Introduction
Apoptosis is a form of programmed cell death defined
by a characteristic set of morphological and biochemical changes
that occurs during embryonic development and tissue homeostasis and
involves eliminating unwanted or damaged cells from eukaryotic
organisms. Apoptosis is mediated by either the extrinsic or
intrinsic pathway, and both pathways converge on the activation of
effector caspases, a family of cysteine-dependent
aspartate-directed proteases, by initiator caspases (1,2). In
the extrinsic apoptosis pathway, interaction between ligands and
death receptors initiates the pathway at the plasma membrane and
subsequently activates initiator caspases such as caspase-8.
Caspase-8 can directly activate downstream effector caspases,
including caspase-3 (3,4). In the intrinsic apoptosis pathway, the
mitochondrial membrane potential (MMP) is altered, and the B cell
lymphoma 2 (Bcl-2) family of proteins plays a central role in
regulating cell death. This process leads to the release of
pro-apoptotic proteins from the mitochondrial intermembrane space,
induces activation of caspase-9, and subsequently activates
effector caspases including caspase-3 (5–7). After
caspase-3 is activated, several specific substrates are cleaved or
other cellular proteins are activated, eventually leading to
apoptosis (8–10). In some cells, caspase-8 also
mediates the intrinsic pathway via cleavage of the pro-apoptotic
Bid, a BH3-only protein (2,11,12).
Because aberrant regulation of apoptosis is representative of
cancer and many therapeutic agents eliminate tumor cells by
inducing apoptotic cell death, induction of apoptotic cell death is
an important mechanism of action of potential anticancer drugs.
Natural products, including plants, microorganisms,
and marine organisms, are valuable sources of therapeutic
compounds. This is particularly evident in cancer therapy, in which
more than 60% of approved drugs are of natural origin (13). Therefore, a new natural source of
anticancer compounds that have relatively fewer side-effects would
be a valuable tool in cancer therapy. In particular, traditional
medicine has been the focus of scientific efforts to discover novel
anticancer agents. Although Oriental medicine has been used
clinically in Asia, including Korea, for thousands of years as an
important alternative remedy for cancer treatment, the molecular
mechanisms underlying the effects of these natural products
obtained from the extracts of medicinal plants are still unclear.
Dendropanax morbifera Leveille is a subtropical,
broad-leaved evergreen tree belonging to the family Araliaceae
endemic to southwestern Korea. The roots, leaves, seeds and stems
of this plant have been widely used in Korea as folk medicine for
treating headache, infectious diseases, skin diseases, and other
maladies. Recently, D. morbifera has been shown to possess
potent anti-diabetic (14),
anti-atherogenic (15),
anti-plasmodial (16) and
anti-complement activity (17,18).
In addition, studies on the anti-inflammatory and anticancer
effects of components isolated from this plant also have been
reported (19–21); however, the molecular mechanisms of
its pro-apoptotic action in cancer cells are not fully
understood.
Thus, in this study, as part of our ongoing
screening program to evaluate the anticancer potential of D.
morbifera, we investigated the pro-apoptotic property of an
ethanol extract of D. morbifera stem bark and the
responsible underlying molecular mechanisms of action in human
leukemia U937 cells.
Materials and methods
Reagents
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT), propidium iodide (PI), 4,6-diamidino-2-phenylindole
(DAPI), and
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide
(JC-1) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Fetal bovine serum (FBS), Roswell Park Memorial Institute
(RPMI)-1640 medium, and penicillin-streptomycin were obtained from
Gibco-BRL (Grand Island, NY, USA). Caspase activity assay kits were
obtained from R&D Systems (Minneapolis, MN, USA). Extracellular
signal-regulated kinase (ERK)-specific inhibitor, PD98059, c-Jun
N-terminal kinase (JNK)-specific inhibitor, SP600125, p38
mitogen-activated protein kinase (MAPK)-specific inhibitor,
SB203580, and Akt inhibitor, LY290042, were purchased from
Calbiochem (San Diego, CA, USA). The DNA staining kit (Cycletest™
Plus Kit) and the enhanced chemiluminescence (ECL) kit were
purchased from Becton-Dickinson (San Jose, CA, USA) and Amersham
Pharmacia Biotech (Arlington Heights, IL, USA), respectively.
Antibodies
Antibodies specific for Fas, Fas ligand (FasL),
tumor necrosis factor-related apoptosis-inducing ligand (TRAIL),
Bcl-2, Bcl-xL, Bax, Bad, Bid, X-linked inhibitor of apoptosis
protein (XIAP), inhibitor of apoptosis protein (IAP)-1, IAP-2,
survivin, caspase-3, -8 and -9, poly(ADP-ribose) polymerases
(PARP), β-catenin, and phospholipase Cγ1 (PLCγ1) were obtained from
Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibodies
specific for ERK, JNK, p38 MAPK, Akt, phospho (p)-ERK, p-JNK, p-38
MAPK and p-Akt were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Antibodies specific for death receptor (DR)4
and DR5 were obtained from Calbiochem. Anti-actin antibody was
obtained from Sigma-Aldrich.
Preparation of an ethanol extract of D.
morbifera stem bark
The stem bark of D. morbifera used in this
research was obtained from Dongeui University Oriental Hospital
(Busan, Korea). To prepare the ethanol extract of D.
morbifera stem bark (EEDM), the dried stem was extracted twice
with 70% ethanol (with 24 h reflux), and the extract was then
concentrated under reduced pressure. The decoction was filtered,
lyophilized, and stored at 4°C until use. The dried extract yield
from the starting crude materials was ~17.8% (w/w). The lyophilized
powder was dissolved in dimethyl sulfoxide (DMSO), filtered through
a 0.22-μm syringe filter to create a stock solution, and then
adjusted to final concentrations using complete RPMI-1640
medium.
Cell culture and MTT assay
U937 human leukemia cells, Chang liver cells, and
WI-38 lung fibroblast cells were purchased from the American Type
Culture Collection (Rockville, MD, USA) and maintained at 37°C in
95% humidified air and 5% CO2 in RPMI-1640 medium that
was supplemented with 10% FBS and 1% penicillin/streptomycin.
Inhibition of cell proliferation was determined using an MTT assay.
Briefly, cells were seeded in 6-well plates and treated with
various concentrations of EEDM for 12 and 24 h. After treatment,
the MTT working solution (0.5 mg/ml) was added to each well and
incubated continuously at 37°C for 3 h. The culture supernatant was
removed from the wells, and DMSO was added to completely dissolve
the formazan crystals. The absorbance of each well was measured at
a 540 nm wavelength with an ELISA reader (Molecular Devices,
Sunnyvale, CA, USA). Morphological observations of cultured cells
were carried out with an inverted microscope (Carl Zeiss,
Germany).
