Introduction
The highly conserved family of 14-3-3 proteins
consisting of seven isoforms (β, γ, ɛ, η, θ, σ and ξ) has been
demonstrated to bind to a wide variety of proteins and to play
important roles in a variety of biological processes, including
cell cycle control, cell survival and cell death through various
signal transduction pathways (1–4). In
normal or tumor cells and tissues, 14-3-3 proteins have been
suggested to participate in a broad spectrum of human diseases such
as cancer (5). However, 14-3-3
proteins exhibit isoform-specific expression in different types of
cells and tissues, and the function of 14-3-3 proteins is complex
and intricate owing to the high sequence homology of its isoforms
(6).
The role of 14-3-3 proteins in carcinogenesis has
been extensively studied. Accumulating evidence has established an
association between 14-3-3 proteins and many types of cancers,
including lung, breast, neck and head cancers (5,7).
However, different isoforms may act as oncogenes or tumor
suppressors in different types of cancers. Abundant expression of
14-3-3ξ is found in breast cancers and promotes cancer progression
via the PI3K/Akt pathway (8,9).
Knockdown of 14-3-3ξ was found to significantly inhibit lung cancer
cell proliferation and promote lung cancer cell sensitivity to
chemotherapy drugs (10,11). In contrast, 14-3-3σ is suggested to
be a tumor suppressor owing to the frequent gene methylation that
occurs in breast cancers (12). In
addition, the β, γ and θ isoforms are also reported to be oncogenic
(13–15). Thus, 14-3-3 proteins can be
potential targets for cancer diagnosis and therapy.
14-3-3 proteins are a group of small and acidic
proteins first identified in brain proteins that are abundant in
total soluble brain extracts (3,16,17).
Thus, dysregulation of 14-3-3 proteins is suggested to be related
to numerous neurological diseases (18–20).
Our previous studies demonstrated that 14-3-3β was highly expressed
in human astrocytomas (21,22). However, the underlying mechanisms
are poorly understood. Research has demonstrated that 14-3-3
proteins bind and regulate glycogen synthase kinase 3 β (GSK3β)
activity in neurons (23). GSK3β is
a serine-threonine kinase that regulates signaling pathways
involved in cell proliferation and the cell cycle (24,25).
GSK3β was also found to contribute to cancers through
Wnt/β-catenin. It was reported that GSK3β promotes the
phosphorylation of β-catenin and its degradation (26). Inhibition of GSK3β by Wnt signaling
leads to the accumulation and nuclear translocation of β-catenin,
which results in the activation of oncogene transcription through
interactions with the T-cell factor/lymphoid enhancer factor
(Tcf/Lef) transcription factors (27). Loss of GSK3β was reported to be
associated with prostate cancer implying dysregulation of GSK3β in
cancers (28). Inhibition of GSK3β
was found to induce the death of glioma cells (29). Given these findings, we hypothesized
that 14-3-3β interacts with GSK3β to regulate β-catenin-mediated
oncogene expression and contributes to tumorigenesis and the
development of astrocytomas.
Astrocytomas are a type of malignant cancer
frequently found in the brains in both adults and children; an
effective therapeutic method is still lacking to date. Therefore,
it is urgent to understand the underlying mechanisms of
astrocytomas (30–32). Our previous investigations
demonstrated that 14-3-3β exhibited an abundant distribution in
astrocytoma tissues and glioma cells, and it was closely related to
the degree of malignancy (21,22).
We hypothesized that 14-3-3β plays important roles in the
development of human astrocytomas and interacts with GSK3β in the
regulation of cell growth and proliferation. In order to test our
hypotheses, gene overexpression and small RNA interference (siRNA)
was performed in normal human astrocytes and glioma cells in the
present study. Co-immunoprecipitation studies showed that 14-3-3β
interacts with GSK3β in glioma cells. Overexpression of 14-3-3β
sequestered GSK3β and enhanced its phosphorylation status, which
resulted in accumulation and nuclear translocation of β-catenin.
Consequently, β-catenin nuclear translocation activated oncogene
transcription including c-myc and cyclin D1, which are responsible
for tumorigenesis and progression. Thus, the delineated mechanism
of 14-3-3β may be responsible for tumorigenesis and progression of
human astrocytomas. Therefore, new therapeutic strategies or drugs
aimed at 14-3-3β may have potential for the treatment of human
astrocytomas.
Materials and methods
Cell lines and cell culture
The human normal astrocyte cell line SVGp12 and
glioma cell line U87 were obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA). All cells were maintained
according to standard protocols. Briefly, cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine
serum to which 100 U/ml of penicillin, 100 μg/ml of streptomycin
and 2 mM of L-glutamate were added. All cells were cultured at 37ºC
with 5% CO2 in an incubator (Life Technologies,
Baltimore, MD, USA).
Recombinant plasmid construction and
transfection
Total RNA was extracted from frozen resected tumor
tissues using TRIzol (Invitrogen, Carlsbad, CA, USA). Total RNA was
isolated using chloroform and precipitated with isopropanol. The
resulting 1 μg of RNA was used as a template for single-stranded
cDNA synthesis with 20 U avian myeloblastosis virus (AMV) reverse
transcriptase (Takara Biotechnology, Dalian, China) according to
the manufacturer’s instructions. Primers with restriction enzyme
sites HindIII and BamHI were used for amplifying cDNA
fragments of 14-3-3β followed by subcloning into the p3XFlag-CMV
expression vector (Sigma Chemical Co., St. Louis, MO, USA).
Small-interfering RNAs (siRNAs) targeting 14-3-3β (sc-29186),
β-catenin (sc-270011) and control siRNA-A (sc-37007) were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cells were
transfected with vectors or siRNA according to the manufacturer’s
instructions. Briefly, cells were seeded in a 6-well culture plate
(2×105 cells/well) and incubated at 37ºC with 5%
CO2 until the cells reached 80% confluence. Plasmid DNA
(1 μg) or siRNA (30 pmol) were diluted in 500 μl of DMEM with 5 μl
Lipofectamine (Invitrogen), mixed and incubated at room temperature
for 15 min. Then, the mixtures were added to the cells with a final
volume of 3 ml of medium and incubated with cells for the indicated
time.
