Introduction
Liver cancer is a significant health issue,
particularly in developing countries where it is inevitably fatal
(1,2), and worldwide it is the third most
common cause of death from cancer. In 2008, an estimated 749,000
newly diagnosed liver cancer cases and 695,000 deaths were reported
(1). The 5-year survival rate of
these patients is between 6.5 and 8.3% (2).
The most common form of liver cancer is
hepatocellular carcinoma (HCC). It is well accepted that the
primary causes of HCC are nonspecific cirrhosis, steatohepatitis
and viral hepatitis, and according to clinical data the incidence
of HCC and the associated mortality continue to increase (3,4).
However, due to the lack of specific early symptoms and symptoms
that overlap with other liver diseases, HCC is often diagnosed at
an advanced stage when curative surgical resection is no longer
viable. At this advanced stage of disease, liver transplantation
may be an option, but it is difficult to carry out due to limited
donor livers, fierce immune rejection and huge expenditure.
Chemotherapy or radiation treatment may be helpful for some
patients, but are usually palliative. Many HCC patients have
concomitant liver cirrhosis or other liver diseases, and the above
therapies have only limited applications. Thus, novel approaches
for HCC patients are urgently needed to provide both preventive and
curative strategies.
Towards this end, the drug metformin, commonly used
to suppress glucose production by the liver (5), has recently been targeted as a
chemoprophylatic agent and for treatment of various human cancers.
Belonging to the biguanide class of drugs, metformin is used as a
first-line therapy for diabetes mellitus type 2 (adult-onset
diabetes), but epidemiological studies have shown that when used
against type 2 diabetes it also reduced the risk of cancer
(6,7). Moreover, according to several clinical
trials, type 2 diabetes is an independent risk factor for HCC
development, and use of metformin appeared specifically to be
associated with a lower risk of liver cancer (8,9).
Features of diabetes such as insulin resistance, glycometabolic
disorder, and hepatic lipid accumulation may also occur in HCC,
precancerous nonalcoholic fatty liver disease and liver cirrhosis
(10–12). Metformin can increase insulin
sensitivity and reduce serum levels of glucose, lipotoxicity and
inflammatory cytokines, which may in turn help to suppress
oncogenesis (13,14).
Metformin inhibits glucose production in the liver
and improves hyperglycemia via activation of AMP-activated protein
kinase (AMPK), an enzyme with an important role in insulin
signaling, whole body energy balance, and the metabolism of glucose
and fat. AMPK is a serine/threonine kinase, which for its
activation requires phosphorylation by upstream kinases at a
specific threonine residue (Thr-172) (15–19).
AMPK has been considered a target for cancer therapy or
prophylaxis.
Previous studies have reported that metformin is
able to inhibit the viability of ovarian cancer cell lines
(8) and to suppress renal carcinoma
in vivo(20). In experiments
with hamsters, the drug also showed preventive activity for
pancreatic cancer (21). The
multiple effects of metformin in cancers such as breast, ovarian,
prostate and colon cancers, appear to act through cell cycle
arrest, apoptosis, prevention of angiogenesis and enhancement of
chemosensitivity (22–24) through AMPK-dependent or -independent
pathways (25,26).
In the present study, we investigated the effects of
metformin on HCC in vitro and in vivo, and the
underlying molecular events.
Materials and methods
Cell culture and cell viability
assay
The human HCC cell lines HepG2 and PLC/PRF/5 were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA) and were cultured in RPMI-1640 medium (Gibco-BRL, Grand
Island, NY, USA) supplemented with 10% fetal calf serum (Sijiqing,
Hangzhou, China), 100 U/ml penicillin and 100 U/ml streptomycin, in
a humidified incubator at 37°C with 5% CO2 and 95%
air.
To assess changes in cell viability, we performed a
methyl thiazolyl tetrazolium (MTT) assay. Briefly, HepG2 and
PLC/PRF/5 cells were seeded at a density of 3×103
cells/well in 96-well plates and cultured for 24 h. The medium was
then replaced with RPMI-1640 medium supplemented with 2.5, 5.0, 10
or 20 mM metformin (American Biomol, Farmingdale, NY, USA) or 100,
250, 500 or 1,000 μM 5-aminoimidazole-4-carboxamide ribonucleoside
(AICAR; Cell Signaling Technology, Inc., Danvers, MA, USA).
Following the above treatments, MTT (Sigma Chemicals, St. Louis,
MO, USA) at 5 mg/ml in phosphate-buffered saline (PBS) was added to
each well, and the cells were incubated for an additional 4 h. The
supernatant was discarded, and 150 μl of dimethyl sulfoxide was
added to each well for 10 min to dissolve the formazan crystals.
The optical density levels of the cell cultures were measured
spectrophotometrically using a dual beam microplate reader at 490
nm.
Flow cytometric assessment of cell cycle
distribution
Cells (5×105) were grown and treated with
metformin (10 mM) or AICAR (500 μM) for 72 h. The cells were
trypsinized and fixed in 70% ethanol overnight. The following day,
the cells were stained with propidium iodide (50 μg/ml; Sigma) for
30 min at 4°C and then analyzed with a flow cytometer. The data
were further analyzed using Windows Multiple Document Interface for
Flow Cytometry (WinMDI) software.
DAPI staining to visualize apoptosis
To assess apoptosis after drug treatment, cells were
stained fluorescently with 4′,6-diamidino-2-phenylindole (DAPI) to
detect nuclear condensation and fragmentation. Cells
(1×105) were cultured in medium with or without
metformin (10 mM) or AICAR (500 μM) for 72 h. The cell culture
medium was then discarded, and 1 μg/ml of DAPI solution (Roche,
Shanghai, China) was added to the cell culture and incubated for 15
min in the dark. After that, the cells were washed with methanol
and examined under a fluorescence microscope.