Nuclear staining with DAPI
For nuclear DAPI staining, the cells were harvested
and washed with ice-cold phosphate-buffered saline (PBS) and fixed
with 3.7% paraformaldehyde (Sigma-Aldrich) in PBS for 10 min at
room temperature. The fixed cells were washed with PBS and stained
with 2.5 μg/ml DAPI solution for 10 min at room temperature. The
cells were then washed twice with PBS and analyzed with a
fluorescence microscope (Carl Zeiss).
Detection of DNA fragmentation with gel
electrophoresis
Following EEDM treatment, the cells were lysed in a
buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA,
and 0.5% Triton X-100 for 1 h at room temperature. The lysates were
vortexed and cleared by centrifugation at 19,000 × g for 30 min at
4°C. A 25:24:1 (v/v/v) equal volume of neutral
phenol:chloroform:isoamyl alcohol (Sigma-Aldrich) was used to
extract DNA in the supernatant, followed by electrophoretic
analysis on 1.5% agarose gels containing 0.1 μg/ml ethidium bromide
(EtBr; Sigma-Aldrich).
Flow cytometric analysis for measuring
the sub-G1 phase and MMP values
The cells were collected, washed with cold PBS, and
fixed in 75% ethanol at 4°C for 30 min. The DNA content of the
cells was measured using a DNA staining kit according to the
manufacturer’s instructions. Then flow cytometric analyses were
carried out using a flow cytometer, and the relative DNA content
was determined using CellQuest software, with the presence of red
fluorescence as a marker. The MMP values were determined with the
dual-emission potential-sensitive probe, JC-1. The cells were
collected and incubated with 10 μM JC-1 for 20 min at 37°C in the
dark. The cells were then washed once with PBS and analyzed with a
flow cytometer (22).
Protein extraction and western blot
analysis
Cells were lysed for 30 min with lysis buffer (20 mM
sucrose, 1 mM EDTA, 20 μM Tris-Cl, pH 7.2, 1 mM DTT, 10 mM KCl, 1.5
mM MgCl2, 5 μg/ml pepstatin A, 10 μg/ml leupeptin, and 2
μg/ml aprotinin) to prepare the total protein. The supernatants
were collected, and the protein concentrations were determined
using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA). For
western blot analysis, an equal amount of protein was subjected to
electrophoresis on sodium dodecyl sulfate (SDS)-polyacrylamide gels
and transferred to a nitrocellulose membrane (Schleicher &
Schuell, Keene, NH, USA) by electroblotting. Blots were probed with
the desired antibodies for 1 h, incubated with the diluted
enzyme-linked secondary antibodies, and visualized with the ECL
solution according to the recommended procedure.
Caspase activity assay
Caspase activity was determined with colorimetric
assay kits, which use synthetic tetrapeptides [Asp-Glu-Val-Asp
(DEAD) for caspase-3, Ile-Glu-Thr-Asp (IETD) for caspase-8,
