1. Introduction
2-Amino-2-[2-(4-octylphenyl)]-1,3-propanediol
hydrochloride (FTY720), also known as fingolimod, is a synthetic
compound produced by modification of a metabolite from Isaria
sinclairii(1,2). FTY720 is a newly developed
immunosuppressant. This drug selectively reduces the number of
lymphocytes in the peripheral circulation, significantly prolongs
the survival of experimental animals and transplanted organs
without prejudice to the immune response to the virus and immune
memory function (3–7). In addition, on September 22, 2010,
FTY720 became the first oral disease-modifying drug approved by the
Food and Drug Administration (FDA) to reduce relapses and delay
disability progression in patients with relapsing forms of multiple
sclerosis. The immunosuppressive activity of FTY720 has been
suggested to be related to its phosphorylation by sphingosine
kinase 2 (SphK2) and subsequent modulation of G protein-coupled
sphingosine-1-phosphate receptors (S1PRs) (S1PR1, S1PR3, S1PR4,
S1PR5) that induce lymphopenia by altering lymphocyte trafficking
(8). In addition to the potent
immunosuppressive effects, evidence suggests that FTY720 has
antitumor efficacy in multiple types of cancer, including breast
(9), glioblastoma (10), prostate (11), lung (12), ovarian (13) and hematopoietic malignancies
(14). This antitumor activity of
FTY720 is reportedly independent of the phosphorylation of FTY720,
which is different from the immunosuppressive effects of FTY720
(15). In the present study, we
described the latest progress of FTY720 in cancer therapy (Table I).
 | Table IEffects of FTY720 on cancer. |
Table I
Effects of FTY720 on cancer.
| Cancer | In
vitro/in vivo | Molecular
targets | Functions | (Ref) |
|---|
| Multiple
myeloma | In
vitro | ROS | Autophagic cell
death and caspase-dependent apoptosis | (21,26) |
| Mantle cell
lymphoma | Both | CD74, Cyclin D1,
PKB | Lysosomal membrane
permeabilization related cell death and prolonged survival in
mouse | (23,28) |
| Acute lymphoblastic
leukemia | In
vitro | ROS | Caspase,
PP2A-independent cell death | (25) |
| Chronic lymphocytic
leukemia and lymphoblastic leukemia/lymphoma | Both | PP2A, ERK1/2 | Induce
caspase-independent cell death and prolong survival in mouse
models | (24) |
| Glioma | Both | FAK, ERK1/2 | Induce
caspase-dependent apoptosis, decrease invasion and inhibit tumor
growth, increase survival time in mouse models | (35,36) |
| Lung carcinoma | Both | S1P, CXCR4 | Antiangiogenic and
reduce tumor size | (12,37,38) |
| Neuroblastoma | Both | SphK2, AKT | Induce cell death
and inhibited the growth of NB xenografts | (39) |
| Prostate
carcinoma | In
vitro | p38MAPK, ERK1/2,
FAK, SphK 1 | Induce
caspase-dependent apoptosis and sensitize cells to
radiotherapy | (40–42) |
| Hepatocellular
carcinoma | Both | Rac, EPCs, CXCL10,
VEGF | Suppress
metastasis | (43,44) |
| Breast cancer | Both | SphK1, JNK | Induce apoptotic
cell death, prevent tumor growth, metastasis and prolong animal
survival | (9,42,45,46) |
| Gastric cancer | Both | PTEN, p53, AKT | Induce
caspase-dependent apoptosis and inhibit tumor growth | (19) |
| Pancreatic
cancer | In
vitro | AKT | Induce
caspase-dependent apoptosis, suppress invasion and migration | (47,48) |
| Colorectal
cancer | In
vitro | JNK |
Antiproliferative | (46) |
2. Anticancer effects of FTY720
In vitro therapeutic activity of
FTY720
Caspase-dependent apoptotic
pathway
During studies assessing the lymphopenic action of
FTY720, it was revealed that FTY720 also induced apoptosis in
peripheral lymphocytes. This observation led to investigations into
the potential of FTY720 to act as an apoptosis-inducing anticancer
agent (8). FTY720 was first
reported to induce early apoptosis in an androgen-independent
prostate cancer cell line dependent on caspase-3 activation
(16). Subsequently, it
demonstrated preclinical antitumor efficacy in various cancer
models. The antitumor activity of FTY720 was reportedly attributed
to different apoptotic pathways. Generally, there are two distinct
pathways for inducing apoptosis, including the mitochondrial death
pathway (intrinsic pathway of apoptosis) and the death receptor
pathway (extrinsic pathway of apoptosis). The mitochondrial death
pathway is controlled by members of the Bcl-2 family, including
Bcl-2, Bad, Bax, Bid and Btf proteins on the mitochondrial
membrane. Death stimuli increase mitochondrial permeability and
release cytochrome c and other factors from the
mitochondria, resulting in caspase-3 activation and then apoptosis.
By contrast, the death receptor pathway is mediated by Fas (CD95)
and Fas-ligand. Binding of Fas and Fas-ligand induces activation of
the caspase cascade via activation of caspase-8 and results in
apoptosis through activation of caspase-3 (17,18).
It has been demonstrated that FTY720 could activate caspase-3,
caspase-9 and poly (ADP-ribose) polymerase (PARP) in gastric cancer
cells, however there was no obvious change in procaspases-8 and
-10, and the cleaved products were not detected (19). In addition, FTY720 was shown to
increase the levels of caspase-2 and -3 and apoptosis-promoting
Bcl-associated proteins, Bad, Bax, Bid and Btf, and decrease the
protein level of Bcl-2 in human renal cancer cells. However, there
was no difference in the protein levels of caspase-8, Fas, FADD and
TRADD, between cells treated with FTY720 and controls (20). These data indicate that FTY720
induces apoptosis through the intrinsic apoptotic pathways, instead
of the extrinsic pathway of apoptosis. By contrast, in multiple
myeloma cells, FTY720 triggered activation of caspase-8, -9, -3,
PARP cleavage, induced alterations in mitochondrial membrane
potential and Bax cleavage, and anti-Fas antibodies augmented
anti-multiple myeloma activity induced by FTY720 (21), demonstrating that FTY720 enhances
death signaling via both the extrinsic and intrinsic pathways. The
difference may be due to the intricate connections between FTY720
and cell apoptosis, depending on cell types and stimulation
manners. Furthermore, there exists a caspase-independent apoptotic
pathway that is mediated by apoptosis-inducing factor (AIF)
(22). However, no reports as yet
have demonstrated that FTY720 could induce cell apoptosis through
this pathway.