Nude mouse tumor model
The Ethics Committee for Animal Experimentation of
the Shantou University Medical College approved the animal
experiments. BALB/c-nu mice were obtained from SLC (Guangzhou,
China). Freshly cultured (human hepatoma) PLC/PRF/5 cells
(1×106) were injected subcutaneously into 6- to
8-week-old male nu/nu mice at 4 sites in both flanks (x2). After 1
week, metformin dissolved in PBS was intragastrically administered
at 250 mg/kg body weight/day. The control group received PBS only.
Tumor formation and growth were measured every 2 days when
xenografts were visible. Mouse body weight was measured the day
before intragastric administration and was repeatedly measured at
week 7. Three hours after intragastric drug administration, serum
glucose was measured with a blood glucose monitor.
Immunohistochemistry
Immunohistochemistry was performed on tumor
xenograft tissues for assessment of gene expression. After animals
were euthanized, tumor xenograft tissues were resected for tissue
processing (fixed in 10% buffered formalin, embedded in paraffin,
and cut into 4-mm sections). For immunohistochemistry, the sections
were deparaffinized, rehydrated, and then incubated in 3% (v/v)
hydrogen peroxide for 10 min at room temperature. The sections were
subjected to antigen retrieval by heating in 10 mM (pH 6.0) citrate
buffer for 20 min in a microwave oven, incubation in 20% normal
serum for 50 min at room temperature, and further incubation with a
rabbit anti-cyclin D1, anti-cyclin E or anti-p27KIP
antibody (Maixin Bio, Fuzhou, China) overnight at 4°C. The
following day, the sections were washed with PBS thrice and then
incubated with a secondary antibody, followed by incubation with
streptavidin-peroxidase solution (both from Vector Laboratories,
Burlingame, CA, USA). The color reaction was performed using
3,39-diaminobenzidine (DAB; Sigma) and counterstaining with
hematoxylin. The negative control was carried out omitting the
primary antibody in the tissue sections.
The stained sections were reviewed and scored under
a light microscope independently by 2 investigators. The images of
the immunostained tissue sections were scanned with an Olympus
charged coupled device (CCD) camera and analyzed by ImageJ
(National Institutes of Health). The positive expression rate was
calculated as the positive cell number/total cell number in a
field. The mean of 5 low-power fields was used.
Protein extraction and western
blotting
Cultured tumor cells and HCC tissues were lysed in
RIPA buffer (Bocai, Shanghai, China) supplemented with a protease
and phosphatase inhibition mixture (10 μl), NaF (10 μl of 100 mM),
sodium orthovanadate (10 μl of 100 mM) and phenylmethylsulfonyl
fluoride (PMSF; 10 μl of 100 mM).
For western blotting, equal amounts of the protein
samples were resolved via SDS-PAGE and transferred to
nitrocellulose membranes (Millipore, Billerica, MA, USA). The
membranes were then blocked with 10% non-fat dried milk in
Tris-buffered saline with Tween-20 (TBST) for 1 h at room
temperature. The membranes were incubated with primary antibodies
overnight at 4°C. The next day, the membranes were washed three
times with TBST and incubated with horseradish
peroxidase-conjugated secondary antibodies (Sigma) for 2 h at room
temperature.
The peroxidase activity was detected using enhanced
chemiluminescence (ECL; Pierce Biotechnology, Inc., Rockford, IL,
USA) and exposed to X-ray film (Kodak, Guangzhou, China). The
positive protein bands were quantitated using laser
densitometry.
Statistical analyses
Data are expressed as means ± standard error and
were analyzed using SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA). The statistical difference between two measurements was
calculated using the Student’s t-test. Results were considered
statistically significant at P<0.05.
Results
Effects of metformin on HCC cell
viability
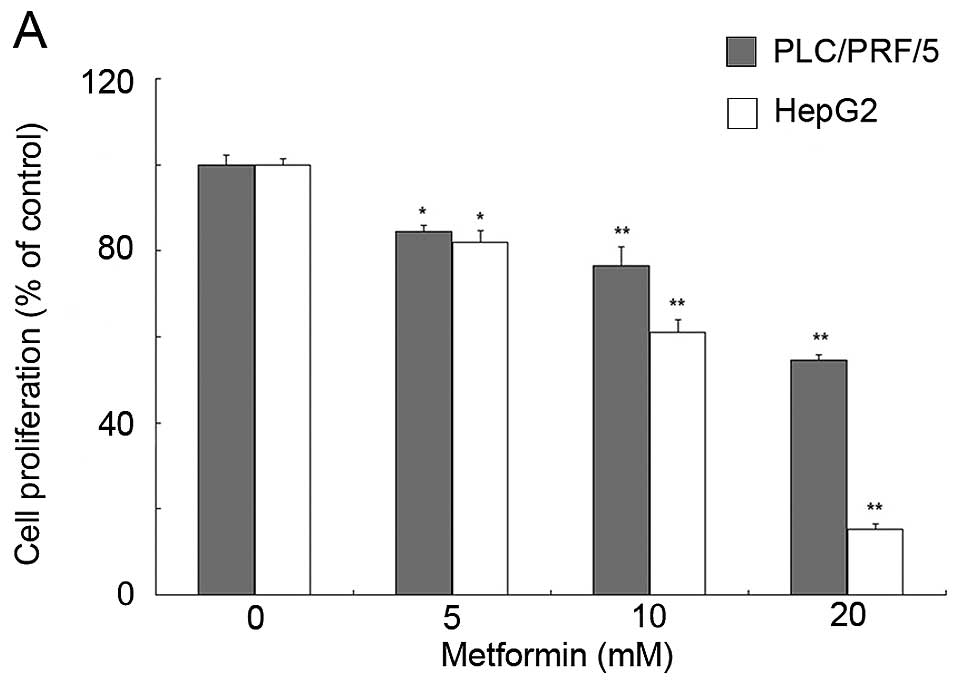
To determine the effects of metformin on HCC cell
viability, the HCC cell lines PLC/PRF/5 and HepG2 were treated with
different concentrations of metformin or the AMPK activator AICAR
for 72 h in vitro. The data showed that metformin
significantly inhibited the viability of HCC cells in a
dose-dependent manner (Fig. 1A),
and AICAR was also able to reduce HCC cell viability (Fig. 1B).
