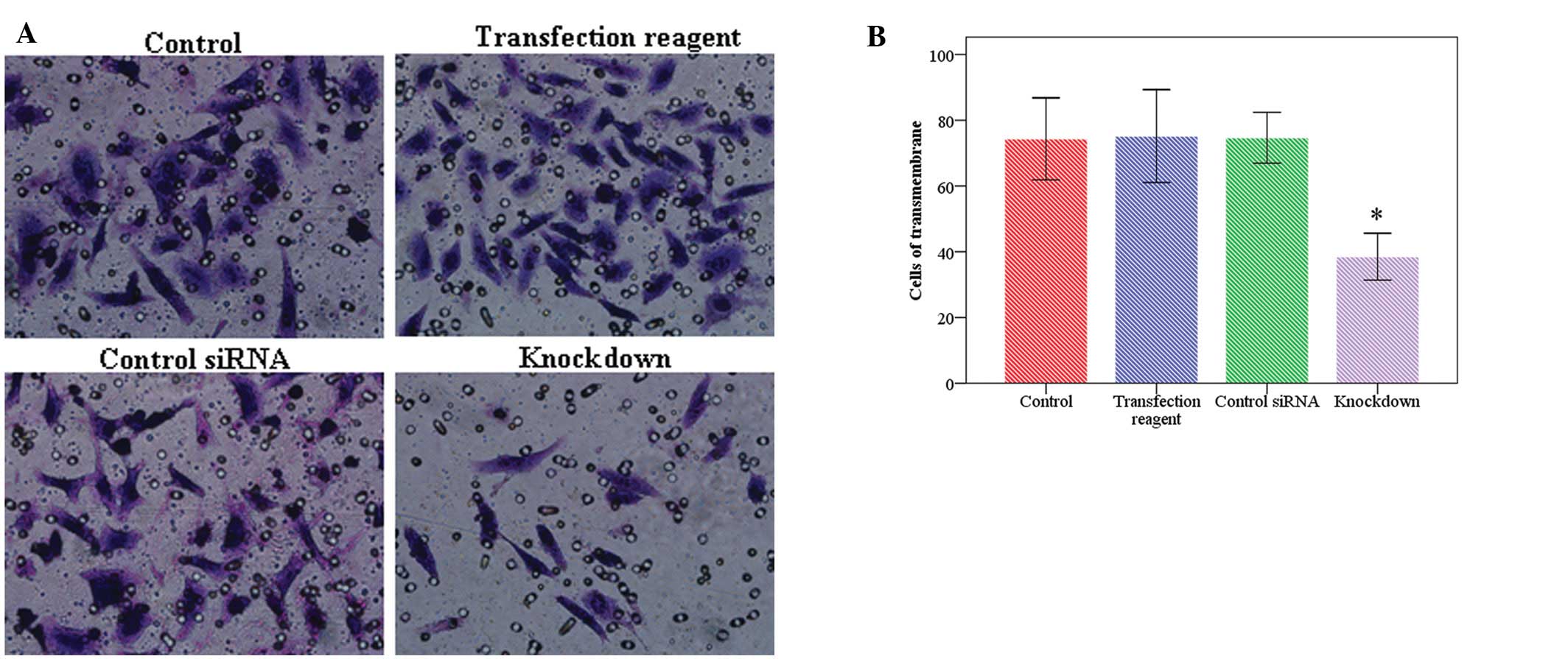
restored (Fig. 3). Transwell assay
showed that following CtBP1 knockdown, the number of invasive cells
was significantly less (38.7±9.2) than the number in the control
cells (73.8±8.1), or the cells treated with transfection reagents
only (72.5±12.9) or with control siRNA (71.8±12.5), thereby
indicating that cell invasive activity was deceased by reduction of
CtBP1 (Fig. 4A and B).