Leu-Glu-His-Asp (LEHD) for caspase-9] labeled with p-nitroaniline
(pNA). Briefly, cells were lysed in the supplied lysis buffer
according to the manufacturer’s protocol. The supernatants were
collected and incubated with the supplied reaction buffer
containing DTT and DEAD-pNA, IETD-pNA, or LEHD-pNA as the substrate
at 37°C. The reactions were measured by changes in absorbance at
405 nm using a microplate reader.
Statistical analysis
All experiments are expressed as the means ±
standard deviations (SDs) of at least 3 separate tests. The
Student’s t-test was used for single-variable comparisons, and a
p-value <0.05 was considered to indicate a statistically
significant result.
Results
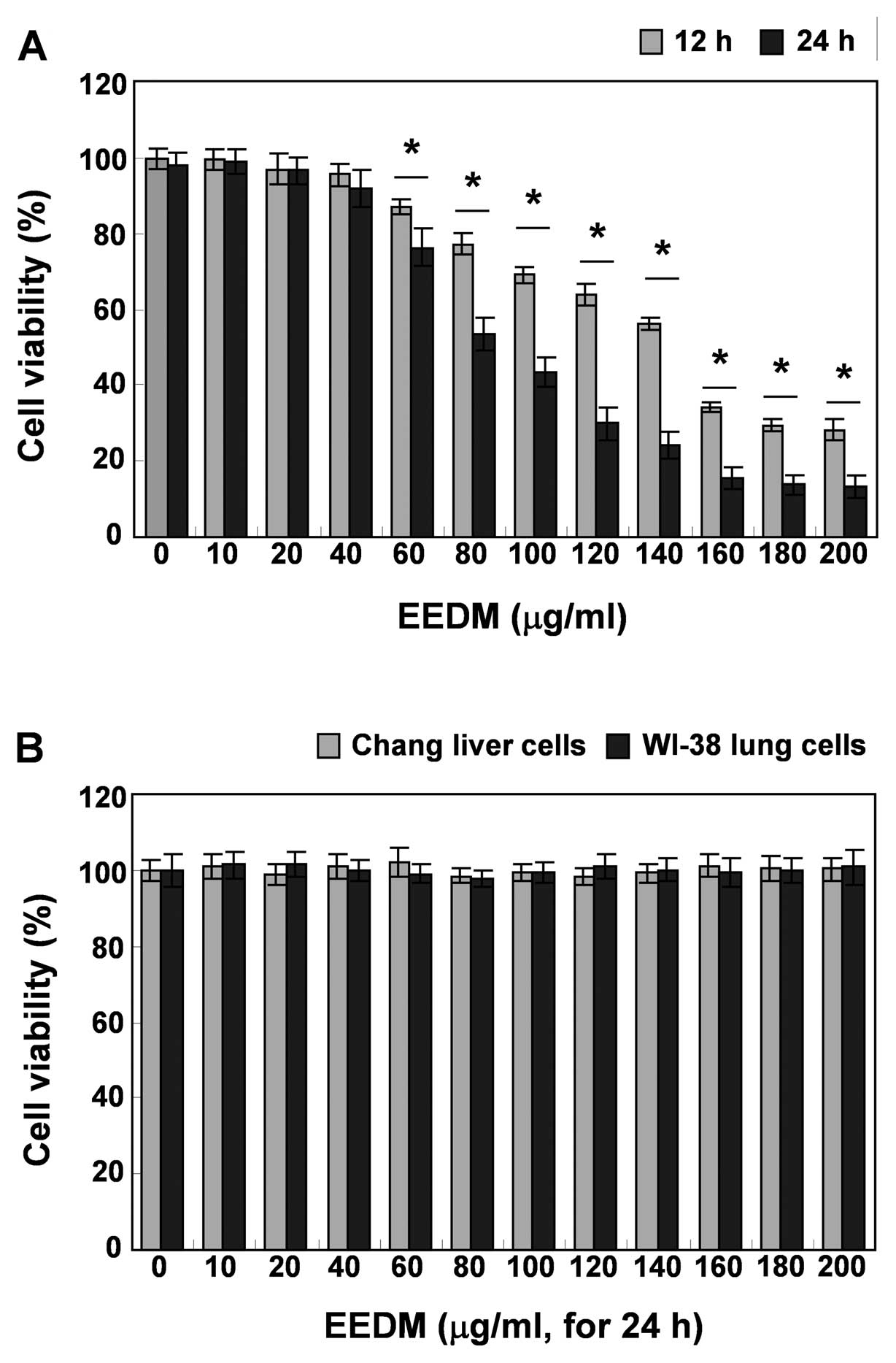
EEDM inhibits cell growth in U937
cells
To investigate whether EEDM inhibits U937 cell
growth, the cells were treated with various concentrations (0–200
μg/ml) of EEDM for 12 and 24 h, and viability was measured with an
MTT assay. Compared to the untreated control, after treatment with
EEDM, as indicated in Fig. 1A, EEDM
had a strong inhibitory effect on cell proliferation of U937 cells
in a dose- and time-dependent manner. However, EEDM had no
antiproliferative effect on Chang normal liver and WI-38
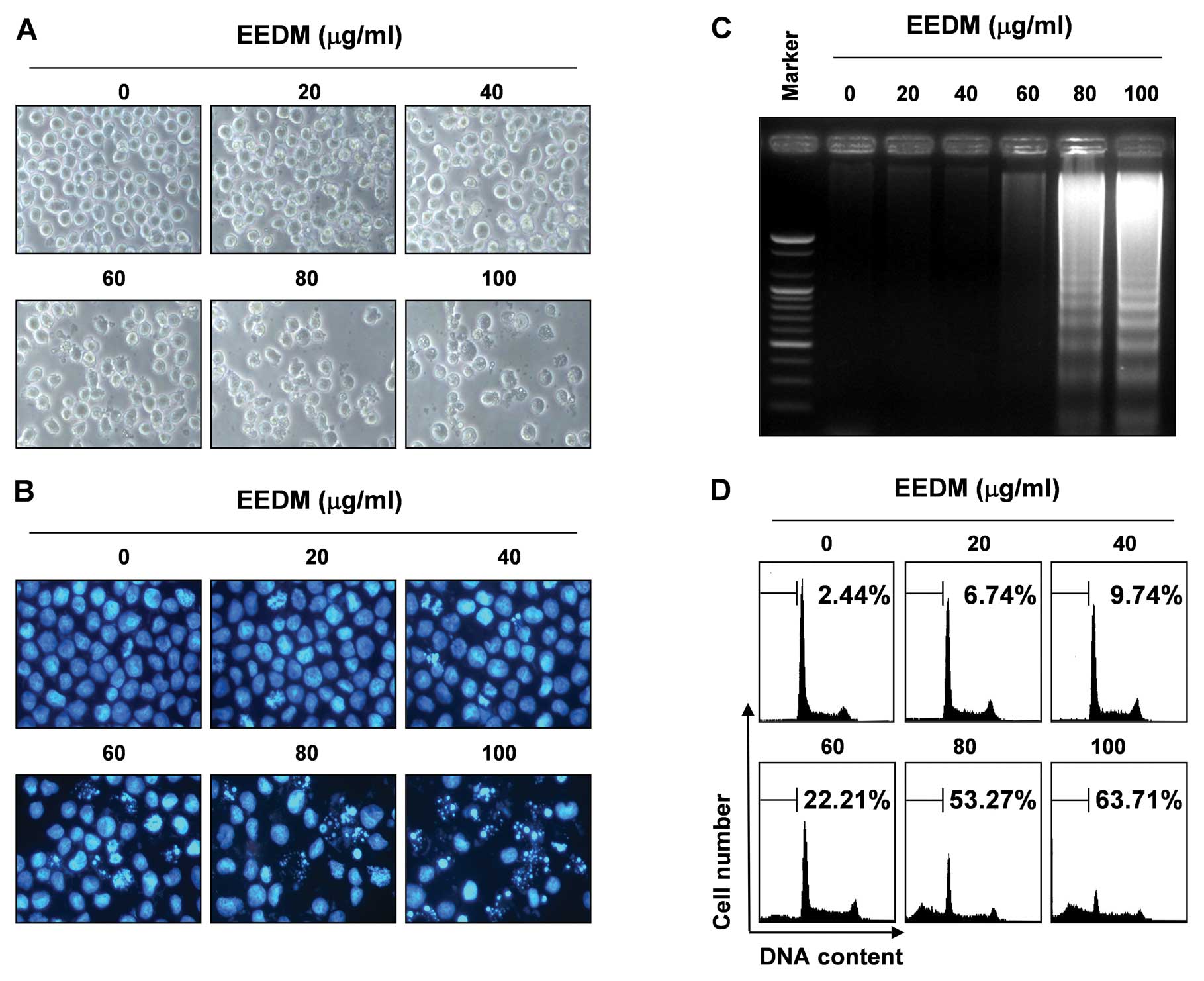
lung-derived cell lines. Direct observation using an inverted
microscope showed that numerous morphological changes occurred in
the U937 cells treated with EEDM. In particular, cell shrinkage and
cytoplasmic condensation appeared in a concentration-dependent
manner after EEDM treatment (Fig.
2A).
EEDM induces apoptosis in U937 cells
Further experiments were conducted to determine
whether the inhibition of cell viability observed in the presence
of EEDM was the result of apoptotic cell death in U937 cells. To
examine the apoptosis morphologically, the nuclei of untreated and
EEDM-treated cells were stained with DAPI solution and then
observed. Following treatment of U937 cells with various
concentrations of EEDM for 24 h, the chromatin stained with DAPI
had a characteristic condensed and fragmented appearance in a
concentration-dependent fashion (Fig.
2B). In addition, following agarose gel electrophoresis of the
DNA from cells treated with EEDM, a characteristic ladder pattern
of discontinuous DNA fragments was observed; however, DNA
fragmentation was barely detected in the control cells (Fig. 2C). We next quantified the apoptotic
dead cells using a flow cytometer. As shown in Fig. 2D, EEDM treatment resulted in a
significant increase in the number of U937 cells in the apoptotic
sub-G1 phase, and this response occurred in a
concentration-dependent manner. These results confirmed that EEDM
inhibits the proliferation of U937 cells by inducing apoptosis.