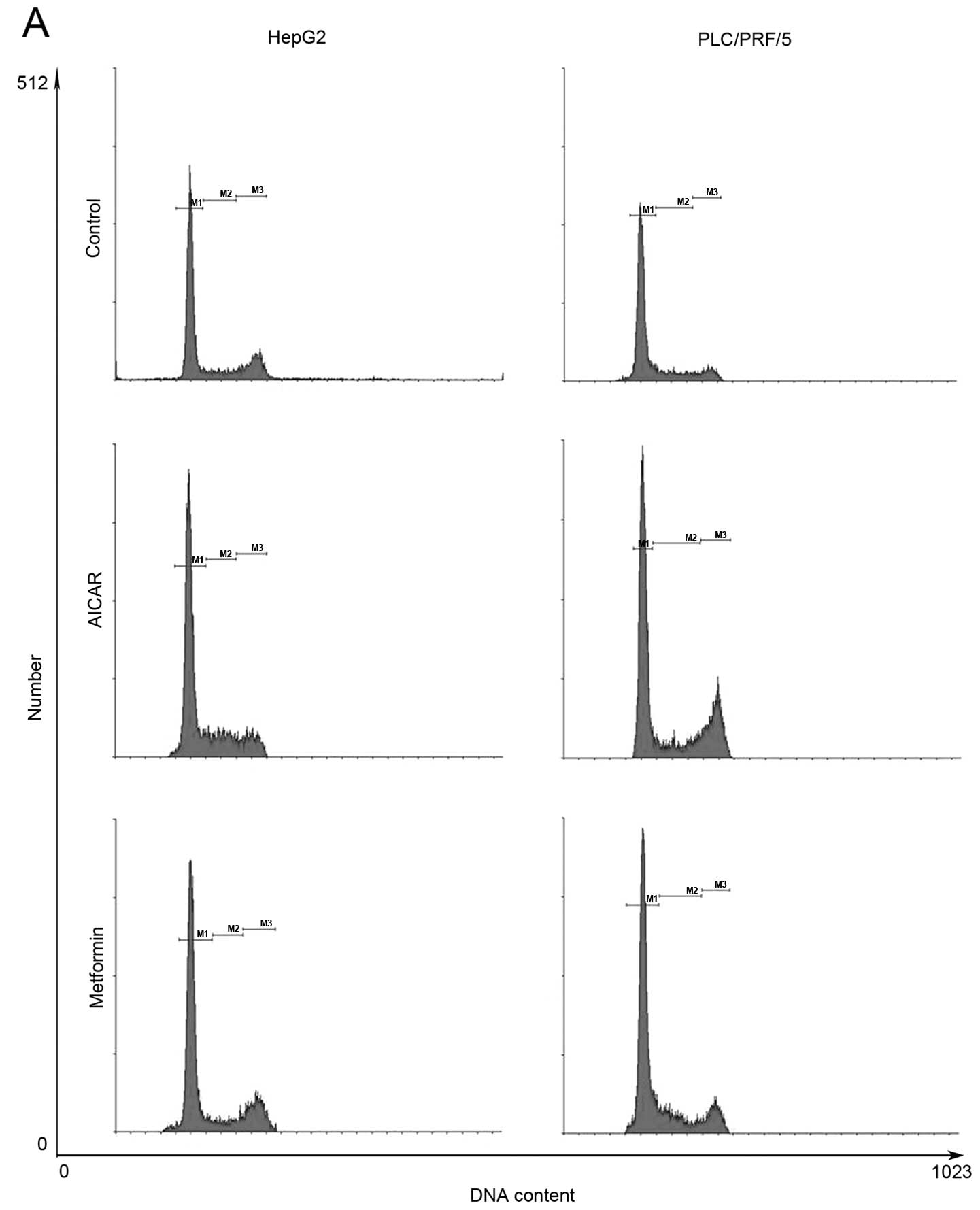
Metformin induces cell cycle arrest at
the G0/G1 phase in HCC cells
Treatment of HCC cells with 10 mM metformin for 72 h
induced HCC cell cycle arrest at the G1/G0 phase (58.99% compared
with 69.28% in HepG2 cells and 50.14% compared with 65.96% in
PLC/PRF/5 cells). Moreover, treatment with 500 μM AICAR for 72 h
replicated the metformin-induced cell cycle arrest phenomenon in
these 2 HCC cell lines (Fig.
2).
Molecularly, treatment of HepG2 cell lines with
metformin or AICAR induced expression of p21CIP and
p27KIP, but downregulated cyclin D1 expression (Fig. 2). These data suggest that metformin
and AICAR modulated expression of cyclin D1, p21CIP and
p27KIP to arrest the cell cycle at the G0/G1 phase, and
that AMPK activation may be responsible for this effect.