Caspase-independent cell death
Due to the differences among cell types, there are
also reports showing that FTY720 induces cell death independent of
apoptosis. For example, in mantle cell lymphoma, FTY720 mediated
time- and dose-dependent cytotoxicity in primary MCL tumor cells
and MCL cell lines; however, FTY720 failed to mediate caspase-3
activation and the expression of Bcl-2 and Mcl-1, two critical
anti-apoptotic proteins in MCL, were not altered in response to
FTY720 (23). In another case,
FTY720 induced toxicity in the Raji cell line and primary CLL B
cells, which was independent of activation of caspases or PARP
processing (24). The mechanisms
underlying the caspase-independent cell death are extremely
complex, and include the downmodulation of cyclin D1 and
phospho-Akt (23), activation of
protein phosphatase 2A (PP2A), dephosphorylation of extracellular
signal regulated kinase (ERK) 1/2 (24), the generation of reactive oxygen
species (ROS) (23,25), and the induction of autophagy
(13,25–28).
However, the precise mechanisms remain unclear and are discussed
later.
Autophagy
Numerous studies have shown that FTY720 was able to
induce autophagy in cancer cells. Autophagy is a process by which
cells conserve and recycle their organelles when in a
nutrient-deprived or stressed state (29). One of the most important functions
of autophagy is to maintain cellular energy subjected to nutrient
deprivation and potentially other forms of stress (30). However, extensive or inappropriate
activation of autophagy can lead to cell death (type II programmed
cell death). Autophagy begins with the formation of an
autophagosome, which travels through the cytoplasm of the cell to a
lysosome, and the two organelles fuse, within the lysosome, the
contents of the autophagosome are degraded via acidic lysosomal
hydrolases (31). It has been
demonstrated that FTY720 induced autophagy in ovarian cancer cells
(8,13), multiple myeloma cells (26), acute lymphoblastic leukemia cells
(25). However, the relationship
between FTY720-induced autophagy and apoptosis or cell death is
controversial. FTY720 could induce both apoptosis and autophagy in
multiple myeloma cells, autophagy induced by FTY720 in multiple
myeloma cells promoted apoptosis, as evidenced by bafilomycin A1,
an autophagy inhibitor, rescued cell death caused by FTY720
(26). Conversely, in ovarian
cancer and acute lymphoblastic leukemia cells, FTY720 induced
autophagy and caspase-independent cell death. Blocking autophagy by
3MA, a known inhibitor that blocks the autophagy pathway at early
stages, or specific siRNA which knocked down the expression of LC3
and Beclin-1 could further enhance FTY720-induced cytotoxicity,
suggesting a protective role of autophagy (13,25).
The discrepancies may be due to the complex and diverse interplays
between autophagy and cell death. Depending on cell types,
environment and stimulus, autophagy and cell death may have
inhibitory, additive or even synergistic effects (32). Therefore, we consider the autophagy
induced by FTY720 in different cell types may lead to either cell
survival or cell death. It should be emphasized that FTY720-induced
autophagy is also cell-specific. Although FTY720 induced
caspase-dependent apoptosis in both MCF7 breast cancer cells and
Huh7 hepatoma cells, no evident induction of autophagy was observed
in these cells following FTY720 treatment (13). In addition to the induction of
autophagy, FTY720 also induced blockage of autophagy. In mantle
cell lymphoma cells, FTY720-treated cells showed characteristics of
autophagy blockage, including accumulation of autolysosomes and
increased LC3-II and p62 levels, which enhanced anticancer efficacy
of milatuzumab (27,28). Thus, the effects of FTY720 on
autophagy are complex and further studies are required.
In vivo therapeutic activity of
FTY720
In addition to the therapeutic activity in
vitro, FTY720 also shows significant treatment effects in
animal models. For example, in nude mice with human gastric cancer
cell xenografts, the tumor growth and cell proliferation in
FTY720-treated mice were significantly suppressed; the apoptotic
index was markedly increased in that of FTY720 groups (19). In addition, in mouse breast cancer
models, tumor growth and metastasis, which were markedly evident in
control mice, were significantly prevented in the FTY720-treated
groups, resulting in a significant prolongation of animal survival
(9). Moreover, FTY720 showed in
vivo anticancer effects in glioblastoma (33), hepatic carcinoma (34), lung cancer (12), renal cancer (20), mantle cell lymphoma (23) and prostate cancer (35). Consequently, FTY720 has promising
effects in cancer therapy both in vitro and in
vivo.
3. Side-effects
In multiple sclerosis models, the used doses of
FTY720 are <0.5 mg/day. However, the doses required for
anticancer effects of FTY720 in animal models (5 or 10 mg/day) are
higher than those used in multiple sclerosis models. Serious
adverse events of FTY720 for multiple sclerosis include
bradycardia, relapse, basal-cell carcinoma. Other adverse events
include macular edema, cancer and laboratory abnormalities
(36). In vivo studies of
the effects of FTY720 on cancer are conducted only on mice. In
animal cancer models, FTY720 treatment did not cause any severe
side-effects at a dosage of ≤10 mg/kg/day in mouse renal cancer
models. In this case, all mice survived with a healthy appearance
and no loss of body weight during the follow up period when
treatment was performed at a dosage of ≤10 mg/kg/day (20). Furthermore, FTY720 prevented tumor
growth and metastasis of mouse breast (9) and bladder (37) xenografts in nude mice without
causing detectable toxicity in vital organs. However, whether these
adverse effects presented in multiple sclerosis would occur during
human cancer treatment, or could be tolerated if the therapy is
efficacious, remains to be determined.