MTT assay
For the MTT assay, cells were plated in 96-well
plates and cultured under regular conditions until they reached 80%
confluence. Plasmid or siRNA was transfected according to standard
protocols, and was continually incubated with cells at 37ºC with 5%
CO2 for 24 or 48 h. Then, the culture medium was
replaced with fresh medium containing MTT (5 mg/ml in PBS, 200
μl/well) (Shanghai Sangon Biological Engineering Co., Ltd,,
Shanghai, China) and incubated with the cells for an additional 4
h. Then formazan was dissolved in DMSO (150 μl/well; Sigma) for 10
min, and the absorbance at 490 nm was determined with an ELISA
reader (BioTek Instruments, Winooski, VT, USA). Each cell viability
assay was performed in quadruplicate and repeated three times. Data
are expressed as mean ± standard error of the mean (SEM) and
differences were analyzed by the Student’s t-test.
Bromodeoxyuridine (BrdU) assay
For the BrdU assay, a BrdU cell proliferation assay
kit (Millipore, Billerica, MA, USA) was used. Transfected cells in
96-well plates were cultured for 24 or 48 h, and 10 μl of BrdU
solution was added per well and incubated for 2 h. The medium was
removed, and 100 μl/well of the Fixing/Denaturing solution was
added and incubated at room temperature for 15 min. Then, the
solution was removed, and 100 μl/well prepared detection antibody
solution was added and incubated for 1 h at room temperature. After
that, plates were washed three times with wash buffer followed by
the addition of 100 μl/well of prepared HRP-conjugated secondary
antibody solution and incubation for 30 min at room temperature.
Then, the plates were washed three times with wash buffer, and 100
μl of TMB substrates was added and incubated for 30 min at room
temperature. The amount of BrdU incorporated into the cells was
determined at 450 nm by a microplate reader (Bio-Rad Laboratories,
Hercules, CA, USA). Each cell proliferation assay was performed in
quadruplicate and repeated three times. Data are expressed as mean
± SEM and differences were analyzed by the Student’s t-test.
Nuclear protein extraction
Nuclear proteins were extracted using an extraction
kit (Shanghai Sangon Biological Engineering) according to the
manufacturer’s protocol. Briefly, cells were lysed in cytoplasmic
buffer containing protease inhibitors, mixed and incubated for 15
min at 4ºC followed by centrifugation at 12,000 rpm for 20 min at
4ºC. Cell sediments were collected and resuspended in nucleus
buffer for 10 min at 4ºC. Then, the sample was centrifuged at
12,000 rpm for 10 min at 4ºC. The supernatant was collected for
analysis.
Co-immunoprecipitation
The transfected cells were harvested at the
indicated time and lysed in RIPA buffer (Bioteke, Beijing, China)
for 30 min at 4ºC followed by centrifugation at 12,000 rpm for 20
min at 4ºC. Protein A-Sepharose beads (50 μl of a 10% suspension;
Amersham Biosciences AB, Uppsala, Sweden) mixed with a mouse
monoclonal anti-Flag (Sigma Chemical), or mouse IgG as a control,
were incubated at 4ºC in 500 μl of lysis buffer for 1 h. Cell
lysates (500 μl) were added to the prepared antibody-bead mixture
and incubated at 4ºC for 2 h. The bead complexes were then
collected by centrifugation and washed with ice-cold lysis buffer
(0.1 M Tris-HCl buffer containing 0.5 M NaCl, pH 8.0, 1 ml each
time) for a total of three times. Then, the protein complex was
eluted from the beads by 200 μl of 0.1 μM glycine buffer (pH 2.5).
The protein complexes were then separated by SDS-PAGE for further
analysis.
Western blot analysis
Proteins from cultured cells or immunoprecipitated
protein complexes were collected, and a total of 20–30 μg of
protein was fractionated by 12% SDS-PAGE electrophoresis and
transferred to nitrocellulose membranes (Amersham, Little Chalfont,
UK). The membranes were treated using the following procedure with
shaking and blocking at room temperature (RT) with 2% non-fat dry
milk in Tris-buffered saline (TBS) for 1 h followed by incubation
in the primary antibodies (rabbit polyclonal 14-3-3β, β-catenin,
GSK3β and phospho-GSK-3β; from Santa Cruz Biotechnology) diluted in
blocking buffer (1:10,000) at 4ºC overnight and washed three times
with TBS and Tween (TBST; 10 mM Tris-HCl, pH 7.5, 150 mM NaCl and
0.05% Tween-20) for 10 min each time at room temperature.
Subsequently, the membranes were incubated in peroxidase-conjugated
secondary antibody goat anti-rabbit IgG (Boster Corp., Wuhan,
Hubei, China; diluted 1:3,000 in blocking buffer) for 1 h. After
washing three times with TBST and once with TBS each for 10 min, 1
ml of 4-chloro-1-naphthol (4-CN) as an HRP substrate with 9 ml of
TBS and 6 μl of H2O2 was used for visualizing
the target protein in the dark for 5–30 min.
Quantitative real-time-polymerase chain
reaction (qRT-PCR) analysis
Total RNA was extracted from the cultured cells
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the
manufacturer’s protocol. Up to 5 μg of the total RNA was
reverse-transcribed into cDNA using M-MLV reverse transcriptase
(Clontech, Palo Alto, CA, USA). The cDNAs were used as templates
for qRT-PCR. For c-myc, the sense primer was
5′-ACACATCAGCACAACTACGC-3′ and the antisense primer was
5′-CCTCTTGACATTCTCCTCG GT-3′. For cyclin D1, the sense primer was
5′-GCCAACCTCC TCAACGACCGG-3′ and the antisense primer was 5′-GTCC
ATGTTCTGCTGGGCCTG-3′. β-actin (sense primer, 5′-CTC
CATCCTGGCCTCGCTGT-3′ and antisense primer, 5′-GCTG
TCACCTTCACCGTTCC-3′) were used as the control. The qRT-PCR mixture
system contained 5 μl SsoFast™ EvaGreen Supermix (Bio-Rad), 1 μl of
cDNA (diluted in 1:50) and 2 μl of each of the forward and reverse
primers (1 μM) to a final volume of 10 μl. The PCR procedure was as
follows: 94ºC for 4 min; 94ºC for 20 sec, 55ºC for 30 sec, and 72ºC
for 20 sec; 2 sec for plate reading for 35 cycles; and melting
curve from 65 to 95ºC. β-actin was used as a control for
normalizing the gene expression. Three independent experiments were
performed. The data obtained were calculated by 2−ΔΔCt
and treated for statistical analysis as previously described
(33), followed by an unpaired
sample t-test.
Statistical analysis
All experiments were performed independently at
least three times. Differences between groups were analyzed by the
Student’s t-test. A P-value <0.01 was considered to indicate a
statistically significant result.