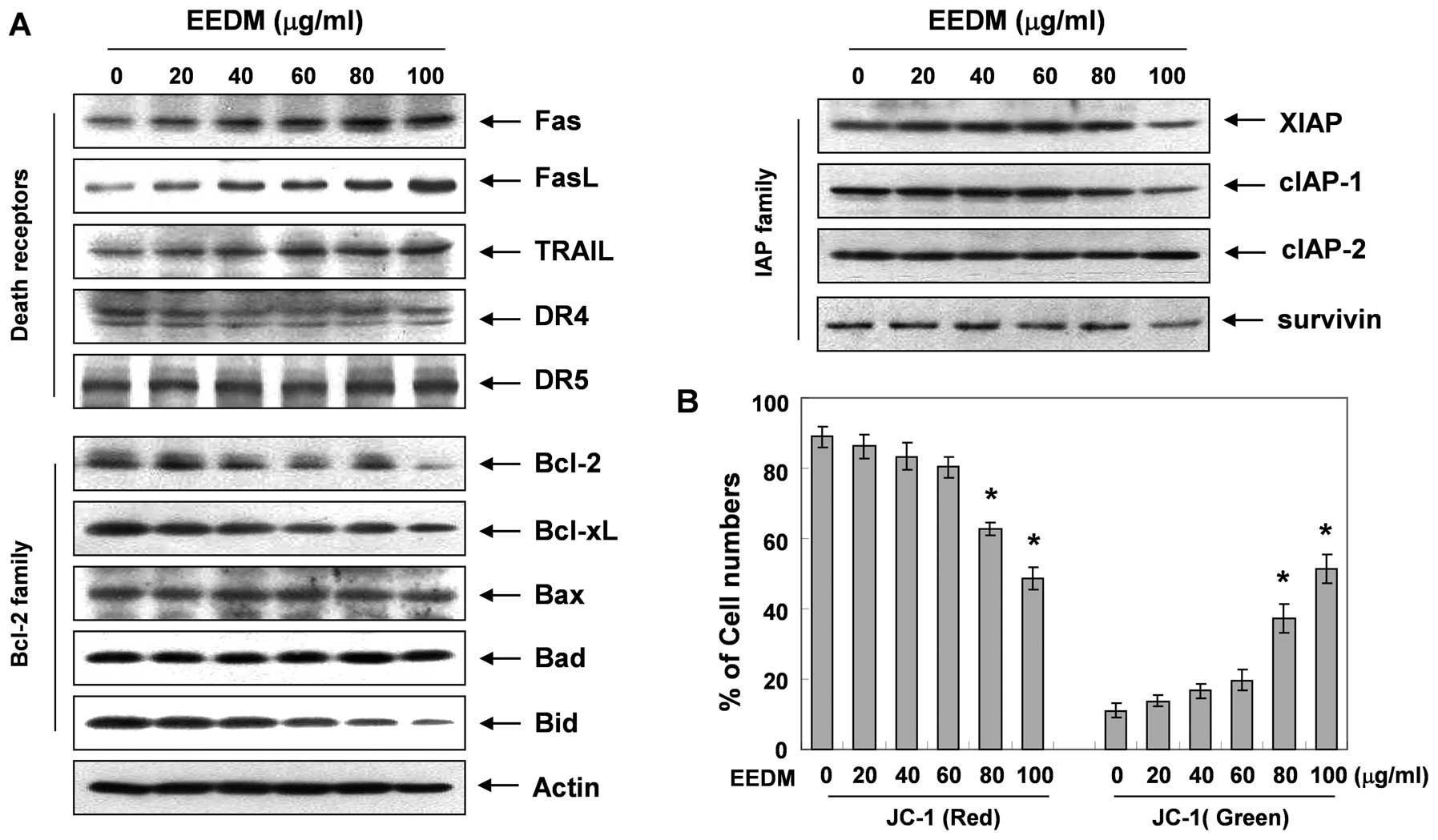
EEDM alters the expression of
apoptosis-related proteins and MMP values in U937 cells
To investigate the mechanisms underlying
EEDM-induced apoptosis, the expression level of several apoptotic
factors was analyzed. Fig. 3A
demonstrates that, after the cells were exposed to EEDM, the levels
of the death receptor-related proteins Fas, FasL and TRAIL, but not
DR4 and DR5, were concentration-dependently increased. Fig. 3A also shows that the levels of the
anti-apoptotic Bcl-2 family of proteins (Bcl-2 and Bcl-xL) were
decreased in response to EEDM; however, the levels of pro-apoptotic
Bax and Bad remained unchanged. In addition, although we did not
detect the truncated form of the pro-apoptotic protein Bid, EEDM
decreased the total form of the Bid proteins, reflecting Bid
cleavage and activation. Under these conditions, levels of the
anti-apoptotic IAP family of proteins such as XIAP, cIAP-1 and
survivin, which bind to caspases and lead to their inactivation,
were inhibited by EEDM treatment in a concentration-dependent
manner.
EEDM induces the loss of MMP
Mitochondrial membrane permeabilization, accompanied
by the collapse of the electrochemical gradient across the
mitochondrial membrane, is a key event during cellular apoptosis.
To investigate whether mitochondrial dysfunction was involved in
EEDM-induced U937 cell apoptosis, we used flow cytometric analysis
with JC-1 staining to examine the change in the MMP values after
EEDM treatment. JC-1 is a lipophilic and cationic dye that
selectively enters mitochondria. In healthy cells with high
mitochondrial potential, JC-1 forms J-aggregates with intense red
fluorescence (590 nm), whereas under apoptotic conditions, the
mitochondrial membrane potential collapses. Thus, JC-1 does not
accumulate within the mitochondria but remains in the cytoplasm in
a monomeric form showing green fluorescence (525 nm). As shown in
Fig. 3B, JC-1 fluorescence shifted
from a JC-1-red-bright/JC-1-green-bright signal in the control
cells to a JC-1-green-bright/JC-1-red-dim signal in the
EEDM-treated cells in a dose-dependent manner, indicating EEDM
induced loss of the MMP in U937 cells.