CtBP1 knockdown inhibits cell growth
through apoptosis instead of cell cycle arrest
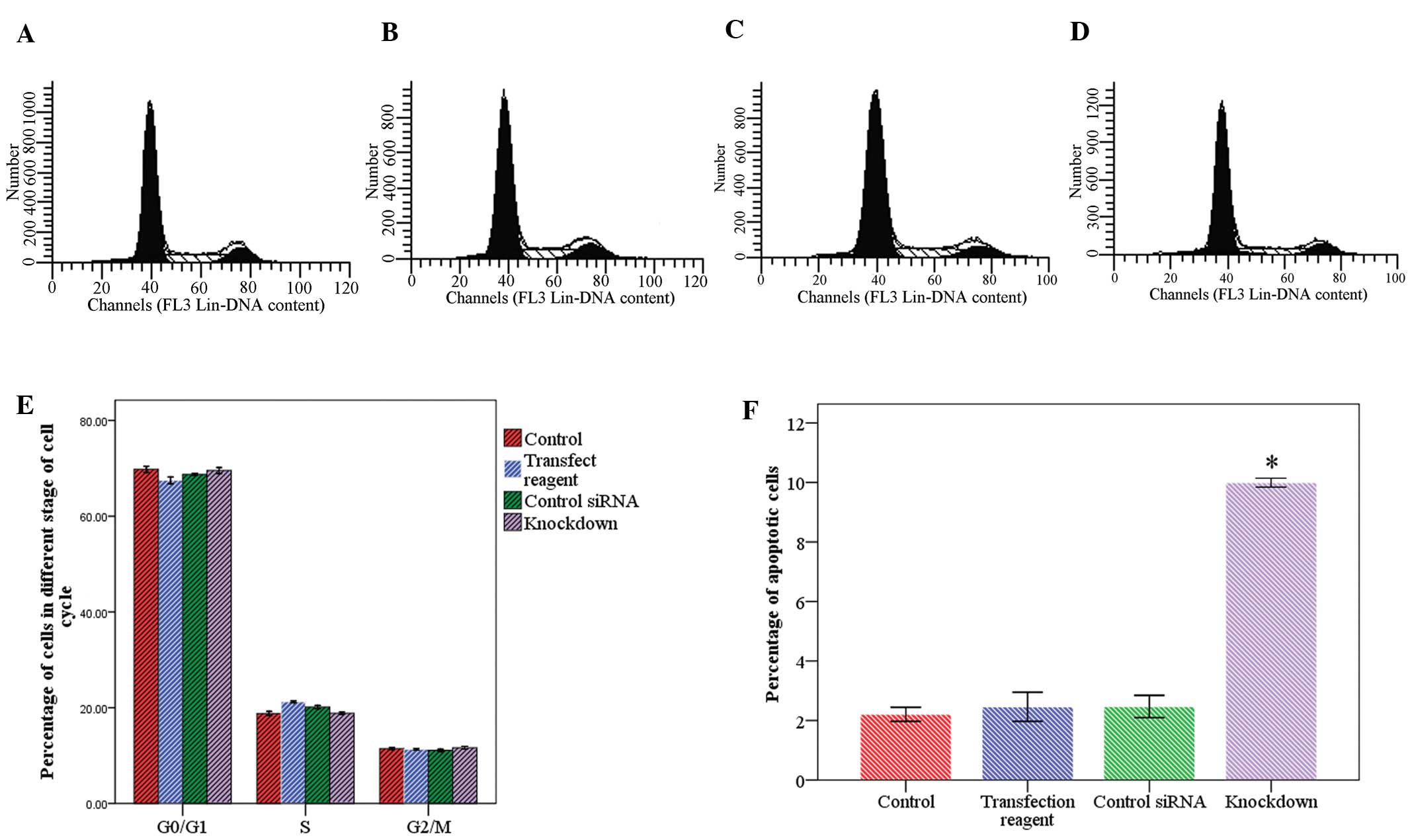
Following CtBP1 knockdown, cell growth was almost
completely inhibited (Fig. 5). Flow
cytometric data showed that the cell distribution in the cell cycle
stages did not differ markedly between the CtBP1 knockdown and the
control cells (Fig. 6E). In
contrast, the precentage of apoptotic cells increased significantly
in the knockdown group (Fig. 6F).