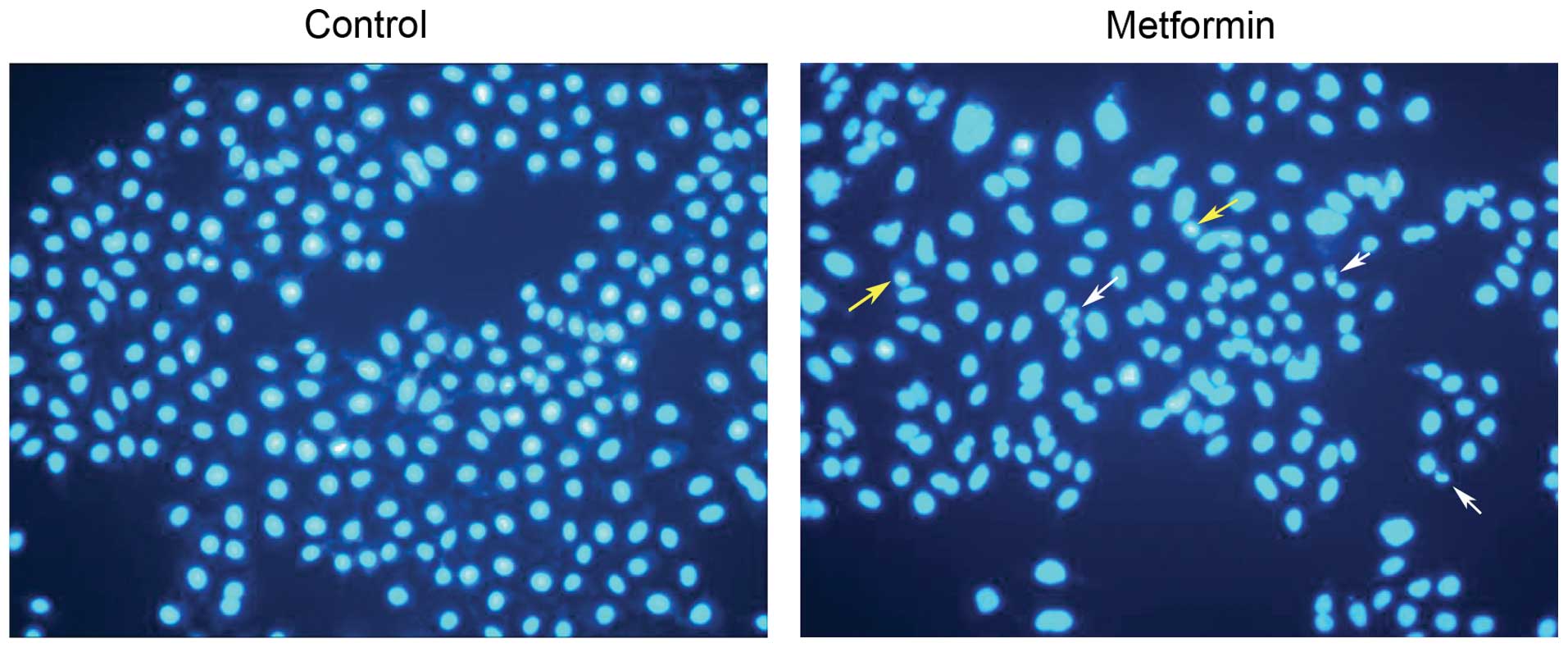
Metformin induces apoptosis of HCC cells
in vitro
We next assessed the effect of metformin on
induction of apoptosis of tumor cells. Treatment of HepG2 cell
lines with 10 mM metformin for 24 h induced apoptosis. Apoptotic
cells with condensed and fragmented nuclei were visualized by
staining with DAPI (Fig. 3).
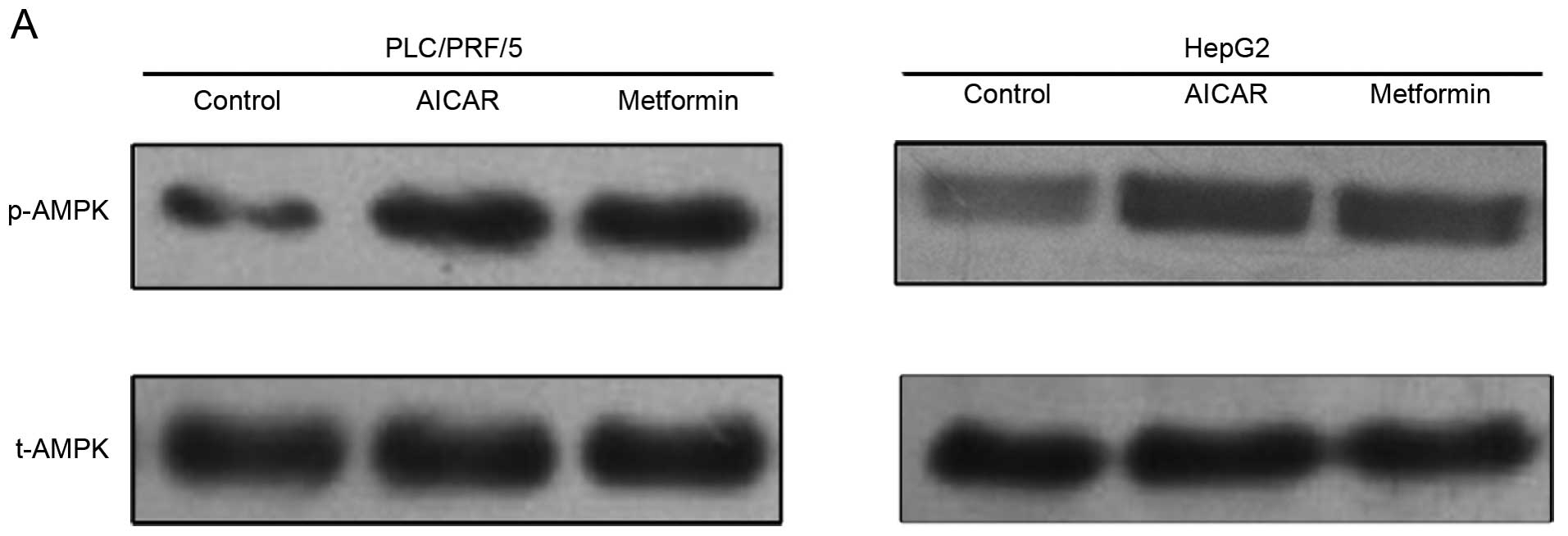
Association of metformin with AMPK
activation in vitro
We further investigated expression of phosphorylated
AMPK (p-AMPK), an active form of AMPK, in HCC cells. Metformin was
able to activate AMPK, similar to AICAR (Fig. 4). This result indicates that AMPK
activation may be involved in the antiproliferative effects of
metformin on HCC cells.