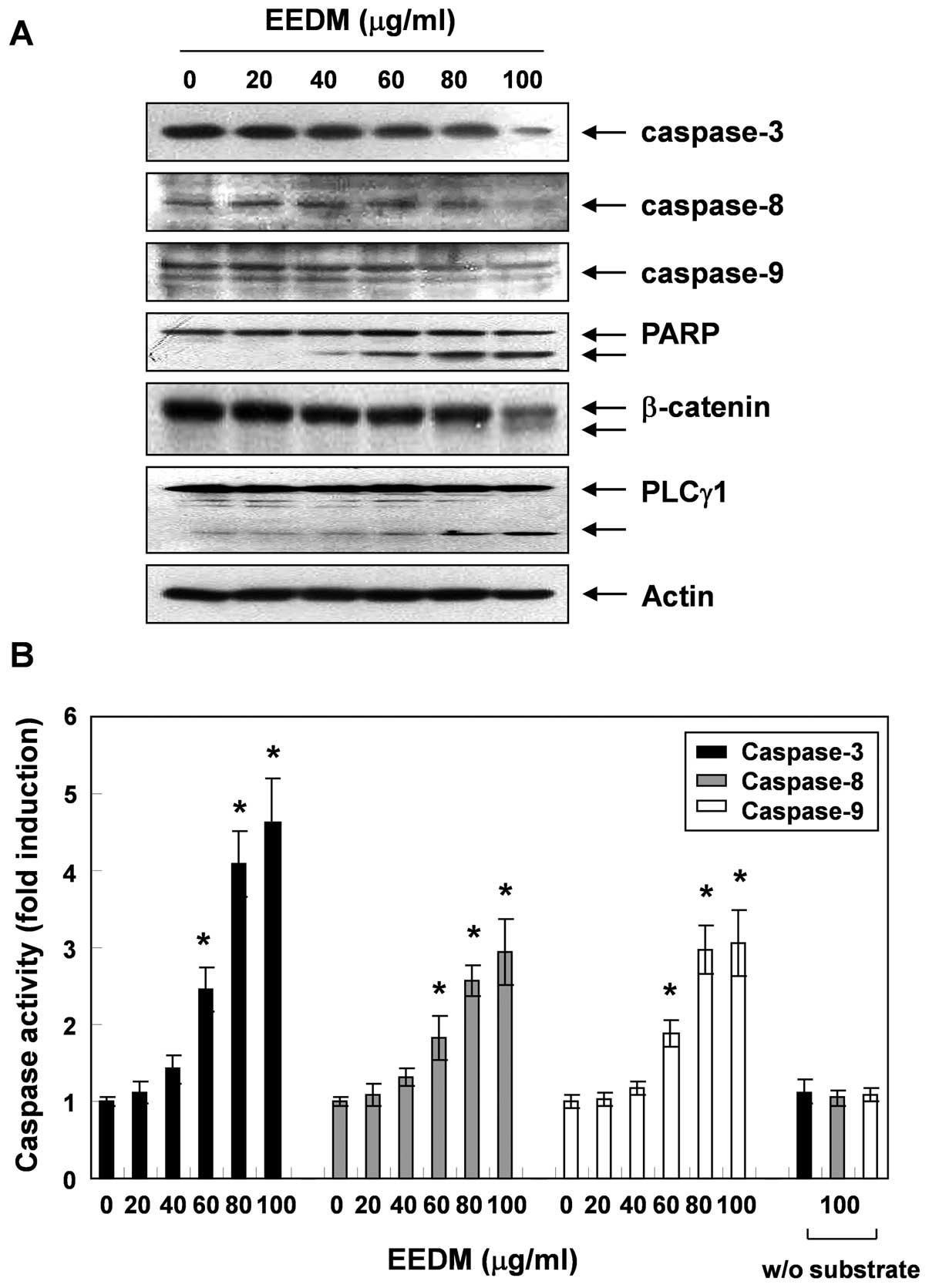
EEDM induces activation of caspases in
U937 cells
To investigate the apoptotic cascade induced by
EEDM, U937 cells were exposed to various concentrations of EEDM for
24 h, after which the expression level and activity of caspase-3,
-8 and -9 were measured. As shown in Fig. 4, the expression of pro-caspase-3, -8
and -9 decreased following EEDM treatment, which was associated
with a significant concentration-dependent increase in the activity
of these caspases compared with the untreated control cells.
Furthermore, subsequent western blot analysis revealed that EEDM
induced proteolytic degradation of PARP, β-catenin, and PLCγ1,
downstream target proteins of activated caspase-3. Cleavage
fragments of these proteins gradually increased over time.
To confirm the role of caspase activation in
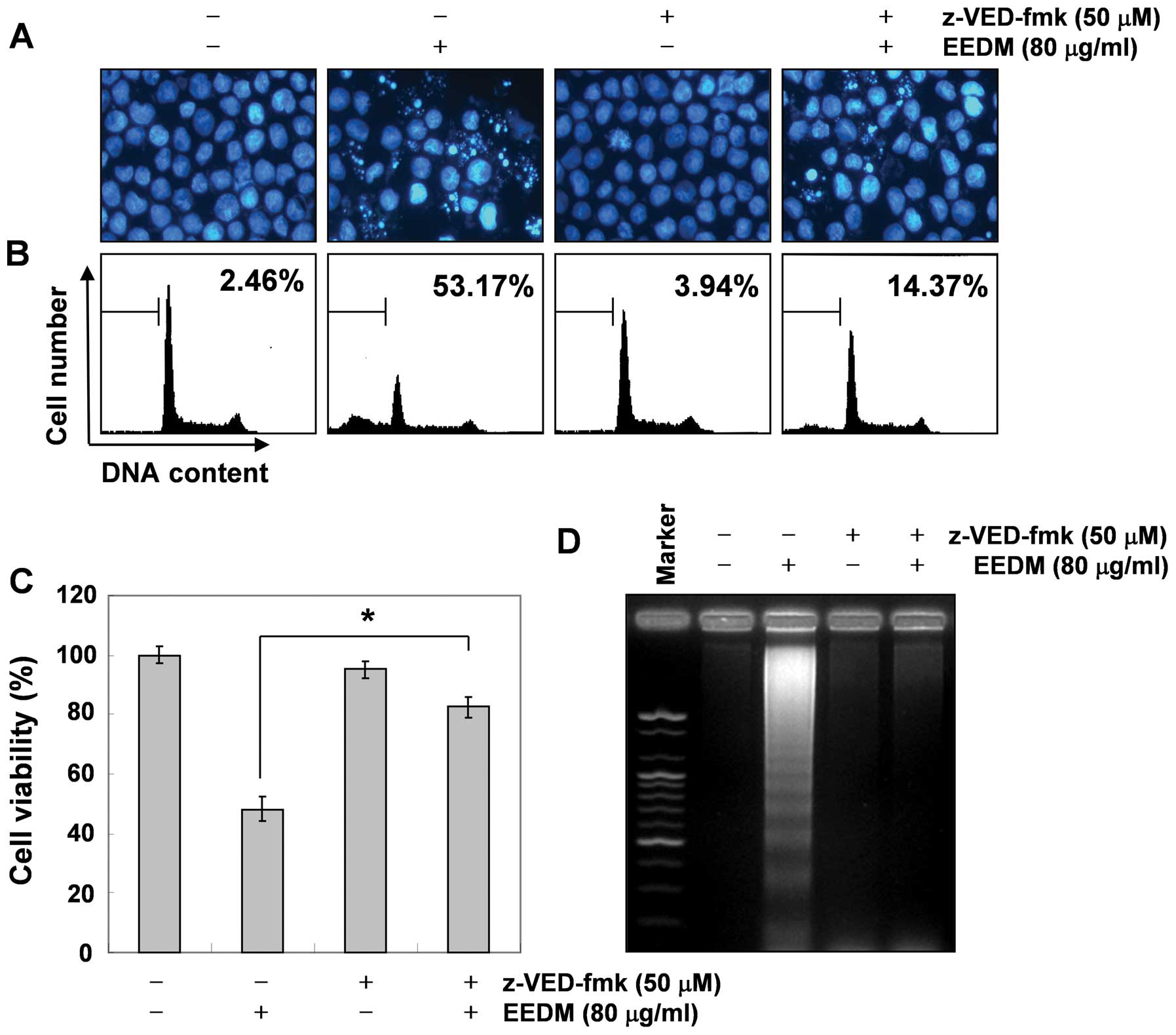
EEDM-induced apoptosis, U937 cells were exposed to a broad-spectrum
caspase inhibitor, z-VED-fmk, for 2 h before EEDM was added. As
shown in Fig. 5, pretreatment with
z-VED-fmk significantly attenuated EEDM-induced chromatin
condensation, the formation of apoptotic bodies, and DNA
fragmentation and restored the increased apoptosis. Furthermore,
z-VED-fmk alone did not affect cell viability but reversed the
anti-proliferative activity of EEDM (Fig. 5C). These data clearly indicate that
EEDM-induced apoptosis in U937 cells was mediated by caspase