4. Molecular targets of unphosphorylated
form of FTY720
The ability of FTY720 to induce cell death in cancer
cells appears largely independent of the phosphorylation of the
drug. FTY720 was able to induce cell death in ovarian cancer
(13) and acute lymphoblastic
leukemia cells (25); however, the
phosphorylated form of FTY720 (FTY720-P) failed to induce cell
death, demonstrating that activation of S1PRS is not sufficient for
cell death. While the mechanisms mediating the cell death action of
FTY720 have yet to be fully clarified, a number of molecular
targets of unphosphorylated form of FTY720 have been proposed which
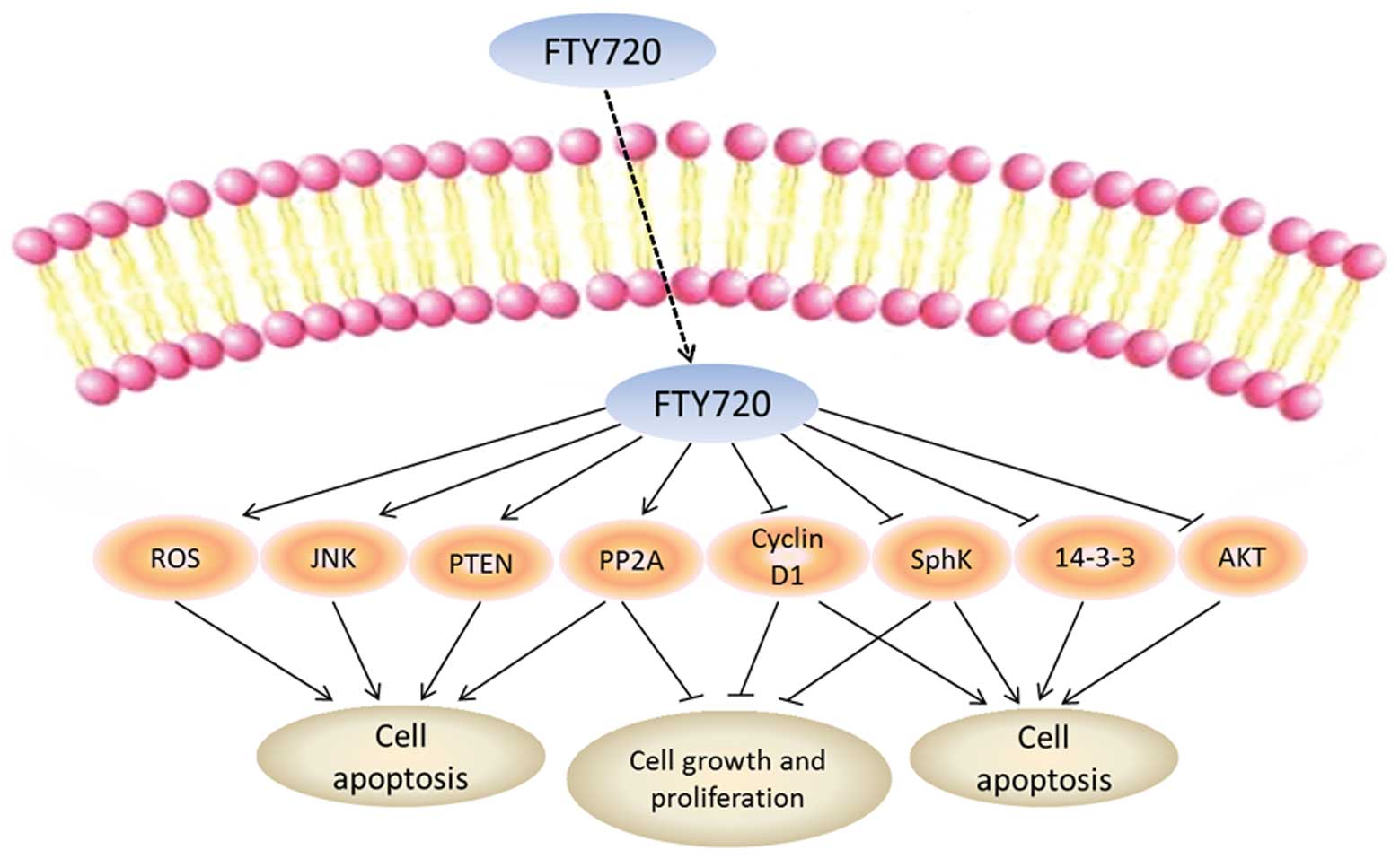
may explain the biological effects of this drug (Fig. 1).
ROS
ROS are chemically reactive molecules containing
oxygen. Examples include oxygen ions and peroxides. ROS form as a
natural byproduct of the normal metabolism of oxygen and have
important roles in cell signaling and homeostasis (38). However, under oxidative stress
conditions, excessive ROS can damage cellular proteins, lipids and
DNA, leading to fatal lesions in cells that contribute to
carcinogenesis.
In acute lymphoblastic leukemia cells, FTY720
increased ROS generation in a time-dependent manner, which then
resulted in a caspase-independent cell death, and the antioxidant
NAC could block the cell death induced by FTY720 (25). The same occurred in mantle cell
lymphoma (23), indicating that ROS
is an upstream of cell death induced by FTY720. However, in
multiple myeloma cells, FTY720-induced ROS activity promoted
autophagy, reduced the expression of Mcl-1, survivin, bcl-2 and
increased cleavage of Bid, ultimately leading to apoptosis of
multiple myeloma cells (26).
Furthermore, in hepatic carcinoma cells, ROS generation induced by
FTY720 culminated in protein kinase C (PKC) δ activation and
subsequent caspase-3-dependent apoptosis (39). In these two cases, ROS generated by
FTY720 promoted cell apoptosis rather than death. Thus, the
difference demonstrates that ROS may be just a mediate between
FTY720 and apoptosis or cell death. However, whether FTY720 induces
apoptosis or cell death depends on cell types. Of note, OSU-2S, a
non-immunosuppressive analogue of FTY720, increased ROS generation,
suppressed cell growth in the same way as FTY720 in hepatocellular
carcinoma (40), suggesting a
direct effect on ROS of unphosphorylated form of FTY720.
Protein phosphatase 2A (PP2A)
PP2A is a ubiquitous and conserved serine/threonine
phosphatase with broad substrate specificity and diverse cellular
functions. PP2A has been shown to be genetically altered or
functionally inactivated in several types of solid cancer and
leukemias, and is therefore a tumor suppressor. Changes in PP2A
subunits or loss of phosphatase activity have been linked to cancer
development and to non-neoplastic diseases (41).
Currently, several studies have demonstrated that
the anticancer activity of FTY720 may depend on its ability to act
as a potent PP2A activator. In ovarian cancer cells, FTY720
activated PP2A, decreased autophagy and increased cell death
(42). In this case, autophagy
acted in a protective role. In acute myeloid leukemia (AML),
FTY720-induced PP2A activity resulted in a dose-dependent growth
inhibition and a significant decrease in viable cells. The specific
PP2A inhibitor OA partially rescued the cells from FTY720-induced
apoptosis (43), indicating that
PP2A activation is required for FTY720-induced apoptosis in AML
cells. FTY720 also induced cell apoptosis in lung tumour (44), CML and Ph-positive B-ALL progenitors
(45,46) potentially by disrupting SET-PP2A
interaction, allowing PP2A activation. Furthermore, animal studies
demonstrated that FTY720 has anticancer effects in vivo by
activating PP2A. Reports highlighted the efficacy of FTY720 in
animal models of AML, reporting restored PP2A activity, decreased
clonogenicity, and suppression of disease (47,48).