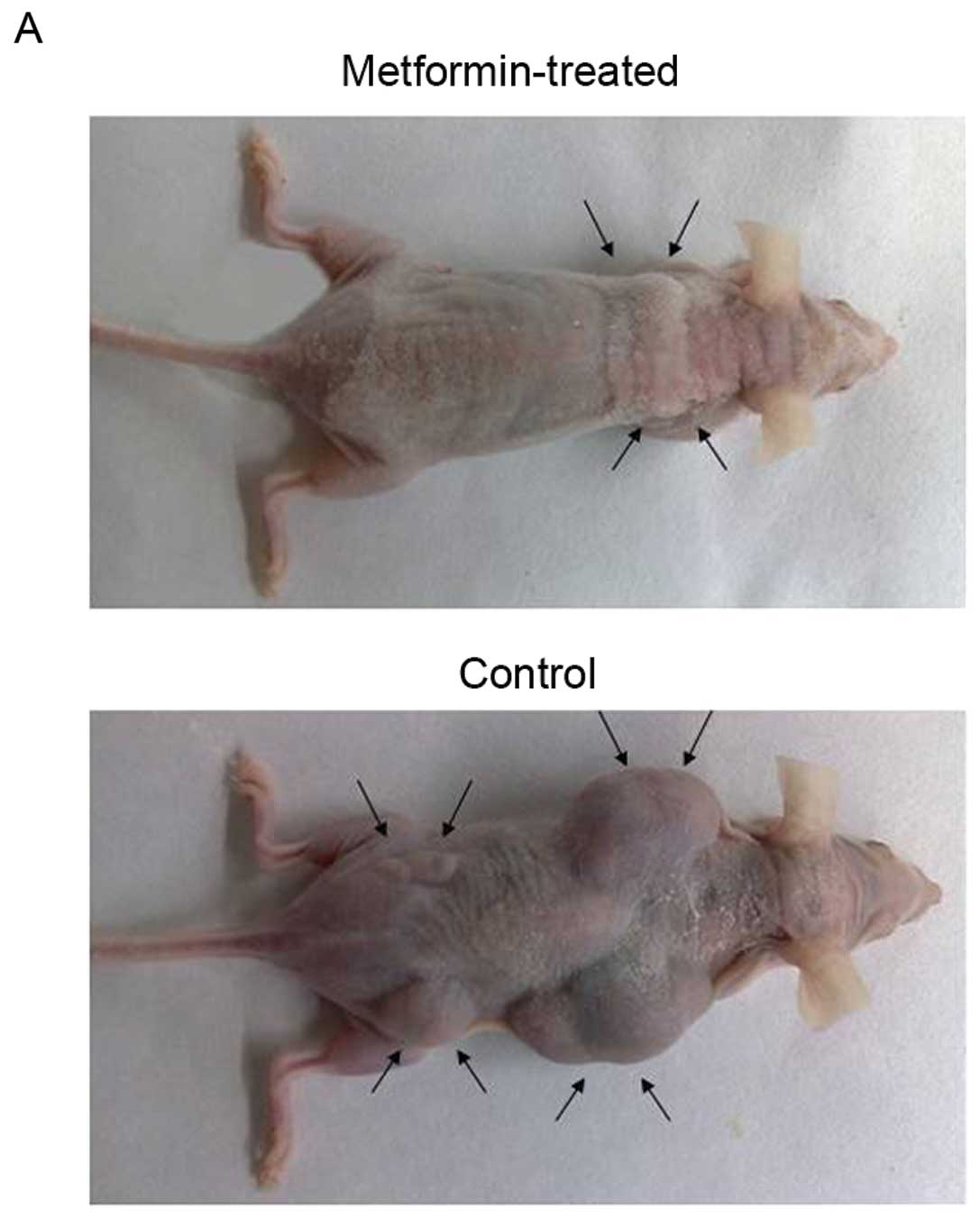
Metformin inhibites HCC cell xenograft
formation and growth in nude mice
To assess the in vivo effects of metformin,
we produced an HCC cell xenograft model in nude mice and then
treated the mice with metformin through intragastric administration
(250 mg/kg body weight/day for 7 weeks) 1 week after subcutaneous
injection of PLC/PRF/5 cells. Our data showed that metformin
markedly suppressed the growth of tumor xenografts when compared to
the isovolumic PBS-treated controls (Fig. 5A and B). In addition, the morbidity
due to HCC in the mouse model was reduced from 41 to 16%, after
metformin treatment (compared with the control, n>10). However,
metformin did not have any effect on mouse body weight and serum
glucose level (Fig. 5C and D).
These data may support the view that metformin is relatively safe
and contributed to the prophylaxis and treatment of HCC.