Results
14-3-3β is involved in cell proliferation
of astrocytes and glioma cells
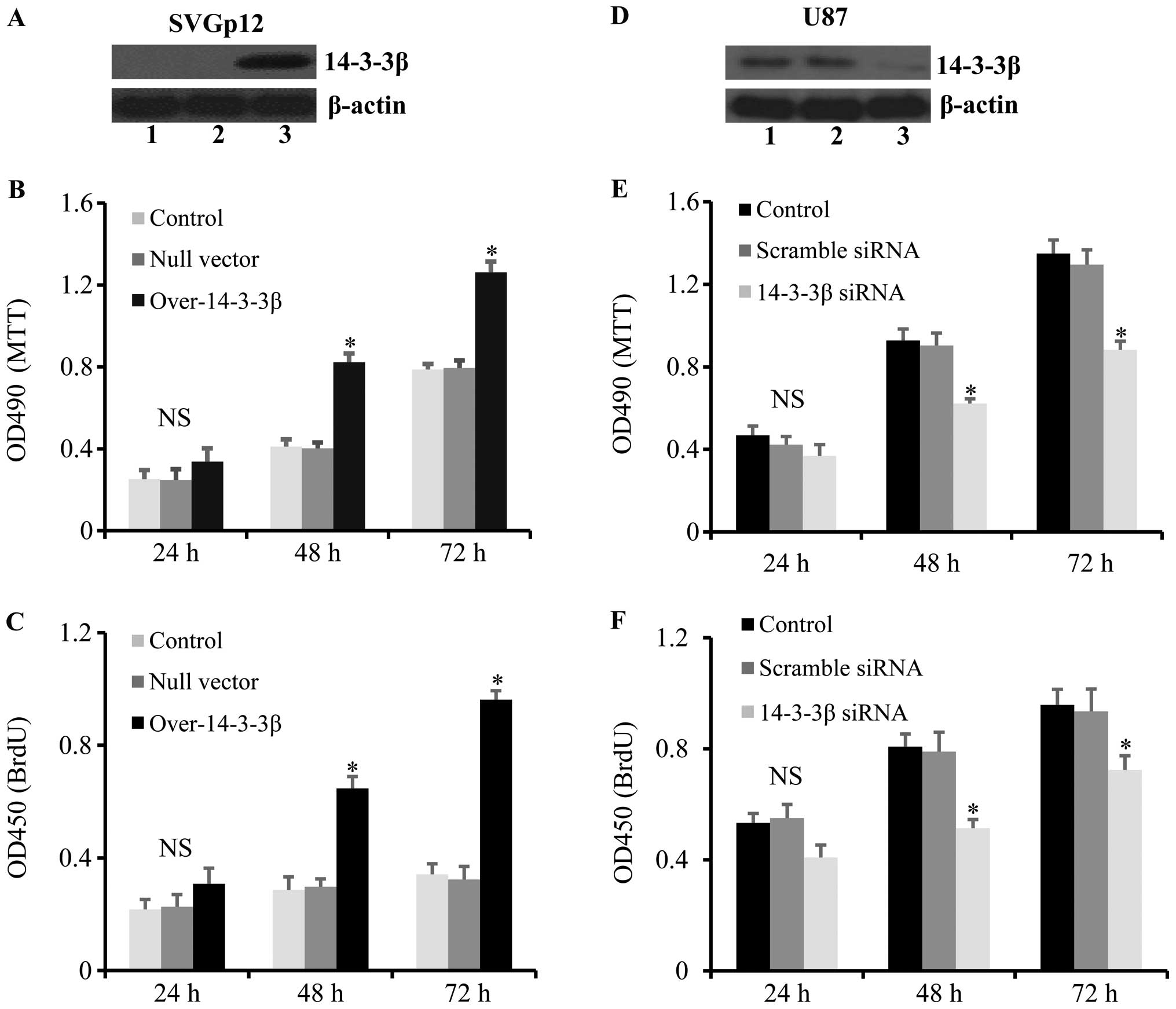
In order to investigate the role of 14-3-3β in
astrocytes, 14-3-3β was overexpressed or silenced by siRNA in the
human normal astrocyte cell line SVGp12 and the glioma cell line
U87, respectively. The results showed that overexpression of
14-3-3β (Fig. 1A) significantly
promoted the growth and proliferation of SVGp12 cells at 48 and 72
h after transfection (Fig. 1B and
C). Furthermore, 14-3-3β was significantly downregulated in U87
cells transfected with 14-3-3β siRNA (Fig. 1D), which resulted in a significant
decrease in cell growth and proliferation of U87 cells at 48 h and
72 h after gene transfection (Fig. 1E
and F). These results demonstrated that 14-3-3β is highly
expressed in glioma cells and 14-3-3β overexpression promotes the
growth and proliferation of human normal astrocytes.