However, it is unclear whether the anticancer effects of FTY720 in
some malignancies should be attributed to activation of PP2A or to
other mechanisms (such as autophagy) (41). For example, although FTY720 induced
PP2A activation and cell death in acute lymphoblastic leukemia
cells, activation of PP2A is not required for FTY720-induced cell
death (25).
Mitogen-activated protein kinase (MAPK)
signaling pathway
MAPKs are serine/threonine-specific protein kinases
belonging to the CMGC (CDK/MAPK/GSK3/CLK) kinase group (49). They regulate proliferation, gene
expression, differentiation, mitosis, cell survival, and apoptosis
in cancer cells. There are several members in the MAPKs family,
among which the ERKs and c-Jun N-terminal kinases (JNKs) are the
most important in regulating cell death and survival.
FTY720 has recently been shown to induce the
activation of ERK1/ERK2 and JNK1/JNK2, but not p38 in breast and
colon cancer cells, which may result in the anti-proliferative
activity of FTY720 (50).
Furthermore, FTY720 increased phosphorylation of ERK in mantle cell
lymphoma (23), but decreased
phosphorylation of ERK in glioblastoma (33) and renal cancer cells (20); notably, they all finally contributed
to apoptosis of cancer cells. The mediators between FTY720 and the
MAPK signaling pathway have not been fully clarified, but recent
literature indicated that ROS may be involved in the activation of
the MAPK pathway. For example, curcumin induced phosphorylation of
ERK1/2 and p38 MAPK, yet the curcumin-induced phosphorylation of
ERK1/2 and p38 MAPK was attenuated in the presence of NAC, a
scavenger of ROS (51).
Furthermore, piperlongumine selectively killed glioblastoma cells
via JNK and p38 activation, and these activations were blocked by
NAC pre-treatment (52). Thus,
FTY720 may also activate MAPK signaling pathway through ROS
generation, but, to date, how FTY720 mediates MAPK has not been
studied. Although the relationship between FTY720 and MAPK
signaling pathway remains unclear, the MAPK signaling pathway may
be a pivotal target of FTY720 in cancer therapy.
PI3K/AKT/mTOR signaling pathway
PI3K/Akt/mTOR is another important intracellular
signaling pathway in regulating cell survival and death. The
mammalian target of rapamycin (mTOR) is a serine/threonine protein
kinase which works downstream of protein kinase B (Akt). Once
activated, mTOR triggers the phosphorylation of the downstream
target p70S6K1, enhances the transcription of certain mRNAs, and
increases the expression of proteins associated with proliferation
(53–55). It has been reported that the
PI3K/Akt/mTOR signaling pathway is overactivated in several types
of cancer, such as non-small cell lung cancer (56,57),
breast cancer (58), colorectal
cancer (59) and cholangiocarcinoma
(60). Therefore, some experimental
cancer drugs aim to inhibit the signaling sequence at some point
(61–63).
FTY720 could also facilitate PI3K/AKT signaling
pathway to induce cell death. In gastric cancer cells, FTY720
induced apparent decrease of phosphorylation of AKT (473) level,
followed by a concentration-dependent reduction of Bcl-2, a
concomitant increase of Bax, cleavage caspase-3, -9, PARP,
resulting in significant apoptosis in cancer cells (19). Moreover, FTY720 induced p-AKT
downmodulation in mantle cell lymphoma, which then caused cell
death (23,28). In addition, in prostate cancer
(64), neuroblastoma (65) and glioma (10,66)
cells, FTY720 also showed anticancer effects by downregulating the
phosphorylation of AKT.
Cyclin D1
Cyclin D1 belongs to the highly conserved cyclin
family. It functions as a regulatory subunit of CDK4 or CDK6, whose
activity is required for cell cycle G1/S transition. This protein
has also been shown to interact with tumor suppressor protein Rb
and the expression of cyclin D1 is regulated positively by Rb.
Mutations, amplification and overexpression of this protein, which
alters cell cycle progression, are frequently observed in a variety
of tumors and may contribute to tumorigenesis. Cyclin D1 has been
reported to be involved in tumorigenesis in breast cancer (67), prostate cancer (68), colorectal cancer (59,69),
ovarian carcinoma (70), non-small
cell lung cancer (71) and gastric
carcinoma (72) cells.
Recently, studies showed that in mantle cell
lymphoma (23,28), human hepatoma cells (14), rat glomerular mesangial cells
(73) and mouse skin
transplantation models (74),
FTY720 reduced the protein level of cyclin D1, induced G1 phase
cell cycle arrest, eventually inhibited cell proliferation and
induced cell death. Meanwhile, recent data have also indicated a
role for cyclin D2 and cyclin D3 in the pathogenesis of cancer
(75–81); however, no reports have studied the
effects of FTY720 on cyclin D2 or cyclin D3 and, thus, remains to
be explored.
Phosphatase and tensin homolog
(PTEN)
PTEN is a protein that, in humans, is encoded by the
PTEN gene (82). It is one of the
most commonly lost tumor suppressors in human cancer. During tumor
development, mutations and deletions of PTEN occur that inactivate
its enzymatic activity leading to increased cell proliferation and
reduced cell death. Frequent genetic inactivation of PTEN occurs in
glioblastoma (83,84), endometrial cancer (85), and prostate cancer (86), and reduced expression is found in
numerous other tumor types such as lung (87) and breast cancer (88).
However, studies on the effects of FTY720 on PTEN
are few and only one reported that FTY720 induced obvious PTEN
expression, consistent with a substantial decrease in p-Akt and
MDM2, finally inhibiting gastric cancer cell proliferation,
inducing G1 phase cell cycle arrest and apoptosis. Suppression of
PTEN expression with siRNA significantly activated Akt, resulting
in decreased apoptosis and increased cell survival (19). Thus, whether the anticancer activity
of FTY720 in some malignancies is related to downmodulation of PTEN
remains to be clarified.
Sphingosine kinase (SphK)
SphK is a conserved lipid kinase that catalyzes
formation sphingosine-1-phoshate (S1P) from the precursor
sphingolipid sphingosine. There are two forms of SphK, SphK1 and
SphK2.