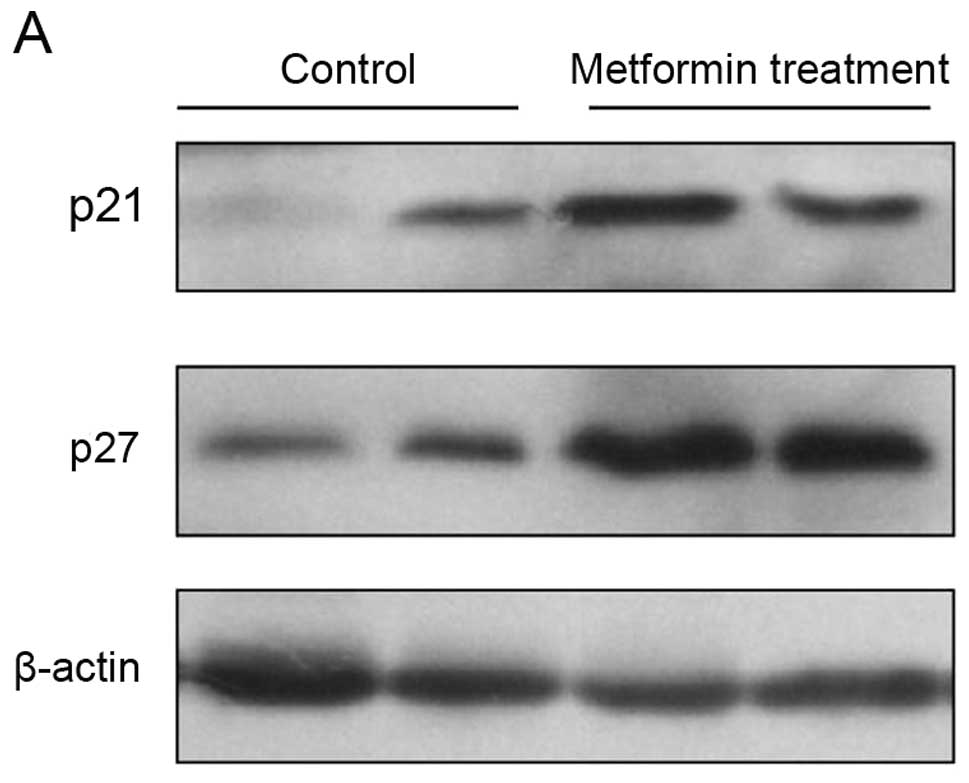
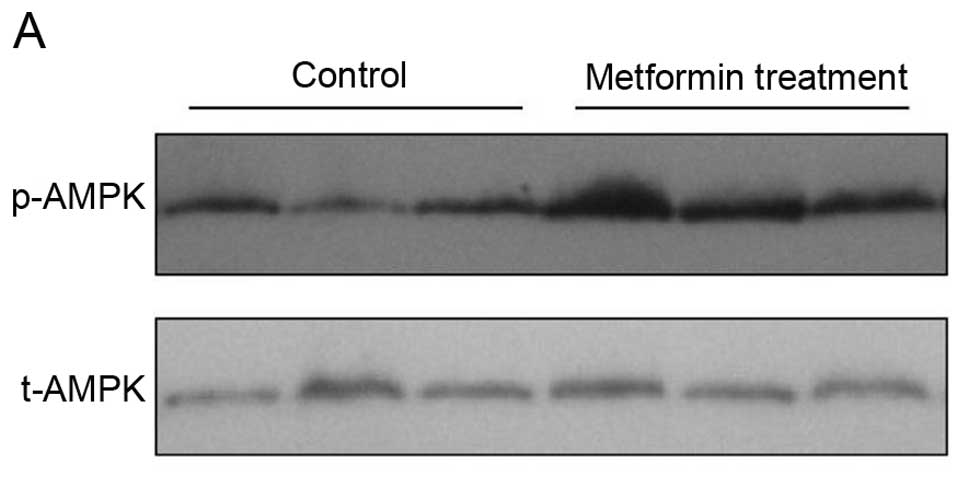
Effects of metformin on expression of
cell cycle regulators and p-AMPK in vivo
To explore the molecular mechanisms responsible for
the metformin-induced antiproliferative effects in vivo,
expression of different cell cycle regulator genes was analyzed in
the PLC/PRF/5 cell tumor xenograft tissues. Western blotting data
showed that the expression of p21CIP and
p27KIP proteins was significantly increased by metformin
treatment when compared with that of the controls (Fig. 6A). Immunohistochemical data showed
that metformin inhibited expression of cyclin D1 and cyclin E
proteins, but upregulated the level of p27KIP in the
metformin-treated tumor xenografts (Fig. 6B–E). Furthermore, we found that
metformin treatment upregulated expression of phosphorylated AMPK
protein (Fig. 7).
Discussion
In the present study, we investigated the effects of
metformin in vitro and in vivo, and the underlying
molecular events. Our data showed that treatment with metformin
in vitro reduced HCC cell viability in a dose-dependent
manner. In vitro, metformin treatment induced HCC cell cycle
arrest at the G1/G0 phase and apoptosis. In the mouse HCC cell
xenograft model, metformin not only blocked tumor progression, but
also reduced tumor morbidity. Molecularly, metformin upregulated
p21CIP and p27KIP expression, but
downregulated cyclin D1 levels both in vitro and in
vivo. We also found evidence to suggest that these effects of
metformin may be through the upregulation of p-AMPK protein. Thus,
metformin warrants further evaluation as a novel strategy for the
treatment and prevention of HCC.
The conventional view of HCC risk factors focused on
viral hepatitis and cirrhosis. Yet recent epidemiological data have
shown an association between diabetes, obesity, and insulin
resistance and the increased incidence of HCC (13). The HCC risk in type 2 diabetics was
found to be as high as 7.1-fold higher than in non-diabetic
patients, depending on the duration of diabetes and the treatment
protocol used (27). Thus,
metformin could be useful for the prevention of HCC, particularly
in type 2 diabetic patients. Our current data that metformin
induces cell cycle arrest at the G1/G0 phase supports this
notion.
Previous studies, including in vitro
experiments, animal models, and epidemiological analyses have shown
that metformin use is associated with a lower rate of cancer
development and that metformin inhibits tumor cell proliferation
(6,20). Previous studies have revealed that
metformin is able to inhibit HCC growth by arresting the tumor cell
cycle at G1 phase, through regulation of the AMPK-dependent pathway
(28). However, Xiong et
al(31) showed that the
induction of HCC cell cycle arrest and apoptosis by metformin was
through an AMPK-independent pathway. In the present study, we
confirmed that metformin treatment upregulated p-AMPK levels in HCC
cells in vitro and in nude mouse xenografts in vivo,
but further study is required to determine whether AMPK activation
is essential for the antitumor effects of metformin.