activation.
Effects of EEDM on the MAPK and PI3K/Akt
signaling pathways in U937 cells
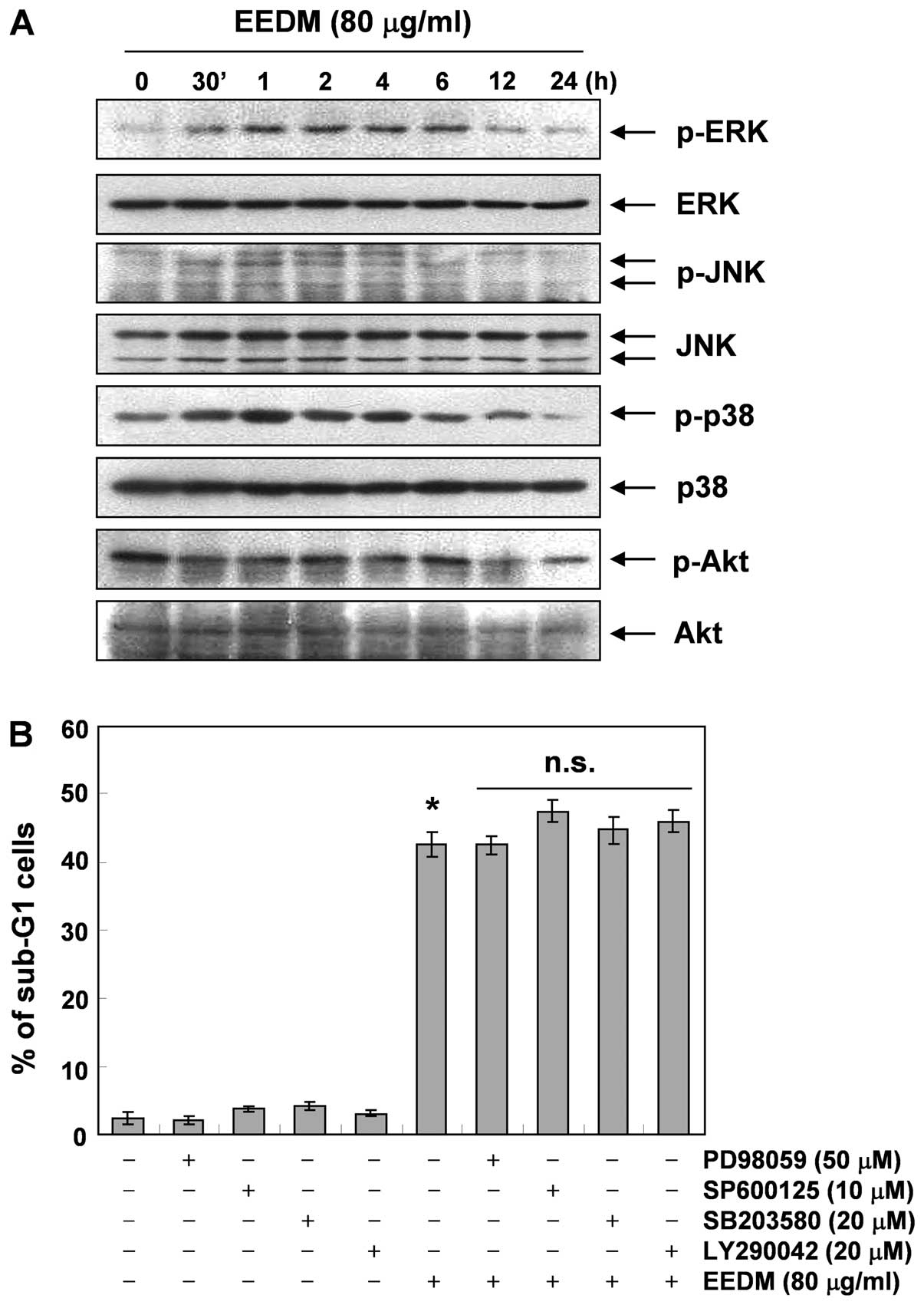
To address whether the activation of the MAPK
signaling pathway is involved in EEDM-induced apoptosis, we
investigated whether members of the MAPK family of proteins are
activated during EEDM-induced apoptosis. As Fig. 6A demonstrates, stimulation of U937
cells with EEDM led to rapid phosphorylation of MAPKs, including
p38 MAPK, ERK and JNK, with peak levels of each phospho-MAPK
observed 0.5 to 6 h after EEDM was added. However, the specific
inhibitors of each protein (PD98059, ERK-specific inhibitor;
SP600125, JNK-specific inhibitor and SB203580, p38 MAPK-specific
inhibitor) did not block EEDM-induced cell death (Fig. 6B), indicating that EEDM-induced
apoptosis is independent of the MAPK pathway. We next examined the
involvement of the phosphatidylinositol-3-kinase (PI3K)/Akt pathway
in EEDM-induced U937 apoptosis. As shown in Fig. 6A, the levels of phosphorylated Akt,
a downstream kinase of PI3K, were time-dependently decreased in
response to EEDM, while the total Akt protein levels remained
constant during EEDM treatment. Similar to MAPK inhibitors,
combination treatment with EEDM and LY294002, a specific inhibitor
of PI3K, did not affect EEDM-induced apoptosis. These results
suggest that EEDM-induced apoptosis is not associated with
downregulation of the Akt signaling pathway.