14-3-3β binds to GSK3β and inhibits GSK3β
activity
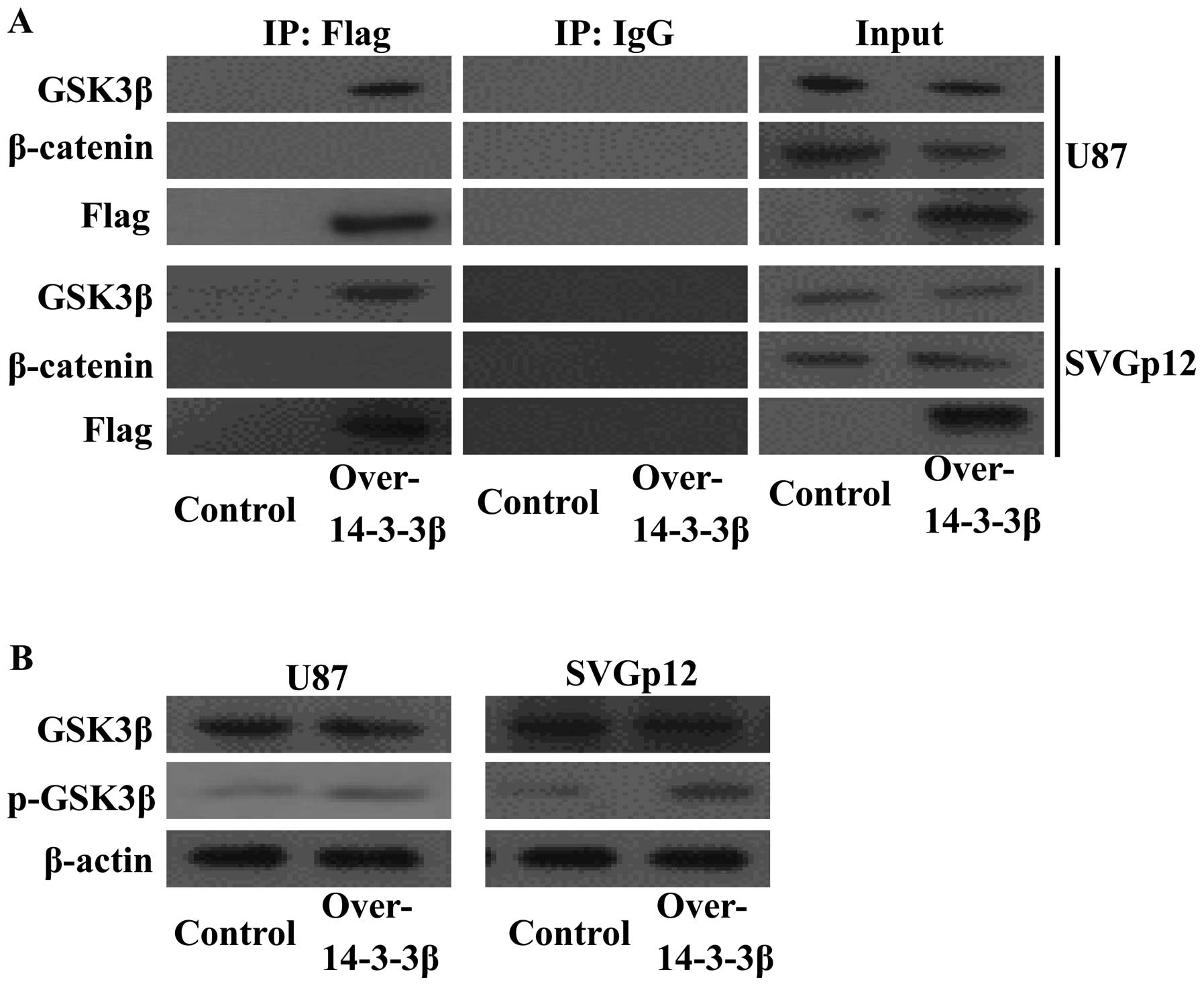
In order to determine whether 14-3-3β and GSK3β
interact, U87 cells were transfected with Flag-tagged 14-3-3β. Flag
antibody was used to bait the 14-3-3β protein complex, and the
interacting proteins were analyzed by western blot analysis. The
14-3-3β protein co-immunoprecipitated with GSK3β in the U87 cell
line (Fig. 2A, upper panels) and in
the SVGp12 cell line (Fig. 2A,
lower panels). The β-catenin protein was not detected in the
protein complex. We speculated that 14-3-3β interacted with GSK3β
and sequestered GSK3β, which enhanced the phosphorylation of GSK3β
and disrupted its interaction with β-catenin. To test these
hypotheses, the phosphorylation of GSK3β was determined by western
blot analysis in transfected cells. As expected, 14-3-3β
overexpression enhanced the phosphorylation of GSK3β (Fig. 2B). These results indicate that
14-3-3β may regulate cell growth and proliferation through the
GSK3β/β-catenin pathway.
14-3-3β increases cell proliferation by
β-catenin
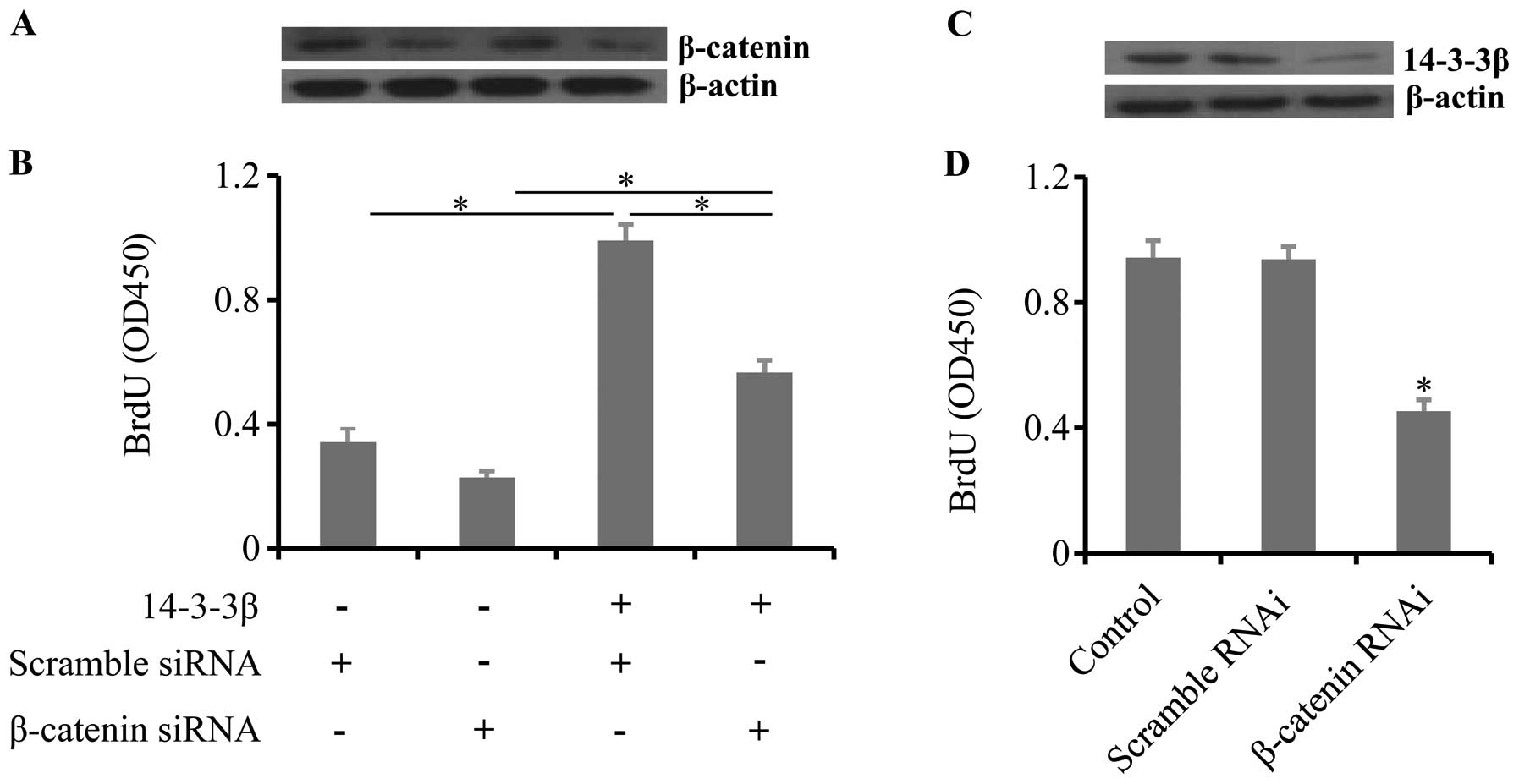
To test the hypotheses that 14-3-3β regulates cell
proliferation though β-catenin, we co-transfected β-catenin siRNA
and 14-3-3β overexpression vectors in SVGp12 cells. Co-transfection
of 14-3-3β overexpression vectors and β-catenin siRNA significantly
inhibited cell proliferation induced by 14-3-3β overexpression
(Fig. 3A and B). To further confirm
the results, β-catenin was knocked down in U87 cells (Fig. 3C). Knockdown of β-catenin also
resulted in a decrease in cell proliferation in U87 cells (Fig. 3D). These results suggest that
14-3-3β regulates cell proliferation through β-catenin.