Several lines of evidence indicate that AMPK
activation may be involved in metformin-induced cell cycle arrest
of tumor cells (22,28). Metformin was found to exert its
antitumor activity through the AMPK-dependent pathway (29). HeLa cells are deficient in the liver
kinase B1 (LKB1; the upstream activator of AMPK) allele and are
insensitive to metformin-induced antitumor effects (25). Our current data showed that
metformin treatment increased the level of AMPK phosphorylation
in vivo and in vitro. Furthermore, AICAR, an AMPK
activator, was able to replicate the other antiproliferative
effects of metformin in HCC cells in vitro. These data
indicate that metformin-activated AMPK contributes to the
inhibitory effects of metformin in HCC cells. However, data that
contradicts this finding has been observed (26,30),
suggesting that the effects of metformin on tumor cells are
AMPK-independent. The reason for this may be because different
experimental conditions were used to test the effects of metformin
(28,31).
We recognize that metformin is not only an AMPK
activator, but also possesses antiproliferative effects, which may
be involved in other gene pathways and multiple regulators.
Therefore, we propose that AMPK-dependent and AMPK-independent
pathways may coexist. In any case, the most destructive feature of
tumor cells is uncontrolled cell cycle progression, and as a
central metabolic sensor AMPK can influence cell proliferation via
the cell cycle, in addition to mediating the metabolism of fatty
acids, glycogen, and proteins (16,28).
From this point of view, AMPK may be a target for tumor
therapy.
The regulation of cell proliferation is dependent on
a balance between cell division and death. In cancer cells, this
balance swings toward proliferation, and cancer cells manifest
dysregulated cell cycle and immortality. In the present study,
metformin arrested HCC cells at the G1 phase of the cell cycle and
induced apoptosis. This is consistent with the findings of Qu et
al(32), and reveals the
diversity of the effects of metformin. Therefore, metformin may be
useful in the control of HCC progression.
In regular cell cycle progression, the transition
from G1 to S phase depends on the regulation of specific cyclins
and cyclin-dependent kinase inhibitors (CDKIs), including cyclin
D1, cyclin E, p21CIP and p27KIP. Cyclin D1
and cyclin E promote cell DNA synthesis and cell growth.
Overexpression of cyclin D1 and cyclin E promotes cancer
progression (33,34). In contrast, downregulation of cyclin
D1 and cyclin E expression restricts the cell cycle progression to
the G0/G1 phase and inhibits tumor cell proliferation (22,35).
Our current data showed that metformin treatment decreased the
levels of cyclin D1 and cyclin E both in HCC cells in vitro
and in nude mouse xenografts.
Moreover, p21CIP downregulates the level
of phosphorylated retinoblastoma (Rb) to induce cell cycle arrest
(36) and combines with
proliferating cell nuclear antigen (PCNA) to reduce DNA replication
(37). The cell cycle-negative
regulator of p27KIP can suppress activation of
cyclin-dependent kinase (CDK) (38), inactivate activated cyclin/CDK
complexes (35,39), and reduce Rb phosphorylation and E2F
release to inhibit gene transcription and control cell cycle
progression (40). Our present
in vivo and in vitro data collectively showed that
metformin inhibited the expression of cyclin D1 protein, but
upregulated p21CIP and p27KIP expression.
These data suggest that metformin can aid in the control of
HCC.
From a clinical perspective, evaluation of
chemotherapeutics must include, not only their antitumor effects,
but also the financial cost and side-effects. As a traditional
antidiabetic agent, metformin is popular since it is relatively
inexpensive and safe. In our in vivo experiment, we found
that metformin did not influence the weight and serum glucose level
of animals. This may be additional support for the use of metformin
in antitumor therapy. However, further investigation is needed to
determine the therapeutic dose range of metformin for antitumor
therapy, and whether metformin treatment has adverse effects within
this range. Recently, there have been several clinical
chemoprevention trials using metformin and we await publication of
the positive data.
Acknowledgements
We would like to thank the members of the Department
of Pathology for their technical support. We also thank the Central
Laboratory (First Affiliated Hospital, Shantou University Medical
College) for their support of our experiments. This study was
supported in part by grants from the National Natural Science
Foundation of China (81070673 and 81172263), the Special Foundation
of Guangdong Province College Talent Introduction (10027425), the
Projects Sponsored by the Scientific Research Foundation for
Returned Overseas Chinese Scholars, the State Education Ministry
(20111568), and the Natural Science Foundation of Guangdong
Province (S2011010005102).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Bosch FX, Ribes J, Díaz M and Cléries R:
Primary liver cancer: worldwide incidence and trends.
Gastroenterology. 127(Suppl 1): S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen DS: Hepatocellular carcinoma in
Taiwan. Hepatol Res. 37(Suppl 2): S101–S105. 2007. View Article : Google Scholar
|
|
5
|
Hundal RS, Krssak M, Dufour S, et al:
Mechanism by which metformin reduces glucose production in type 2
diabetes. Diabetes. 49:2063–2069. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: a cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bowker SL, Majumdar SR, Veugelers P and
Johnson JA: Increased cancer-related mortality for patients with
type 2 diabetes who use sulfonylureas or insulin. Diabetes Care.
29:254–258. 2006. View Article : Google Scholar
|
|
8
|
Gotlieb WH, Saumet J, Beauchamp MC, et al:
In vitro metformin anti-neoplastic activity in epithelial ovarian
cancer. Gynecol Oncol. 110:246–250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang ZJ, Zheng ZJ, Shi R, Su Q, Jiang Q
and Kip KE: Metformin for liver cancer prevention in patients with
type 2 diabetes: a systematic review and meta-analysis. J Clin
Endocrinol Metab. 97:2347–2353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vernon G, Baranova A and Younossi ZM:
Systematic review: the epidemiology and natural history of
non-alcoholic fatty liver disease and non-alcoholic steatohepatitis
in adults. Aliment Pharmacol Ther. 34:274–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Starley BQ, Calcagno CJ and Harrison SA:
Nonalcoholic fatty liver disease and hepatocellular carcinoma: a
weighty connection. Hepatology. 51:1820–1832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baig NA, Herrine SK and Rubin R: Liver
disease and diabetes mellitus. Clin Lab Med. 21:193–207.