SphK1 is a lipid enzyme with oncogenic properties
that converts the proapoptotic lipids ceramide and sphingosine into
the antiapoptotic lipid S1P and activates the signal transduction
pathways that lead to cell proliferation, migration, the activation
of the inflammatory response, and the impairment of apoptosis
(89). There is compelling evidence
that SphK1 activation contributes to tumorigenesis in prostate
cancer (90), colorectal cancer
(91) and breast cancer (92) cells. However, until recently, the
absence of clinically applicable SphK1 inhibitors limited the
translation of these findings into patients (89). Recent studies shed further insight
into the mode of action of FTY720 by demonstrating that it directly
inhibits SphK1 activity both in vitro and in vivo.
For instance, FTY720 could resensitize human colorectal cancer to
cetuximab by inhibiting SphK1 both in vitro and in
vivo, with inhibition of tumor growth, interference with signal
transduction, induction of cancer cell apoptosis, and prolongation
of mouse survival (91). In
addition to directly inhibiting SphK1 activity, FTY720 has been
demonstrated to induce SphK1 degradation via ubiquitination and
subsequent proteasomal processing (93); however, the precise mechanism has
not been defined.
There exist two mammalian isoforms of SphK, SphK1
and SphK2, which share a considerable degree of sequence
similarity, but differ in their developmental expression,
subcellular localization and ability to phosphorylate artificial
substrates (94). Moreover, FTY720
becomes phosphorylated mostly by SphK2. In neuroblastoma, FTY720,
but not P-FTY720, induced neuroblastoma cell death, inhibited the
growth of neuroblastoma xenografts and enhanced the
tumor-suppressive effect of topotecan both in vitro and
in vivo. Notably, FTY720 significantly decreased SphK2 mRNA
and protein expression instead of SphK1, disrupted the
ceramide-sphingosine-S1P balance (65). This may be due to the fact that
neuroblastoma predominantly expresses SphK2 (95) and overexpression of SphK2 in
neuroblastoma cells promoted tumor growth and cell
proliferation.
Thus, FTY720 appears to target SphK via multiple
mechanisms. With the well described role of SphK in oncogenesis,
such effects of FTY720 on this enzyme provide a mechanistic
explanation for its anticancer properties.
14-3-3
14-3-3 proteins are a family of conserved regulatory
molecules expressed in all eukaryotic cells. 14-3-3 proteins have
the ability to bind a multitude of functionally diverse signaling
proteins, including kinases, phosphatases and transmembrane
receptors. For example, 14-3-3 interacted with both the dynein
intermediate chain (DIC) and an Hsp70 co-chaperone Bcl-2-associated
athanogene 3 (BAG3), thereby recruiting chaperone-associated
protein cargos to dynein motors for their transport to aggresomes
(96). Through binding to apoptosis
signal-regulating kinase-1 (ASK-1), 14-3-3 negatively regulated the
kinase activity of ASK-1, thereby blocking activation of
stress-activated protein kinases (SAPK) such as p38 and JNK
(97).
It was recently shown that FTY720 straightly bound
to 14-3-3 proteins to render them phosphorylatable on Ser58 (in
14-3-3ζ) at the dimer interface. Phosphorylation of 14-3-3 induced
by FTY720 could facilitate, at least in part, the apoptotic
activity of this drug, as cells expressing non-phosphorylatable
14-3-3 exhibited attenuated apoptosis in response to FTY720
(98). Therefore, 14-3-3 proteins
are key intermediates linking FTY720 and mitochondria, apoptotic
commitment.
5. Conclusion
FTY720 was originally developed as an
immunosuppressive agent and is currently undergoing multiple
clinical trials, including prevention of kidney graft rejection and
treatment of relapsing multiple sclerosis. However, over the past
ten years, FTY720 has emerged as a key player in cancer therapy.
FTY720 has shown preclinical antitumor efficacy in cancer models,
including those of breast, bladder, glioblastoma, ovarian,
prostate, lung, liver and hematopoietic malignancies. In these
malignancies, the anticancer effects of FTY720 are reportedly
attributed to its cytotoxicity towards cancer cells through
caspase-dependent, caspase-independent or autophagic cell death
pathways. Moreover, in most instances, phosphorylation of FTY720 is
not required for its cytotoxic effect. To date, numerous molecular
targets have been proposed for the unphosphorylated form of FTY720,
including ROS, PP2A, cyclin D1, SphK1 and 14-3-3. These downstream
targets appear to be associated with the ability of FTY720 to
suppress cell growth and/or induce cell death. These observations
make FTY720 an attractive therapeutic drug for developing
alternative treatment protocols, and, possibly, for combining with
other anticancer drugs to overcome drug resistance and achieve
better outcomes. Thus, it is clear that further studies are
required to elucidate the full spectrum of direct and downstream
cellular targets of this FTY720. Ultimately, novel molecular
targets will be found and may hold promise for clinical cancer
therapy.
Acknowledgements
The present study was supported by grants from the
Key Subject of Jiangsu Province (X4200722).
References
|
1
|
Billich A, Bornancin F, Dévay P,
Mechtcheriakova D, Urtz N and Baumruker T: Phosphorylation of the
immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem.
278:47408–47415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paugh SW, Payne SG, Barbour SE, Milstien S
and Spiegel S: The immunosuppressant FTY720 is phosphorylated by
sphingosine kinase type 2. FEBS Lett. 554:189–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suzuki S, Enosawa S, Kakefuda T, et al: A
novel immunosuppressant, FTY720, with a unique mechanism of action,
induces long-term graft acceptance in rat and dog
allotransplantation. Transplantation. 61:200–205. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Enosawa S, Suzuki S, Kakefuda T, Li XK and
Amemiya H: Induction of selective cell death targeting on mature
T-lymphocytes in rats by a novel immunosuppressant, FTY720.
Immunopharmacology. 34:171–179. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suzuki S, Li XK, Shinomiya T, et al:
Induction of lymphocyte apoptosis and prolongation of allograft
survival by FTY720. Transplant Proc. 28:2049–2050. 1996.PubMed/NCBI
|
|
6
|
Suzuki S, Enosawa S, Kakefuda T, Amemiya
H, Hoshino Y and Chiba K: Long-term graft acceptance in allografted
rats and dogs by treatment with a novel immunosuppressant, FTY720.
Transplant Proc. 28:1375–1376. 1996.PubMed/NCBI
|
|
7
|
Suzuki S, Enosawa S, Kakefuda T, et al:
Immunosuppressive effect of a new drug, FTY720, on lymphocyte
responses in vitro and cardiac allograft survival in rats.