14-3-3β promotes the activity of
β-catenin
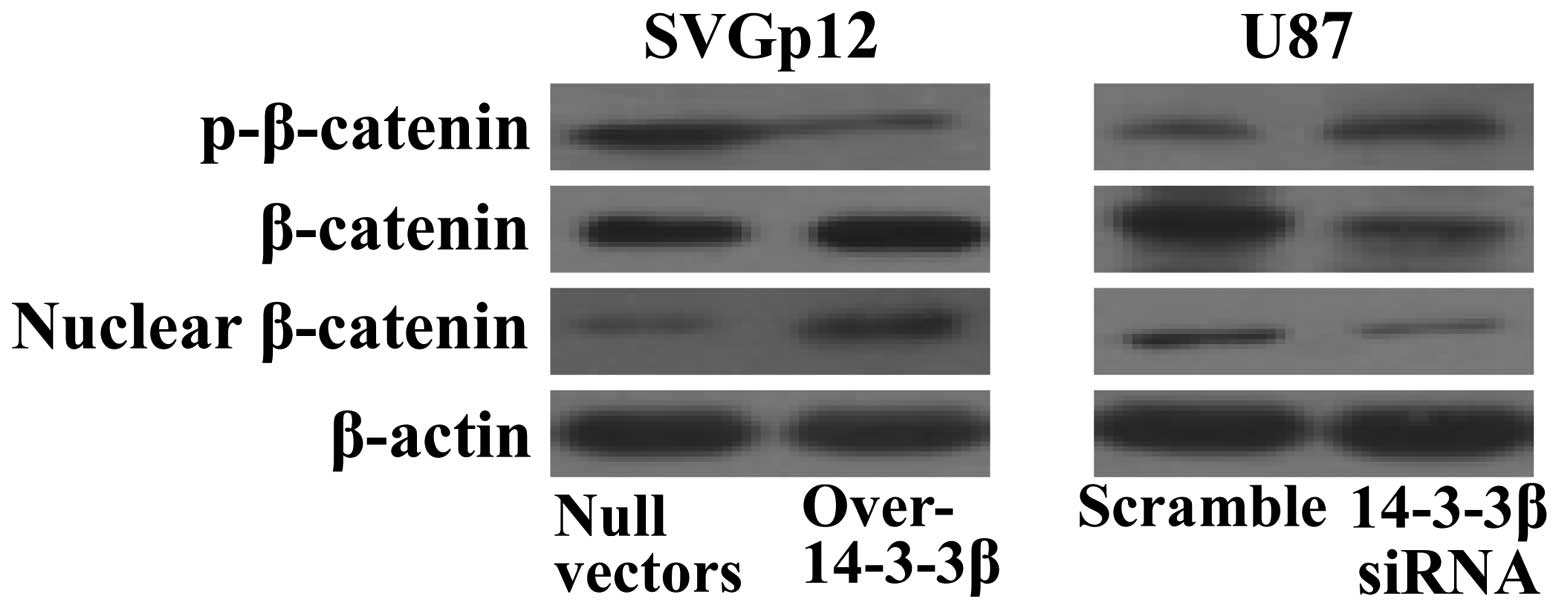
To further explore the underlying mechanisms of
14-3-3β and β-catenin in regulating cell proliferation, the
activity of β-catenin was analyzed in transfected cells. Western
blot analysis showed that phosphorylation of β-catenin was
decreased after 14-3-3β overexpression in SVGp12 cells, which led
to an increase in total β-catenin protein levels. In addition,
there was also more β-catenin protein detected in the nucleus
(Fig. 4, left panels). In contrast,
knockdown of 14-3-3β in U87 cells decreased both the total protein
and nuclear levels of β-catenin (Fig.
4, right panels). These results suggest that 14-3-3β may
augment β-catenin stability and nuclear translocation through
sequestering GSK3β.
