Introduction
Pancreatic carcinoma is a refractory disease with
few effective therapies (1). Only
18% of patients survive 1 year after diagnosis and the 5-year
survival rate is 4%. By the time patients exhibit symptoms and the
disease is diagnosed, it is already beyond the early stages
(2,3). The capacity of a tumor to grow and
proliferate is dependent on a small subset of cells, the cancer
stem cells (CSCs), which are immature cells that can replicate or
self-renew, and differentiate or grow into different types of
cancer cells (4).
VP16 is an important chemotherapeutic agent that is
used to treat a wide spectrum of human cancers. It has been in
clinical use for more than 2 decades and remains one of the most
highly prescribed anticancer drugs in the world (5). This drug is a pro-apoptosis agent
which triggers cell death pathways (6).
In the present study, we examined the effect of VP16
on human pancreatic cancer cells and the change of pancreatic
CSCs.
Materials and methods
Cell lines
The Panc-1 human pancreatic cancer cell line was
purchased from the Shanghai Cell Bank (Shanghai, China).
Cell growth inhibition assays
The inhibitory effect of VP16 (etoposide; Qilu
Pharmaceutical Co., Ltd., Shandong, China) on the growth of Panc-1
cells was evaluated by CCK-8 kit (Sigma-Aldrich, Ireland) (7). In brief, 200 μl medium/well containing
2×103 cells were seeded in 96-well microtiter plates for
the CCK-8 assays. After treatment with VP16, cells in the 96-well
plates were used for CCK-8 assays. The optical density was measured
at 490 nm using a microplate reader (SpectraMax 340; Molecular
Devices Co., Sunnyvale, CA, USA).
The cell viability ratio was calculated using the
following formula: Inhibitory ratio (%) = (ODcontrol −
ODtreated)/ODcontrol × 100% (8). Results from 3 independent experiments
in triplicates are presented.
Cell cycle analysis
Cell cycle distribution was analyzed as previously
described (9). In brief,
1×106 cells were incubated with the indicated amounts
(0–300 μg/ml) of VP16 for 48 h. Cells were collected and then fixed
in 70% ethanol at 4°C for 24 h. Cells were washed again with PBS
and incubated with PI (10 μg/ml) with simultaneous RNase treatment
at 37°C for 30 min. Cell DNA content was measured using a FACStar
flow cytometer (Becton-Dickinson, San Jose, CA, USA).
Annexin V/PI analysis
Apoptosis was determined by staining cells with
Annexin V-fluorescein isothiocyanate as previously described
(10). In brief, 1×106
cells were incubated with the indicated concentrations (0–300
μg/ml) of VP16 for 48 h. Five microliters Annexin V-FITC and 5 μl
PI were added. Cells were vortexed and incubated for 15 min in the
dark. Binding buffer (200 μl) was added to each tube. Flow
cytometric analysis was performed immediately after staining. All
assays were independently repeated for 3 times.
Mitochondrial membrane potential (Δψm)
assay
The loss of Δψm in the cells is one of the
mechanisms of induction of apoptosis, which has been linked to the
initiation and activation of apoptotic cascades (11). With a variety of stimuli, including
the translocation of Bax from the cytosol to the mitochondria, this
event occurs, which triggers the release of cytochrome c
from the mitochondria to the cytosol (12). In brief, 1×106 cells were
incubated with the indicated amounts (0–300 μg/ml) of VP16. Cells
were incubated with JC-1 dye for 15 min at 37°C, and then
resuspended in 200 μl PBS for FACS analysis.
Protein extraction and western blot
assays
The expressions of proteins were evaluated using
western blot analysis, as previously described (13). In brief, cells were treated with
different concentrations of VP16 (0–300 μg/ml) for 48 h. Cells were
collected and suspended in 5 volumes of lysis buffer (20 mM HEPES,
pH 7.9, 20% glycerol, 200 mM KCl, 0.5 mM EDTA, 0.5% NP-40, 0.5 mM
DTT, 1% protease inhibitor cocktail). Lysates were collected.
Supernatant samples containing 40 μg total protein were loaded on
SDS-PAGE gel for electrophoresis. Membranes were blocked for 1 h
with 5% milk. Membranes were then incubated overnight at 4°C with
primary antibodies (caspases-3 and -9, cytochrome c, PARP,
Bcl-2, Bax and β-actin) and with horseradish peroxidase-conjugated
secondary antibodies for 1.5 h at room temperature. The experiment
was repeated 3 times. Nuclear and Cytoplasmic Protein Extraction
kit was used to extract protein of cytochrome c.
Quantification of caspase-3 activity
Caspase-3 activity was analyzed using a caspase-3
activity assay kit (Beyotime, China). Cells were treated with the
indicated reagents for 24 h and cell lysates were prepared for the
following experiment according to the manufacturer’s
instructions.
Change of antigens of CSCs by flow
cytometry
Previous studies found that malignant tumors were
heterogeneous. This subpopulation of CSCs has been demonstrated to
be responsible for tumor initiation, proliferation, recurrence and
resistance to chemotherapy (14–18).