Transplant Immunol. 4:252–255. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pitman MR, Woodcock JM, Lopez AF and
Pitson SM: Molecular targets of FTY720 (fingolimod). Curr Mol Med.
12:1207–1219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Azuma H, Takahara S, Ichimaru N, et al:
Marked prevention of tumor growth and metastasis by a novel
immunosuppressive agent, FTY720, in mouse breast cancer models.
Cancer Res. 62:1410–1419. 2002.PubMed/NCBI
|
|
10
|
Sonoda Y, Yamamoto D, Sakurai S, et al:
FTY720, a novel immunosuppressive agent, induces apoptosis in human
glioma cells. Biochem Biophys Res Commun. 281:282–288. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chua CW, Lee DT, Ling MT, et al: FTY720, a
fungus metabolite, inhibits in vivo growth of androgen-independent
prostate cancer. Int J Cancer. 117:1039–1048. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schmid G, Guba M, Papyan A, et al: FTY720
inhibits tumor growth and angiogenesis. Transplant Proc.
37:110–111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang N, Qi Y, Wadham C, et al: FTY720
induces necrotic cell death and autophagy in ovarian cancer cells:
a protective role of autophagy. Autophagy. 6:1157–1167. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee TK, Man K, Ho JW, et al: FTY720
induces apoptosis of human hepatoma cell lines through
PI3-K-mediated Akt dephosphorylation. Carcinogenesis. 25:2397–2405.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vadas M, Xia P, McCaughan G and Gamble J:
The role of sphingosine kinase 1 in cancer: oncogene or
non-oncogene addiction? Biochim Biophys Acta. 1781:442–447. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang JD, Takahara S, Nonomura N, et al:
Early induction of apoptosis in androgen-independent prostate
cancer cell line by FTY720 requires caspase-3 activation. Prostate.
40:50–55. 1999. View Article : Google Scholar
|
|
17
|
Hu S, Vincenz C, Buller M and Dixit VM: A
novel family of viral death effector domain-containing molecules
that inhibit both CD-95- and tumor necrosis factor
receptor-1-induced apoptosis. J Biol Chem. 272:9621–9624. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cohen GM: Caspases: the executioners of
apoptosis. Biochem J. 326(Pt 1): 1–16. 1997.
|
|
19
|
Zheng T, Meng X, Wang J, et al: PTEN- and
p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric
cancer cells and nude mice. J Cell Biochem. 111:218–228. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ubai T, Azuma H, Kotake Y, et al: FTY720
induced Bcl-associated and Fas-independent apoptosis in human renal
cancer cells in vitro and significantly reduced in vivo tumor
growth in mouse xenograft. Anticancer Res. 27:75–88.
2007.PubMed/NCBI
|
|
21
|
Yasui H, Hideshima T, Raje N, et al:
FTY720 induces apoptosis in multiple myeloma cells and overcomes
drug resistance. Cancer Res. 65:7478–7484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Susin SA, Lorenzo HK, Zamzami N, et al:
Molecular characterization of mitochondrial apoptosis-inducing
factor. Nature. 397:441–446. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu Q, Alinari L, Chen CS, et al: FTY720
shows promising in vitro and in vivo preclinical activity by
downmodulating Cyclin D1 and phospho-Akt in mantle cell lymphoma.
Clin Cancer Res. 16:3182–3192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Q, Zhao X, Frissora F, et al: FTY720
demonstrates promising preclinical activity for chronic lymphocytic
leukemia and lymphoblastic leukemia/lymphoma. Blood. 111:275–284.
2008. View Article : Google Scholar
|
|
25
|
Wallington-Beddoe CT, Hewson J, Bradstock
KF and Bendall LJ: FTY720 produces caspase-independent cell death
of acute lymphoblastic leukemia cells. Autophagy. 7:707–715. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liao A, Hu R, Zhao Q, et al: Autophagy
induced by FTY720 promotes apoptosis in U266 cells. Eur J Pharm
Sci. 45:600–605. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alinari L, Baiocchi RA and Praetorius-Ibba
M: FTY720-induced blockage of autophagy enhances anticancer
efficacy of milatuzumab in mantle cell lymphoma: is FTY720 the next
autophagy-blocking agent in lymphoma treatment? Autophagy.
8:416–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alinari L, Mahoney E, Patton J, et al:
FTY720 increases CD74 expression and sensitizes mantle cell
lymphoma cells to milatuzumab-mediated cell death. Blood.
118:6893–6903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ogata M, Hino S, Saito A, et al: Autophagy
is activated for cell survival after endoplasmic reticulum stress.
Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wirawan E, Vanden Berghe T, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: for better or for worse.
Cell Res. 22:43–61. 2012. View Article : Google Scholar
|
|
33
|
Estrada-Bernal A, Palanichamy K, Ray
Chaudhury A and Van Brocklyn JR: Induction of brain tumor stem cell
apoptosis by FTY720: a potential therapeutic agent for
glioblastoma. Neuro Oncol. 14:405–415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ng KT, Man K, Ho JW, et al: Marked
suppression of tumor growth by FTY720 in a rat liver tumor model:
The significance of downregulation of cell survival Akt pathway.
Int J Oncol. 30:375–380. 2007.PubMed/NCBI
|
|
35
|
Zhou C, Ling MT, Kin-Wah Lee T, Man K,
Wang X and Wong YC: FTY720, a fungus metabolite, inhibits invasion
ability of androgen-independent prostate cancer cells through
inactivation of RhoA-GTPase. Cancer Lett. 233:36–47. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kappos L, Radue EW, O’Connor P, et al: A
placebo-controlled trial of oral fingolimod in relapsing multiple
sclerosis. N Engl J Med. 362:387–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Azuma H, Takahara S, Horie S, Muto S,
Otsuki Y and Katsuoka Y: Induction of apoptosis in human bladder
cancer cells in vitro and in vivo caused by FTY720 treatment. J
Urol. 169:2372–2377. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Devasagayam TP, Tilak JC, Boloor KK, Sane
KS, Ghaskadbi SS and Lele RD: Free radicals and antioxidants in
human health: current status and future prospects. J Assoc
Physicians India. 52:794–804. 2004.PubMed/NCBI
|
|
39
|
Hung JH, Lu YS, Wang YC, et al: FTY720
induces apoptosis in hepatocellular carcinoma cells through
activation of protein kinase C delta signaling. Cancer Res.