2001.PubMed/NCBI
|
|
13
|
Giovannucci E, Harlan DM, Archer MC, et
al: Diabetes and cancer: a consensus report. CA Cancer J Clin.
60:207–221. 2010. View Article : Google Scholar
|
|
14
|
Stickel F and Hellerbrand C: Non-alcoholic
fatty liver disease as a risk factor for hepatocellular carcinoma:
mechanisms and implications. Gut. 59:1303–1307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hardie DG: Minireview: the AMP-activated
protein kinase cascade: the key sensor of cellular energy status.
Endocrinology. 144:5179–5183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kyriakis J: At the crossroads:
AMP-activated kinase and the LKB1 tumor suppressor link cell
proliferation to metabolic regulation. J Biol. 2:262003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baas AF, Kuipers J, van der Wel NN, et al:
Complete polarization of single intestinal epithelial cells upon
activation of LKB1 by STRAD. Cell. 116:457–466. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Corradetti MN, Inoki K, Bardeesy N,
DePinho RA and Guan KL: Regulation of the TSC pathway by LKB1:
evidence of a molecular link between tuberous sclerosis complex and
Peutz-Jeghers syndrome. Genes Dev. 18:1533–1538. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Slattery ML and Fitzpatrick FA:
Convergence of hormones, inflammation, and energy-related factors:
a novel pathway of cancer etiology. Cancer Prev Res. 2:922–930.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Li M, Song B, et al: Metformin
inhibits renal cell carcinoma in vitro and in vivo xenograft. Urol
Oncol. 31:264–270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schneider MB, Matsuzaki H, Haorah J, et
al: Prevention of pancreatic cancer induction in hamsters by
metformin. Gastroenterology. 120:1263–1270. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhuang Y and Miskimins WK: Cell cycle
arrest in Metformin treated breast cancer cells involves activation
of AMPK, downregulation of cyclin D1, and requires
p27Kip1 or p21Cip1. J Mol Signal. 3:182008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rattan R, Graham RP, Maguire JL, Giri S
and Shridhar V: Metformin suppresses ovarian cancer growth and
metastasis with enhancement of cisplatin cytotoxicity in vivo.
Neoplasia. 13:483–491. 2011.PubMed/NCBI
|
|
24
|
Algire C, Amrein L, Zakikhani M, Panasci L
and Pollak M: Metformin blocks the stimulative effect of a
high-energy diet on colon carcinoma growth in vivo and is
associated with reduced expression of fatty acid synthase. Endocr
Relat Cancer. 17:351–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zakikhani M, Dowling RJO, Sonenberg N and
Pollak MN: The effects of adiponectin and metformin on prostate and
colon neoplasia involve activation of AMP-activated protein kinase.
Cancer Prev Res. 1:369–375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yasmeen A, Beauchamp MC, Piura E, Segal E,
Pollak M and Gotlieb WH: Induction of apoptosis by metformin in
epithelial ovarian cancer: involvement of the Bcl-2 family
proteins. Gynecol Oncol. 121:492–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hassan MM, Curley SA, Li D, et al:
Association of diabetes duration and diabetes treatment with the
risk of hepatocellular carcinoma. Cancer. 116:1938–1946. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen HP, Shieh JJ, Chang CC, et al:
Metformin decreases hepatocellular carcinoma risk in a
dose-dependent manner: population-based and in vitro studies. Gut.
62:606–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ouyang J, Parakhia RA and Ochs RS:
Metformin activates AMP kinase through inhibition of AMP deaminase.
J Biol Chem. 286:1–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bhalla K, Hwang BJ, Dewi RE, et al:
Metformin prevents liver tumorigenesis by inhibiting pathways
driving hepatic lipogenesis. Cancer Prev Res. 5:544–552. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiong Y, Lu QJ, Zhao J and Wu GY:
Metformin inhibits growth of hepatocellular carcinoma cells by
inducing apoptosis via mitochondrion-mediated pathway. Asian Pac J
Cancer Prev. 13:3275–3279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qu Z, Zhang Y, Liao M, Chen Y, Zhao J and
Pan Y: In vitro and in vivo antitumoral action of metformin on
hepatocellular carcinoma. Hepatol Res. 42:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nielsen NH, Arnerlöv C, Emdin SO and
Landberg G: Cyclin E overexpression, a negative prognostic factor
in breast cancer with strong correlation to oestrogen receptor
status. Br J Cancer. 74:874–880. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Biliran HJ, Wang Y, Banerjee S, et al:
Overexpression of cyclin D1 promotes tumor cell growth and confers
resistance to cisplatin-mediated apoptosis in an elastase-myc
transgene-expressing pancreatic tumor cell line. Clin Cancer Res.
11:6075–6086. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Polyak K, Kato JY, Solomon MJ, et al:
p27Kip1, a cyclin-Cdk inhibitor, links transforming growth
factor-beta and contact inhibition to cell cycle arrest. Genes Dev.
8:9–22. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng F, McLaughlin P, Verderame M and
Zagon I: The OGF-OGFr axis utilizes the p21 pathway to restrict
progression of human pancreatic cancer. Mol Cancer. 7:52008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cayrol C, Knibiehler M and Ducommun B: p21
binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient
cells. Oncogene. 16:311–320. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gardner LB, Li Q, Park MS, Flanagan WM,
Semenza GL and Dang CV: Hypoxia inhibits G1/S transition through
regulation of p27 expression. J Biol Chem. 276:7919–7926. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|