The percentage of apoptotic cells was 2.2±0.2, 2.5±0.3, 2.5±0.3 and
9.9±0.2 in the control cells, cells treated with transfection
reagent only, cells transfected with control siRNA and cells
transfected with CtBP1 siRNA, respectively.
Discussion
The CtBP family proteins play a modulatory role in
several essential cellular processes. Vertebrate genomes code for
two related proteins, CtBP1 and CtBP2, which function as
transcriptional corepressors (18).
CtBP1 acts as a tumor suppressor by binding to E1A resulting in
restraint of tumorigenesis (19,20).
E1A introduced into cancer cell lines was found to reverse EMT by
antagonizing the activity of CtBP1, resulting in the expression of
several epithelial genes (21,22).
CtBP1 functions as an antagonist of the epithelial phenotype,
repressing the gene expression of epithelial cell adhesion
molecules such as E-cadherin, desmoglein-2 and plakoglobin
(23). Thus, CtBP1-mediated
repression of adhesion molecules promotes EMT, a process involved
in tumorigenesis and tumor progression which contributes to motile
and invasive phenotypes, and resistance to apoptosis (24).
We found that the CtBP1 protein was upregulated in
HCC when compared with the paired non-tumor tissues, and was
correlated to the loss of E-cadherin. To further confirm the role
of CtBP1 in malignant progression of HCC, we employed siRNA to
knock down the CtBP1 gene in HepG2 cells, a human HCC cell line
highly expressing CtBP1 protein. Knockdown of CtBP1 significantly
elevated the expression of the epithelial adhesion molecule
E-cadherin and reduced the invasive capacity of HepG2 cells. These
data were consistent with the data obtained from the human HCC
samples. The results from clinical samples and the cultured HepG2
cells imply that CtBP1 is a a repressor of E-cadherin expression
and hence is a contributor of EMT in HCC.
Cell survival data showed that knockdown of CtBP1
inhibited the growth of HepG2 cells through apoptosis instead of
cell cycle arrest. This resulted from restored E-cadherin protein
in the HepG2 cells. In fact, restoration of E-cadherin has been
shown to sensitize human melanoma cells to drug-induced apoptosis
(25). Similarly, restoration of
E-cadherin expression in pancreatic cancer cells has been shown to
inhibit growth by induction of apoptosis (26). How CtBP1 functions in apoptosis in
HCC requires further investigation.
In summary, CtBP1 upregulation and the resulting
E-cadherin downregulation were correlated with the progression of
human HCC by increased proliferation and increased invasion,
characteristic of the features of EMT induction which contribute to
cell proliferation and invasion. CtBP1 may constitute a potential
novel therapeutic target for human HCC.
Acknowledgements
Supported by the Program for Innovative Research
Team of Guilin Medical University.
References
|
1
|
Clavien PA, Lesurtel M, Bossuyt PM, et al:
Recommendations for liver transplantation for hepatocellular
carcinoma: an international consensus conference report. Lancet
Oncol. 13:e11–e22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schrader J, Gordon-Walker TT, Aucott RL,
et al: Matrix stiffness modulates proliferation, chemotherapeutic
response, and dormancy in hepatocellular carcinoma cells.
Hepatology. 53:1192–1205. 2011. View Article : Google Scholar
|
|
4
|
Osada T, Sakamoto M, Ino Y, et al:
E-cadherin is involved in the intrahepatic metastasis of
hepatocellular carcinoma. Hepatology. 24:1460–1467. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wei Y, Van Nhieu JT, Prigent S,
Srivatanakul P, Tiollais P and Buendia MA: Altered expression of
E-cadherin in hepatocellular carcinoma: correlations with genetic
alterations, β-catenin expression, and clinical features.