68:1204–1212. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Omar HA, Chou CC, Berman-Booty LD, et al:
Antitumor effects of OSU-2S, a nonimmunosuppressive analogue of
FTY720, in hepatocellular carcinoma. Hepatology. 53:1943–1958.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Perrotti D and Neviani P: Protein
phosphatase 2A: a target for anticancer therapy. Lancet Oncol.
14:e229–e238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yin X, Zhang N and Di W: Regulation of
LC3-dependent protective autophagy in ovarian cancer cells by
protein phosphatase 2A. Int J Gynecol Cancer. 23:630–641. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang Y, Huang Q, Lu Y, Li X and Huang S:
Reactivating PP2A by FTY720 as a novel therapy for AML with C-KIT
tyrosine kinase domain mutation. J Cell Biochem. 113:1314–1322.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saddoughi SA, Gencer S, Peterson YK, et
al: Sphingosine analogue drug FTY720 targets I2PP2A/SET and
mediates lung tumour suppression via activation of
PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 5:105–121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Neviani P, Santhanam R, Trotta R, et al:
The tumor suppressor PP2A is functionally inactivated in blast
crisis CML through the inhibitory activity of the BCR/ABL-regulated
SET protein. Cancer Cell. 8:355–368. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Neviani P, Santhanam R, Oaks JJ, et al:
FTY720, a new alternative for treating blast crisis chronic
myelogenous leukemia and Philadelphia chromosome-positive acute
lymphocytic leukemia. J Clin Invest. 117:2408–2421. 2007.
View Article : Google Scholar
|
|
47
|
Cristobal I, Garcia-Orti L, Cirauqui C,
Alonso MM, Calasanz MJ and Odero MD: PP2A impaired activity is a
common event in acute myeloid leukemia and its activation by
forskolin has a potent anti-leukemic effect. Leukemia. 25:606–614.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Roberts KG, Smith AM, McDougall F, et al:
Essential requirement for PP2A inhibition by the oncogenic receptor
c-KIT suggests PP2A reactivation as a strategy to treat
c-KIT+ cancers. Cancer Res. 70:5438–5447. 2010.
View Article : Google Scholar
|
|
49
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nagaoka Y, Otsuki K, Fujita T and Uesato
S: Effects of phosphorylation of immunomodulatory agent FTY720
(fingolimod) on antiproliferative activity against breast and colon
cancer cells. Biol Pharm Bull. 31:1177–1181. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lee YJ, Kim NY, Suh YA and Lee C:
Involvement of ROS in curcumin-induced autophagic cell death.
Korean J Physiol Pharmacol. 15:1–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu JM, Pan F, Li L, et al: Piperlongumine
selectively kills glioblastoma multiforme cells via reactive oxygen
species accumulation dependent JNK and p38 activation. Biochem
Biophys Res Commun. 437:87–93. 2013. View Article : Google Scholar
|
|
53
|
Borders EB, Bivona C and Medina PJ:
Mammalian target of rapamycin: biological function and target for
novel anticancer agents. Am J Health Syst Pharm. 67:2095–2106.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yap TA, Garrett MD, Walton MI, Raynaud F,
de Bono JS and Workman P: Targeting the PI3K-AKT-mTOR pathway:
progress, pitfalls, and promises. Curr Opin Pharmacol. 8:393–412.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Alvarez M, Roman E, Santos ES and Raez LE:
New targets for non-small-cell lung cancer therapy. Expert Rev
Anticancer Ther. 7:1423–1437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rosell R, Felip E, Garcia-Campelo R and
Balana C: The biology of non-small-cell lung cancer: identifying
new targets for rational therapy. Lung Cancer. 46:135–148. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zheng J, Zou X and Yao J: The antitumor
effect of GDC-0941 alone and in combination with rapamycin in
breast cancer cells. Chemotherapy. 58:273–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liao WT, Li TT, Wang ZG, et al:
MicroRNA-224 promotes cell proliferation and tumor growth in human
colorectal cancer by repressing PHLPP1 and PHLPP2. Clin Cancer Res.
19:4662–4672. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yothaisong S, Dokduang H, Techasen A, et
al: Increased activation of PI3K/AKT signaling pathway is
associated with cholangiocarcinoma metastasis and PI3K/mTOR
inhibition presents a possible therapeutic strategy. Tumour Biol.
Jul 6–2013.(Epub ahead of print).
|
|
61
|
Wu P and Hu YZ: PI3K/Akt/mTOR pathway
inhibitors in cancer: a perspective on clinical progress. Curr Med
Chem. 17:4326–4341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ghayad SE and Cohen PA: Inhibitors of the
PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent
Pat Anticancer Drug Discov. 5:29–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cortot A, Armand JP and Soria JC:
PI3K-AKT-mTOR pathway inhibitors. Bull Cancer. 93:19–26. 2006.(In
French).
|
|
64
|
Chua CW, Chiu YT, Yuen HF, et al:
Suppression of androgen-independent prostate cancer cell
aggressiveness by FTY720: validating Runx2 as a potential
antimetastatic drug screening platform. Clin Cancer Res.
15:4322–4335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li MH, Hla T and Ferrer F: FTY720 inhibits
tumor growth and enhances the tumor-suppressive effect of topotecan
in neuroblastoma by interfering with the sphingolipid signaling
pathway. Pediatr Blood Cancer. 60:1418–1423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chang CY, Ma KH, Wang JK, Tung YL and
Chueh SH: Inhibition of protein kinase C promotes differentiation
of neuroblastoma x glioma NG108–15 hybrid cells. Eur J Neurosci.
34:1074–1084. 2011.
|
|
67
|
Weng JR, Bai LY, Chiu CF, Hu JL, Chiu SJ
and Wu CY: Cucurbitane triterpenoid from Momordica charantia
induces apoptosis and autophagy in breast cancer cells, in part,
through peroxisome proliferator-activated receptor gamma
activation. Evid Based Complement Alternat Med.
2013:9356752013.PubMed/NCBI
|
|
68
|
Tolba MF, Esmat A, Al-Abd AM, et al:
Caffeic acid phenethyl ester synergistically enhances docetaxel and
paclitaxel cytotoxicity in prostate cancer cells. IUBMB Life.
65:716–729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li X, Pu J, Jiang S, et al: Henryin, an
ent-kaurane diterpenoid, inhibits Wnt signaling through
interference with beta-catenin/TCF4 interaction in colorectal
cancer cells. PLoS One. 8:e685252013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chung YC, Lu LC, Tsai MH, et al: The
inhibitory effect of ellagic acid on cell growth of ovarian
carcinoma cells. Evid Based Complement Alternat Med.