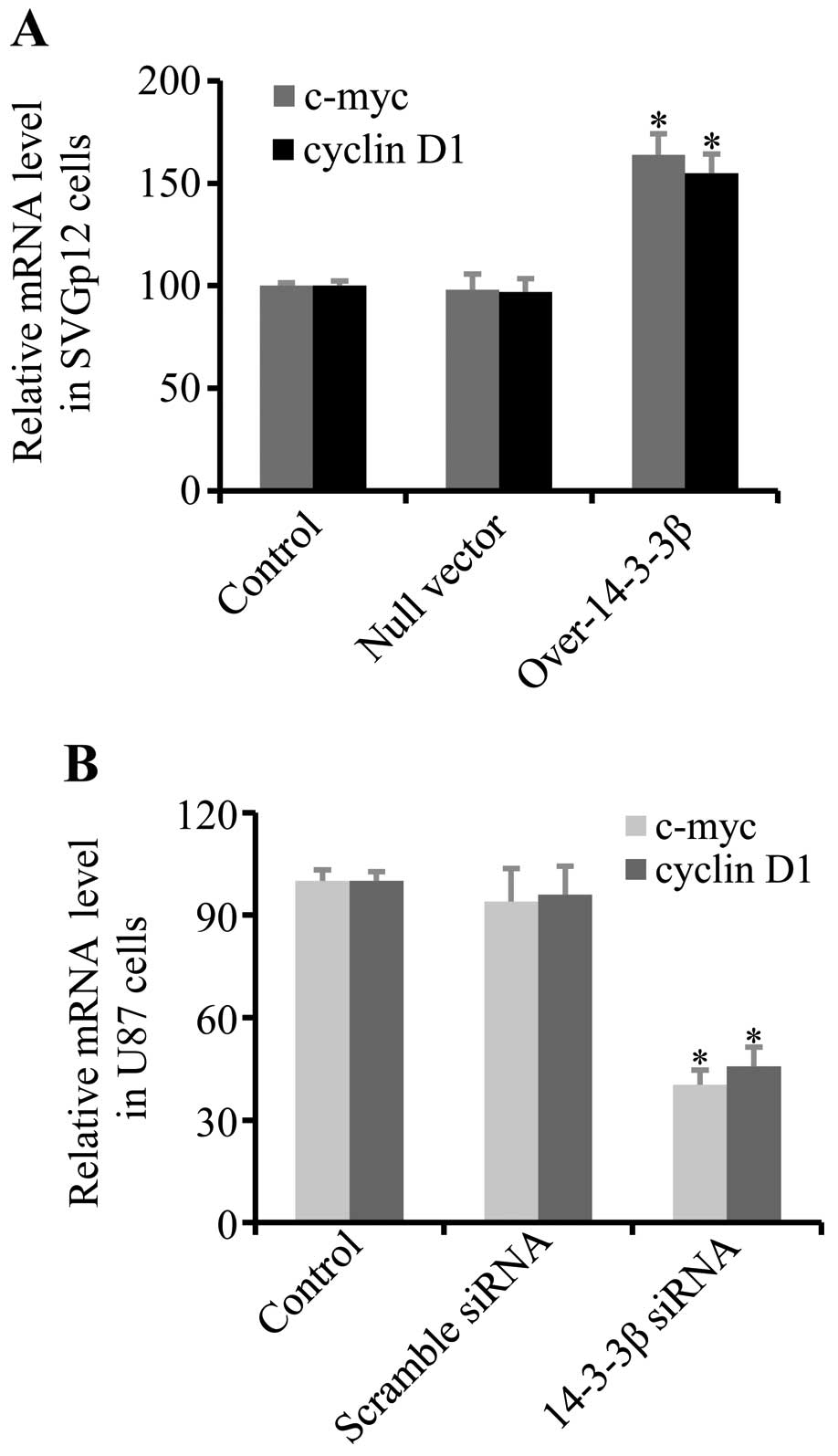
14-3-3β increases oncogene transcription
mediated by β-catenin
To further confirm that 14-3-3β augments the
activity of β-catenin, the transcription levels of the c-myc
oncogene and cyclin D1, which were activated by β-catenin nuclear
translocation (34,35), were analyzed by qRT-PCR.
Overexpression of 14-3-3β in human normal SVGp12 astrocytes
increased the transcription level of c-myc and cyclin D1 (Fig. 5A). In contrast, knockdown of 14-3-3β
in U87 glioma cells decreased the transcription levels of c-myc and
cyclin E (Fig. 5B). These results
suggest that 14-3-3β promotes oncogene expression mediated by
β-catenin.
Discussion
In general, we demonstrated that 14-3-3β regulated
the proliferation of glioma cells through sequestering GSK3β, which
augmented the nuclear translocation of β-catenin leading to an
increase in oncogene expression. The present study provides a
mechanism of 14-3-3β in regulating human astrocytomas. However,
further investigation in vitro and in vivo of 14-3-3β
in astrocytomas is required.
Our previous studies showed that the mRNA and
protein levels of 14-3-3β were closely related to the pathological
grades of astrocytoma implying critical roles of 14-3-3β in
tumorigenesis. In the present study, we found that overexpression
of 14-3-3β in normal human astrocytes significantly increased cell
proliferation, while silencing 14-3-3β in glioma cells inhibited
cell proliferation. The function of 14-3-3β in regulating cell
proliferation through various pathways is well known. It has been
reported that 14-3-3β regulates the G2/M phase transition via
interaction with integrin, testicular protein kinase 1 and Wee1
(36–38). 14-3-3β has been found to be
oncogenic in fibroblasts, and can promote tumorigenesis in nude
mice (39). Knockdown of 14-3-3β in
liver cancer cells suppressed cell proliferation and decreased
oncogenicity in nude mice (40).
Upregulation of 14-3-3β promoted cell proliferation and tumor
formation by the mitogen-activated protein kinase (MAPK)-dependent
signaling pathway in NIH3T3 cells (39). Increased expression of 14-3-3β was
observed in Kaposi’s sarcoma and papillary thyroid carcinomas and
promoted cell proliferation and tumor progression (41,42).
Our previous studies demonstrated that 14-3-3β expression increased
with the degree of human astrocytoma. Thus, in accordance with
previous studies, our present study suggests that 14-3-3β plays
important roles in glioma cells implying that targeting 14-3-3β for
human astrocytoma therapy may be a promising method.
14-3-3β is expressed in tumor tissues and cell lines
of many types of cancers including lung, prostate and breast cancer
(42,43). However, the mechanism of 14-3-3β in
the regulation of cancer cells is quite complicated. 14-3-3β is
reported to be involved in cell apoptosis through interaction with
apoptotic factors, such as Bcl and Bax. 14-3-3β can disturb the
complex of Bax and Bcl, which promotes apoptosis upon Bax
phosphorylation (39,44). In the present study, we demonstrated
that 14-3-3β interacts with GSK3β in the regulation of cell
proliferation. GSK3β regulates a wide range of cellular processes
including cell cycle control, cell growth and cell survival via
diverse signaling pathways (45).
GSK3β activity depends on its phosphorylation of serine 9. Both
PI3K/Akt and Wnt signaling are required for the phosphorylation of
GSK3β. Activation of PI3K/Akt phosphorylates and inhibits GSK3β,
which frequently occurs in cancers (25,46).
Under normal conditions, GSK3β is unphosphorylated and active, and
could phosphorylate and interact with β-catenin leading to
β-catenin degradation (24,28). On the contrary, the accumulation of
β-catenin can lead to the activation of oncogene expression. 14-3-3
proteins have the ability to bind phospho-serine-containing
sequence motifs. In the present study, we found that serine-9
phosphorylation of GSK3β was enhanced in 14-3-3β-overexpressing
cells implying that 14-3-3β interacted with GSK3β, which
sequestered GSK3β and increased phosphorylation of GSK3β.
Inhibition of GSK3β has been suggested to facilitate cancer cell
proliferation (47,48).