Hepatology. 36:692–701. 2002.PubMed/NCBI
|
|
6
|
Marsit CJ, Posner MR, McClean MD and
Kelsey KT: Hypermethylation of E-cadherin is an independent
predictor of improved survival in head and neck squamous cell
carcinoma. Cancer. 113:1566–1571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maeda G, Chiba T, Aoba T and Imai K:
Epigenetic inactivation of E-cadherin by promoter hypermethylation
in oral carcinoma cells. Odontology. 95:24–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiao W, Miyazaki K and Kitajima Y: Inverse
correlation between E-cadherin and Snail expression in
hepatocellular carcinoma cell lines in vitro and in vivo. Br J
Cancer. 86:98–101. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Evans AJ, Russell RC, Roche O, et al: VHL
promotes E2 box-dependent E-cadherin transcription by HIF-mediated
regulation of SIP1 and snail. Mol Cell Biol. 27:157–169. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohshima K, Haraoka S, Yoshioka S, et al:
Chromosome 16q deletion and loss of E-cadherin expression in
Hodgkin and Reed-Sternberg cells. Int J Cancer. 92:678–682. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pan Y, Matsuyama H, Wang N, et al:
Chromosome 16q24 deletion and decreased E-cadherin expression:
possible association with metastatic potential in prostate cancer.
Prostate. 36:31–38. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu YC, Shen CY, Wu HS, et al: Mechanisms
inactivating the gene for E-cadherin in sporadic gastric
carcinomas. World J Gastroenterol. 12:2168–2173. 2006.PubMed/NCBI
|
|
13
|
Deng Y, Deng H, Liu J, et al:
Transcriptional down-regulation of Brca1 and E-cadherin by CtBP1 in
breast cancer. Mol Carcinog. 51:500–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moreno-Bueno G, Portillo F and Cano A:
Transcriptional regulation of cell polarity in EMT and cancer.
Oncogene. 27:6958–6969. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pena C, Garcia JM, Garcia V, et al: The
expression levels of the transcriptional regulators p300 and CtBP
modulate the correlations between SNAIL, ZEB1, E-cadherin and
vitamin D receptor in human colon carcinomas. Int J Cancer.
119:2098–2104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu W, Dong J, Huang Z, Guo D, Liu Y and
Shi S: Comparison of four current staging systems for Chinese
patients with hepatocellular carcinoma undergoing curative
resection: Okuda, CLIP, TNM and CUPI. J Gastroenterol Hepatol.
23:1874–1878. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gotte M, Kersting C, Radke I, Kiesel L and
Wulfing P: An expression signature of syndecan-1 (CD138),
E-cadherin and c-met is associated with factors of angiogenesis and
lymphangiogenesis in ductal breast carcinoma in situ. Breast Cancer
Res. 9:R82007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chinnadurai G: CtBP, an unconventional
transcriptional corepressor in development and oncogenesis. Mol
Cell. 9:213–224. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schaeper U, Subramanian T, Lim L, Boyd JM
and Chinnadurai G: Interaction between a cellular protein that
binds to the C-terminal region of adenovirus E1A (CtBP) and a novel
cellular protein is disrupted by E1A through a conserved PLDLS
motif. J Biol Chem. 273:8549–8552. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao LJ, Subramanian T, Vijayalingam S and
Chinnadurai G: PLDLS-dependent interaction of E1A with CtBP:
regulation of CtBP nuclear localization and transcriptional
functions. Oncogene. 26:7544–7551. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frisch SM: Antioncogenic effect of
adenovirus E1A in human tumor cells. Proc Natl Acad Sci USA.
88:9077–9081. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frisch SM: Tumor suppression activity of
adenovirus E1a protein: anoikis and the epithelial phenotype. Adv
Cancer Res. 80:39–49. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grooteclaes ML and Frisch SM: Evidence for
a function of CtBP in epithelial gene regulation and anoikis.
Oncogene. 19:3823–3828. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kippenberger S, Loitsch S, Thaci D, et al:
Restoration of E-cadherin sensitizes human melanoma cells for
apoptosis. Melanoma Res. 16:393–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lowy AM, Knight J and Groden J:
Restoration of E-cadherin/β-catenin expression in pancreatic cancer
cells inhibits growth by induction of apoptosis. Surgery.
132:141–148. 2002.
|