2013:3067052013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li H, Sun L, Xu Y, et al: Overexpression
of MTA3 correlates with tumor progression in non-small cell lung
cancer. PLoS One. 8:e666792013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chang MS, Kim DH, Roh JK, et al:
Epstein-Barr virus-encoded BARF1 promotes proliferation of gastric
carcinoma cells through regulation of NF-kappaB. J Virol.
87:10515–10523. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jiang J, Huang X, Wang Y, Deng A and Zhou
J: FTY720 induces cell cycle arrest and apoptosis of rat glomerular
mesangial cells. Mol Biol Rep. 39:8243–8250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li QY, Chi YY and Liu SQ: Cell cycle
arrest effects of large-dose FTY720 on lymphocytes in mouse skin
transplantation models. Immunopharmacol Immunotoxicol. 30:365–381.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gulappa T, Reddy RS, Suman S, Nyakeriga AM
and Damodaran C: Molecular interplay between cdk4 and p21 dictates
G/G cell cycle arrest in prostate cancer cells. Cancer Lett.
337:177–183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bermudez O, Hennen E, Koch I, Lindner M
and Eickelberg O: Gli1 mediates lung cancer cell proliferation and
Sonic Hedgehog-dependent mesenchymal cell activation. PLoS One.
8:e632262013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Aigelsreiter A, Ress AL, Bettermann K, et
al: Low expression of the putative tumour suppressor spinophilin is
associated with higher proliferative activity and poor prognosis in
patients with hepatocellular carcinoma. Br J Cancer. 108:1830–1837.
2013. View Article : Google Scholar
|
|
78
|
Guo J, Gao J, Li Z, et al: Adenovirus
vector-mediated Gli1 siRNA induces growth inhibition and apoptosis
in human pancreatic cancer with Smo-dependent or Smo-independent Hh
pathway activation in vitro and in vivo. Cancer Lett. 339:185–194.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yoon JS, Kim HM, Yadunandam AK, et al:
Neferine isolated from Nelumbo nucifera enhances anti-cancer
activities in Hep3B cells: Molecular mechanisms of cell cycle
arrest, ER stress induced apoptosis and anti-angiogenic response.
Phytomedicine. 20:1013–1022. 2013. View Article : Google Scholar
|
|
80
|
Kurokawa K, Akaike Y, Masuda K, et al:
Downregulation of serine/arginine-rich splicing factor 3 induces G1
cell cycle arrest and apoptosis in colon cancer cells. Oncogene.
March 18–2013.(Epub ahead of print).
|
|
81
|
Xing Z, Zhang Y, Zhang X, Yang Y, Ma Y and
Pang D: Fangchinoline induces G1 arrest in breast cancer cells
through cell-cycle regulation. Phytother Res. Feb 11–2013.(Epub
ahead of print).
|
|
82
|
Steck PA, Pershouse MA, Jasser SA, et al:
Identification of a candidate tumour suppressor gene, MMAC1, at
chromosome 10q23.3 that is mutated in multiple advanced cancers.
Nat Genet. 15:356–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Santoni M, Burattini L, Nabissi M, et al:
Essential role of gli proteins in glioblastoma multiforme. Curr
Protein Pept Sci. 14:133–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yang P, Wang Y, Peng X, et al: Management
and survival rates in patients with glioma in China (2004–2010): a
retrospective study from a single-institution. J Neurooncol.
113:259–266. 2013.PubMed/NCBI
|
|
85
|
Brucka A and Szyłło K: Immunoexpression of
the PTEN protein and matrix metalloproteinase-2 in endometrial
cysts, endometrioid and clear cell ovarian cancer. Ginekol Pol.
84:344–351. 2013.PubMed/NCBI
|
|
86
|
Abdulkareem IH and Blair M: Effects of
indomethacin on expression of PTEN tumour suppressor in human
cancers. Niger Med J. 54:100–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
McQuitty E, Zhang W, Hendrickson H, et al:
Lung adenocarcinoma biomarker incidence in Hispanic versus
non-Hispanic white patients. Arch Pathol Lab Med. Jun 26–2013.(Epub
ahead of print).
|
|
88
|
Filippini SE and Vega A: Breast cancer
genes: beyond BRCA1 and BRCA2. Front Biosci. 18:1358–1372. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Alshaker H, Sauer L, Monteil D, et al:
Therapeutic potential of targeting SK1 in human cancers. Adv Cancer
Res. 117:143–200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lim KG, Tonelli F, Berdyshev E, et al:
Inhibition kinetics and regulation of sphingosine kinase 1
expression in prostate cancer cells: functional differences between
sphingosine kinase 1a and 1b. Int J Biochem Cell Biol.
44:1457–1464. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Rosa R, Marciano R, Malapelle U, et al:
Sphingosine kinase 1 overexpression contributes to cetuximab
resistance in human colorectal cancer models. Clin Cancer Res.
19:138–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lim KG, Tonelli F, Li Z, et al: FTY720
analogues as sphingosine kinase 1 inhibitors: enzyme inhibition
kinetics, allosterism, proteasomal degradation, and actin
rearrangement in MCF-7 breast cancer cells. J Biol Chem.
286:18633–18640. 2011. View Article : Google Scholar
|
|
93
|
Tonelli F, Lim KG, Loveridge C, et al:
FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1
and promote its proteasomal degradation in human pulmonary artery
smooth muscle, breast cancer and androgen-independent prostate
cancer cells. Cell Signal. 22:1536–1542. 2010. View Article : Google Scholar
|
|
94
|
Pitson SM: Regulation of sphingosine
kinase and sphingolipid signaling. Trends Biochem Sci. 36:97–107.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Li MH, Hla T and Ferrer F: Sphingolipid
modulation of angiogenic factor expression in neuroblastoma. Cancer
Prev Res (Phila). 4:1325–1332. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Xu Z, Graham K, Foote M, et al: 14-3-3
targets chaperone-associated misfolded proteins to aggresomes. J
Cell Sci. 126:4173–4186
|
|
97
|
Zhang L, Chen J and Fu H: Suppression of
apoptosis signal-regulating kinase 1-induced cell death by 14-3-3
proteins. Proc Natl Acad Sci USA. 96:8511–8515. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Woodcock JM, Ma Y, Coolen C, et al:
Sphingosine and FTY720 directly bind pro-survival 14-3-3 proteins
to regulate their function. Cell Signal. 22:1291–1299. 2010.
View Article : Google Scholar : PubMed/NCBI
|