Discussion
The induction of apoptotic cell death is a promising
emerging strategy for the treatment of cancer, and many studies
have screened for pro-apoptotic natural compounds. Moreover,
natural products, such as traditional herbal medicines, have
relatively fewer side-effects compared to modern chemotherapeutics
and have long been used clinically to treat various diseases,
including cancer. Therefore, discovering naturally occurring agents
with pro-apoptotic activity in cancer cells is a promising approach
for developing novel cancer chemotherapies. Although the findings
from recent studies have demonstrated that the components of D.
morbifera have diverse pharmacological properties, including
anti-inflammatory and anticancer effects (14–21),
the mode of action of its antitumor property is still largely
unknown. Therefore, before the extract of D. morbifera can
be further developed as an anticancer agent, the antitumor activity
and the underlying molecular mechanism must be elucidated. The
results of this study clearly demonstrate that EEDM inhibited U937
leukemic cell growth, but not normal cells, by induction of
apoptosis. After the cells were exposed to EEDM, chromatin
condensation, apoptotic bodies and DNA fragmentation were clearly
observed. Furthermore, the data obtained from flow cytometric
analysis after PI staining confirmed EEDM-induced apoptosis in U937
cells (Figs. 1 and 2).
Apoptosis manifests as two major pathways that
initiate apoptosis designated as intrinsic (i.e. mitochondrial) and
extrinsic (i.e. death receptor) pathways. The intrinsic pathway is
controlled by the Bcl-2 family proteins, which play important
regulatory roles in apoptosis, by either inhibiting or promoting
apoptosis (1,2). Heterodimerization between the pro-
(such as Bcl-2 and Bcl-xL) and anti-apoptotic proteins (such as Bax
and Bad) of this family and the balance of Bcl-2 family members may
determine the susceptibility to a given apoptotic stimulus and cell
fate (23). In the extrinsic
pathway, the roles of death legends and their receptors are
well-characterized. When death legends engage death receptors, a
protein complex forms, and caspase-8 activation leads to apoptosis
(2,12). Apoptosis may also be inhibited by
various proteins, including members of the IAP family, which are
largely overexpressed by most tumors. These proteins promote tumor
cell survival due to direct inhibition by binding to several
caspases after a wide variety of apoptotic stimuli elicited via the
intrinsic and extrinsic pathways (24,25).
In this study, EEDM did not alter the expression of pro-apoptotic
Bax and Bad in U937 cells but downregulated the expression of
anti-apoptotic Bcl-2 and Bcl-xL, resulting in an increase in the
ratio of Bax/Bad:Bcl-2 and Bax/Bad:Bcl-xL. Additionally, an
increase in the Fas, FasL and TRAIL proteins, crucial members of
the extrinsic pathway, was observed in the EEDM-treated U937 cells
(Fig. 3). Our results also revealed
that EEDM-induced apoptosis was related to downregulation of IAP
family proteins such as XIAP, cIAP-1 and survivin (Fig. 3). These results indicate that
EEDM-induced apoptosis correlates strongly with the downregulation
of anti-apoptotic proteins.
Cellular demolition in apoptosis is carried out by
caspases, which are key executioners of apoptosis and play
important roles in drug-induced apoptosis in a large variety of
cancer cells. Two members of this group of enzymes are known as
‘initiator’ and ‘effector’ caspases (5,7). Upon
activation, extrinsic and intrinsic caspases such as caspase-8 and
-9, respectively, trigger the proteolytic activation of executioner
caspases such as caspase-3 and -7. In general, caspase-3 is the
common effector for most apoptotic pathways, and its active form is
responsible for the cleavage and breakdown of several cellular
components related to DNA repair and regulation. Once activated,
caspase-3 also cleaves numerous important cellular substrates and
causes membrane blebbing, disassembly of the cell structure, and
DNA fragmentation, which eventually lead to cell death (8–10). In
addition, activated caspase-8 can convert Bid to truncated Bid
(tBid), leading to mitochondrial depolarization and the release of
many apoptogenic proteins, such as cytochrome c, from the
mitochondrial intermembrane space after translocation of tBid to
the mitochondria (11). This leads
finally to the activation of caspase-3 and induction of apoptosis
via a complex of apoptotic protease activating factor-1 (Apaf-1),
pro-caspase-9, and cytochrome c(2,12).
During the process, the electrochemical gradient across the
mitochondrial membrane collapses; therefore, the loss of MMP is
another hallmark of apoptosis. Our data clearly showed that
treatment with EEDM led to the collapse of the MMP, which was
associated with the reduction of whole Bid proteins, which may
relate to the activation of tBid (Fig.
3). The data also revealed that exposing U937 cells to EEDM
resulted in the proteolytic activation of caspase-8 and -9, which
involves initiator caspases of the extrinsic and intrinsic
pathways, respectively (Fig. 4),
suggesting that extrinsic and intrinsic pathways may have
contributed, at least in part, to EEDM-induced apoptosis of U937
cells. Furthermore, EEDM markedly activated the key executioner,
caspase-3, and the concomitant degradation of PARP, β-catenin, and
PLCγ1 proteins (Fig. 4). However,
under the same experimental conditions, significant protection of
EEDM-induced apoptosis was observed following pretreatment with a
pan caspase inhibitor, z-VED-fmk (Fig.
5). These data indicate that caspases are the key molecules
mediating EEDM-induced apoptosis, and EEDM-induced apoptosis in
U937 cells may be mediated through a caspase-dependent pathway.