For a long time, 14-3-3 proteins were thought to be
brain-specific proteins due to their high abundance in brain
tissues (49). At present, 14-3-3
proteins have been found to be expressed in all eukaryotic cells
(50). The interaction of 14-3-3
proteins with GSK3β in other cell types has been demonstrated in
various studies (51–53). GSK3β and β-catenin form a
destruction complex along Axin and adenomatous polyposis coli. The
complex is cytoplasmic and leads to β-catenin phosphorylation by
GSK3β and subsequent ubiquitination and degradation. Once the
destruction complex is degraded, β-catenin is stabilized leading to
its accumulation in the cytoplasm, resulting in its subsequent
translocation into the nucleus (54). In the present study, we demonstrated
that overexpression of 14-3-3β is associated with the sequestration
of GSK3β and causes β-catenin release. The nuclear translocated
β-catenin along with the Tcf/Lef complex activates oncogene
expression including c-myc and cyclin D1, which are highly
upregulated in human tumors and induce cell proliferation (34,35).
In the present study, we demonstrated that overexpression of
14-3-3β increased the transcription levels of c-myc and cyclin D1.
This may be responsible for the formation and development of
astrocytomas.
In conclusion, the present study revealed that
14-3-3β mediated the cell proliferation of glioma cells through
GSK3β/β-catenin. Overexpression of 14-3-3β sequestered GSK3β
leading to an increase in β-catenin nuclear translocation and
activation of oncogene transcription. Given the high abundance of
14-3-3β in astrocytoma tissues, one can speculate that novel
therapeutic strategies or drugs aimed at 14-3-3β may have potential
for the treatment of human astrocytomas. However, further in
vitro and in vivo studies should be conducted for
verifying the precise mechanisms of 14-3-3β in astrocytomas.
Abbreviations:
|
GSK3β
|
glycogen synthase kinase 3β
|
|
siRNA
|
small interfering RNA
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
BrdU
|
bromodeoxyuridine
|
References
|
1
|
van Hemert MJ, Steensma HY and van Heusden
GP: 14-3-3 proteins: key regulators of cell division, signalling
and apoptosis. Bioessays. 23:936–946. 2001.PubMed/NCBI
|
|
2
|
Baldin V: 14-3-3 proteins and growth
control. Prog Cell Cycle Res. 4:49–60. 2000. View Article : Google Scholar
|
|
3
|
Fu H, Subramanian RR and Masters SC:
14-3-3 proteins: structure, function, and regulation. Annu Rev
Pharmacol Toxicol. 40:617–647. 2000.
|
|
4
|
Aitken A: Functional specificity in 14-3-3
isoform interactions through dimer formation and phosphorylation.
Chromosome location of mammalian isoforms and variants. Plant Mol
Biol. 50:993–1010. 2002.
|
|
5
|
Hermeking H: The 14-3-3 cancer connection.
Nat Rev Cancer. 3:931–943. 2003.
|
|
6
|
Wilker E and Yaffe MB: 14-3-3 proteins - a
focus on cancer and human disease. J Mol Cell Cardiol. 37:633–642.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tzivion G, Gupta VS, Kaplun L and Balan V:
14-3-3 proteins as potential oncogenes. Semin Cancer Biol.
16:203–213. 2006. View Article : Google Scholar
|
|
8
|
Neal CL, Yao J, Yang W, et al: 14-3-3ζ
overexpression defines high risk for breast cancer recurrence and
promotes cancer cell survival. Cancer Res. 69:3425–3432. 2009.
|
|
9
|
Neal CL, Xu J, Li P, et al: Overexpression
of 14-3-3ζ in cancer cells activates PI3K via binding the p85
regulatory subunit. Oncogene. 31:897–906. 2012.
|
|
10
|
Zang D, Li X and Zhang L: 14-3-3ζ
overexpression and abnormal β-catenin expression are associated
with poor differentiation and progression in stage I non-small cell
lung cancer. Clin Exp Med. 10:221–228. 2010.
|
|
11
|
Li Z, Zhao J, Du Y, et al: Down-regulation
of 14-3-3ζ suppresses anchorage-independent growth of lung cancer
cells through anoikis activation. Proc Natl Acad Sci USA.
105:162–167. 2008.
|
|
12
|
Luo J, Feng J, Lu J, et al: Aberrant
methylation profile of 14-3-3 sigma and its reduced
transcription/expression levels in Chinese sporadic female breast
carcinogenesis. Med Oncol. 27:791–797. 2010. View Article : Google Scholar
|
|
13
|
Frasor J, Chang EC, Komm B, et al: Gene
expression preferentially regulated by tamoxifen in breast cancer
cells and correlations with clinical outcome. Cancer Res.
66:7334–7340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun B, Zhang S, Zhang D, et al:
Identification of metastasis-related proteins and their clinical
relevance to triple-negative human breast cancer. Clin Cancer Res.
14:7050–7059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ramirez JL, Rosell R, Taron M, et al:
14-3-3σ methylation in pretreatment serum circulating DNA of
cisplatin-plus-gemcitabine-treated advanced non-small-cell lung
cancer patients predicts survival: the Spanish Lung Cancer Group. J
Clin Oncol. 23:9105–9112. 2005.
|
|
16
|
Muslin AJ and Xing H: 14-3-3 proteins:
regulation of subcellular localization by molecular interference.
Cell Signal. 12:703–709. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pawson T and Nash P: Protein-protein
interactions define specificity in signal transduction. Genes Dev.
14:1027–1047. 2000.PubMed/NCBI
|
|
18
|
Chohan G, Pennington C, Mackenzie JM, et
al: The role of cerebrospinal fluid 14-3-3 and other proteins in
the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a
10-year review. J Neurol Neurosurg Psychiatry. 81:1243–1248.
2010.PubMed/NCBI
|
|
19
|
Ladogana A, Sanchez-Juan P, Mitrova E, et
al: Cerebrospinal fluid biomarkers in human genetic transmissible
spongiform encephalopathies. J Neurol. 256:1620–1628. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Larska M, Polak MP, Zmudzinski JF and
Torres JM: Comparison of mRNA expression levels of selected genes
in the brain stem of cattle naturally infected with classical and
atypical BSE. Brain Res. 1351:13–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao L, Cao W, Zhang W, et al:
Identification of 14-3-3 protein isoforms in human astrocytoma by
immunohistochemistry. Neurosci Lett. 432:94–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang X, Cao W, Lin H, et al:
Isoform-specific expression of 14-3-3 proteins in human
astrocytoma. J Neurol Sci. 276:54–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mwangi S, Anitha M, Fu H, Sitaraman SV and
Srinivasan S: Glial cell line-derived neurotrophic factor-mediated
enteric neuronal survival involves glycogen synthase kinase-3β
phosphorylation and coupling with 14-3-3. Neuroscience.
143:241–251. 2006.PubMed/NCBI
|
|
24
|
Doble BW and Woodgett JR: GSK-3: tricks of
the trade for a multi-tasking kinase. J Cell Sci. 116:1175–1186.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grimes CA and Jope RS: The multifaceted
roles of glycogen synthase kinase 3beta in cellular signaling. Prog
Neurobiol. 65:391–426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ciani L and Salinas PC: WNTs in the
vertebrate nervous system: from patterning to neuronal
connectivity. Nat Rev Neurosci. 6:351–362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Polakis P: The oncogenic activation of
β-catenin. Curr Opin Genet Dev. 9:15–21. 1999.
|
|
28
|
Mulholland DJ, Dedhar S, Wu H and Nelson
CC: PTEN and GSK3β: key regulators of progression to
androgen-independent prostate cancer. Oncogene. 25:329–337.
2006.
|
|
29
|
Kotliarova S, Pastorino S, Kovell LC, et
al: Glycogen synthase kinase-3 inhibition induces glioma cell death
through c-MYC, nuclear factor-κB, and glucose regulation. Cancer
Res. 68:6643–6651. 2008.PubMed/NCBI
|
|
30
|
Davis FG, McCarthy BJ, Freels S, Kupelian
V and Bondy ML: The conditional probability of survival of patients
with primary malignant brain tumors: surveillance, epidemiology,
and end results (SEER) data. Cancer. 85:485–491. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DeAngelis LM: Brain tumors. N Engl J Med.
344:114–123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van den Bent MJ, Hegi ME and Stupp R:
Recent developments in the use of chemotherapy in brain tumours.
Eur J Cancer. 42:582–588. 2006.PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krieghoff E, Behrens J and Mayr B:
Nucleo-cytoplasmic distribution of β-catenin is regulated by
retention. J Cell Sci. 119:1453–1463. 2006.
|
|
35
|
Polakis P: Wnt signaling in cancer. Cold
Spring Harb Perspect Biol. 4:pii: a008052. 2012. View Article : Google Scholar
|
|
36
|
Han DC, Rodriguez LG and Guan JL:
Identification of a novel interaction between integrin β1 and
14-3-3β. Oncogene. 20:346–357. 2001.
|
|
37
|
Toshima JY, Toshima J, Watanabe T and
Mizuno K: Binding of 14-3-3β regulates the kinase activity and
subcellular localization of testicular protein kinase 1. J Biol
Chem. 276:43471–43481. 2001.
|
|
38
|
Wang Y, Jacobs C, Hook KE, et al: Binding
of 14-3-3β to the carboxyl terminus of Wee1 increases Wee1
stability, kinase activity, and G2-M cell population. Cell Growth
Differ. 11:211–219. 2000.
|
|
39
|
Takihara Y, Matsuda Y and Hara J: Role of
the β isoform of 14-3-3 proteins in cellular proliferation and
oncogenic transformation. Carcinogenesis. 21:2073–2077. 2000.
|
|
40
|
Sugiyama A, Miyagi Y, Komiya Y, et al:
Forced expression of antisense 14-3-3β RNA suppresses tumor cell
growth in vitro and in vivo. Carcinogenesis. 24:1549–1559.
2003.
|
|
41
|
Musholt TJ, Brehm C, Hanack J, von
Wasielewski R and Musholt PB: Identification of differentially
expressed genes in papillary thyroid carcinomas with and without
rearrangements of the tyrosine kinase receptors RET and/or NTRK1. J
Surg Res. 131:15–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zeng Y, Li Y, Chen RS, et al:
Overexpression of xCT induces up-regulation of 14-3-3β in Kaposi’s
sarcoma. Biosci Rep. 30:277–283. 2010.PubMed/NCBI
|
|
43
|
Qi W, Liu X, Qiao D and Martinez JD:
Isoform-specific expression of 14-3-3 proteins in human lung cancer
tissues. Int J Cancer. 113:359–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chiang CW, Harris G, Ellig C, et al:
Protein phosphatase 2A activates the proapoptotic function of BAD
in interleukin-3-dependent lymphoid cells by a mechanism requiring
14-3-3 dissociation. Blood. 97:1289–1297. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kockeritz L, Doble B, Patel S and Woodgett
JR: Glycogen synthase kinase-3 - an overview of an over-achieving
protein kinase. Curr Drug Targets. 7:1377–1388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaidanovich-Beilin O, Milman A, Weizman A,
Pick CG and Eldar-Finkelman H: Rapid antidepressive-like activity
of specific glycogen synthase kinase-3 inhibitor and its effect on
β-catenin in mouse hippocampus. Biol Psychiatry. 55:781–784.
2004.PubMed/NCBI
|
|
47
|
Li J, Mizukami Y, Zhang X, Jo WS and Chung
DC: Oncogenic K-ras stimulates Wnt signaling in colon cancer
through inhibition of GSK-3β. Gastroenterology. 128:1907–1918.
2005.
|
|
48
|
Welsh GI and Proud CG: Glycogen synthase
kinase-3 is rapidly inactivated in response to insulin and
phosphorylates eukaryotic initiation factor eIF-2B. Biochem J.
294:625–629. 1993.PubMed/NCBI
|
|
49
|
Berg D, Holzmann C and Riess O: 14-3-3
proteins in the nervous system. Nat Rev Neurosci. 4:752–762. 2003.
View Article : Google Scholar
|
|
50
|
Mhawech P: 14-3-3 proteins - an update.
Cell Res. 15:228–236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chang TC, Liu CC, Hsing EW, et al: 14-3-3σ
regulates β-catenin-mediated mouse embryonic stem cell
proliferation by sequestering GSK-3β. PLoS One. 7:e401932012.
|
|
52
|
Goñi-Oliver P, Avila J and Hernández F:
Calpain regulates N-terminal interaction of GSK-3β with 14-3-3ζ,
p53 and PKB but not with axin. Neurochem Int. 59:97–100.
2011.PubMed/NCBI
|
|
53
|
Gurusamy N, Watanabe K, Ma M, et al:
Glycogen synthase kinase 3β together with 14-3-3 protein regulates
diabetic cardiomyopathy: effect of losartan and tempol. FEBS Lett.
580:1932–1940. 2006.
|
|
54
|
Kikuchi A, Yamamoto H, Sato A and
Matsumoto S: New insights into the mechanism of Wnt signaling
pathway activation. Int Rev Cell Mol Biol. 291:21–71. 2011.
View Article : Google Scholar : PubMed/NCBI
|