In contrast, the PI3K/Akt and MAPK signaling
pathways play critical roles in cell survival and apoptosis in
various cancer cells. The activation of PI3K/Akt and ERK pathways
leads to cell survival through the phosphorylation of various
anti-apoptotic downstream effectors, whereas the p38 MAPK and JNK
pathways are more often associated with the induction of apoptosis
(26–29). Our data indicated that
phosphorylation of all three MAPKs increased rapidly in U937 cells
in response to EEDM and peaked after 0.5 to 6 h; whereas,
phosphorylation of Akt, downstream of PI3K, decreased in response
to EEDM treatment after 12 h. However, all specific inhibitors of
the PI3K/Akt and MAPK signaling pathways did not inhibit
EEDM-induced apoptosis of U937 cells (Fig. 6). Although more experiments are
required to find the relationship between EEDM and these signaling
pathways, the results indicate that EEDM-induced apoptosis in U937
cells is not associated with the PI3K/Akt and MAPK signaling
pathways.
In conclusion, the present study clearly
demonstrates that EEDM significantly inhibits cell proliferation
and induces apoptosis in U937 cells via the extrinsic and intrinsic
pathways. EEDM-induced apoptosis was mediated by caspase activation
and was triggered by mitochondrial dysfunction and modulation in
Bcl-2 and IAP family protein levels. These novel phenomena have not
been previously described and provide important new insights into
the biological effects of EEDM. These results warrant further
investigation of D. morbifera as a source of
pharmacologically active agents as well as the identification and
purification of the active compounds in the present EEDM. In
vivo studies using preclinical tumor models are also needed to
fully establish the potential of EEDM as a chemopreventive and
therapeutic agent in cancer.
Acknowledgements
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
grant funded by the Korean government (no. 2012046358).
References
|
1
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163.
2005.PubMed/NCBI
|
|
2
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brenner D and Mak TW: Mitochondrial cell
death effectors. Curr Opin Cell Biol. 21:871–877. 2009. View Article : Google Scholar
|
|
4
|
Röder C, Trauzold A and Kalthoff H: Impact
of death receptor signaling on the malignancy of pancreatic ductal
adenocarcinoma. Eur J Cell Biol. 90:450–455. 2011.PubMed/NCBI
|
|
5
|
Fulda S and Kroemer G: Mitochondria as
therapeutic targets for the treatment of malignant disease.
Antioxid Redox Signal. 15:2937–2949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim IH, Kim SW, Kim SH, Lee SO, Lee ST,
Kim DG, Lee MJ and Park WH: Parthenolide-induced apoptosis of
hepatic stellate cells and anti-fibrotic effects in an in vivo rat
model. Exp Mol Med. 44:448–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kwon TK: Phorbol myristate acetate
inhibits okadaic acid-induced apoptosis and downregulation of
X-linked inhibitor of apoptosis in U937 cells. Biochem Biophys Res
Commun. 287:135–141. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fukuda K: Apoptosis-associated cleavage of
β-catenin in human colon cancer and rat hepatoma cells. Int J
Biochem Cell Biol. 31:519–529. 1999.
|
|
11
|
Eskes R, Desagher S, Antonsson B and
Martinou JC: Bid induces the oligomerization and insertion of Bax
into the outer mitochondrial membrane. Mol Cell Biol. 20:929–935.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shamas-Din A, Brahmbhatt H, Leber B and
Andrews DW: BH3-only proteins: orchestrators of apoptosis. Biochim
Biophys Acta. 1813:508–520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Newman DJ and Cragg GM: Natural products
as sources of new drugs over the 30 years from 1981 to 2010. J Nat
Prod. 75:311–335. 2012.PubMed/NCBI
|
|
14
|
Moon HI: Antidiabetic effects of
dendropanoxide from leaves of Dendropanax morbifera Leveille
in normal and streptozotocin-induced diabetic rats. Hum Exp
Toxicol. 30:870–875. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chung IM, Kim MY, Park WH and Moon HI:
Antiatherogenic activity of Dendropanax morbifera essential
oil in rats. Pharmazie. 64:547–549. 2009.
|
|
16
|
Chung IM, Kim MY, Park SD, Park WH and
Moon HI: In vitro evaluation of the antiplasmodial activity of
Dendropanax morbifera against chloroquine-sensitive strains
of Plasmodium falciparum. Phytother Res. 23:1634–1637.
2009.PubMed/NCBI
|
|
17
|
Chung IM, Song HK, Kim SJ and Moon HI:
Anticomplement activity of polyacetylenes from leaves of
Dendropanax morbifera Leveille. Phytother Res. 25:784–786.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park BY, Min BS, Oh SR, Kim JH, Kim TJ,
Kim DH, Bae KH and Lee HK: Isolation and anticomplement activity of
compounds from Dendropanax morbifera. J Ethnopharmacol.
90:403–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Richmond JD, Agius BR, Wright BS, Haber
WA, Moriarity DM and Setzer WN: Essential oil compositions and
cytotoxic activities of Dendropanax capillaris, Oreopanax
nubigenus, and Schefflera rodrigueziana from Monteverde,
Costa Rica. Nat Prod Commun. 4:271–274. 2009.PubMed/NCBI
|
|
20
|
Yu HY, Kim KS, Lee YC, Moon HI and Lee JH:
Oleifolioside A, a new active compound, attenuates LPS-stimulated
iNOS and COX-2 expression through the downregulation of NF-κB and
MAPK activities in RAW 264.7 macrophages. Evid Based Complement
Alternat Med. 2012:6375122012.PubMed/NCBI
|
|
21
|
Yu HY, Jin CY, Kim KS, Lee YC, Park SH,
Kim GY, Kim WJ, Moon HI, Choi YH and Lee JH: Oleifolioside A
mediates caspase-independent human cervical carcinoma HeLa cell
apoptosis involving nuclear relocation of mitochondrial apoptogenic
factors AIF and EndoG. J Agric Food Chem. 60:5400–5406. 2012.
View Article : Google Scholar
|
|
22
|
Tak JK, Lee JH and Park JW: Resveratrol
and piperine enhance radiosensitivity of tumor cells. BMB Rep.
45:242–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nechushtan A, Smith CL, Hsu YT and Youle
RJ: Conformation of the Bax C-terminus regulates subcellular
location and cell death. EMBO J. 18:2330–2341. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Danson S, Dean E, Dive C and Ranson M:
IAPs as a target for anticancer therapy. Curr Cancer Drug Targets.
7:785–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Altieri DC: Survivin and IAP proteins in
cell-death mechanisms. Biochem J. 430:199–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ashkenazi A: Targeting the extrinsic
apoptosis pathway in cancer. Cytokine Growth Factor Rev.
19:325–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aksamitiene E, Kiyatkin A and Kholodenko
BN: Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt
pathways: a fine balance. Biochem Soc Trans. 40:139–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar : PubMed/NCBI
|