On the basis of the above findings, we hypothesized that the ratio
of CSCs and non-CSCs must change during treatment of VP16. Cells
were treated with VP16 (100 μg/ml) for 48 h, and then flow
cytometry was performed to detect expression of CSC markers, such
as CD133, CD44, CD24 and ESA antigens (BioLegend Inc., San Diego,
CA, USA). Each analysis included 100,000 events. Isotype-matched
mouse IgG was used as a control.
Statistical analysis
All data are expressed as means ± SD. Statistical
comparisons of results were made using analysis of variance
(ANOVA). Differences between groups were determined using the
Student’s t-test for unpaired observations and P-values <0.05
were considered to indicate statistically significant
differences.
Results
VP16 induces apoptosis of Panc-1
cells
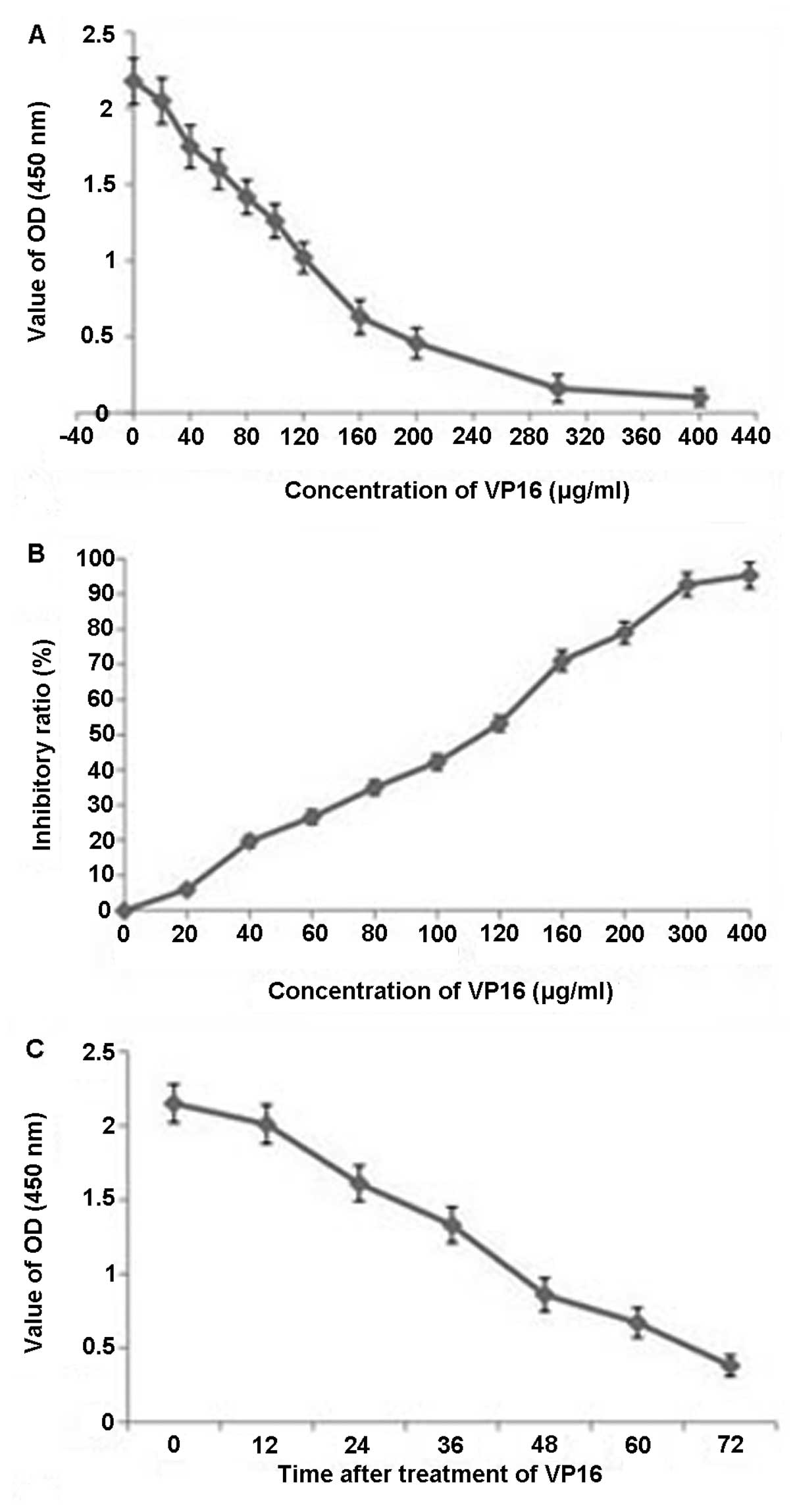
A dose-dependent reduction in cell growth was
observed. The ratio of inhibition with different concentrations of
VP16 is shown in Fig. 1. These
results indicated that VP16 inhibited Panc-1 cell proliferation in
a dose- and time-dependent manner.
Effects of VP16 on cell cycle and
apoptosis in Panc-1 cells
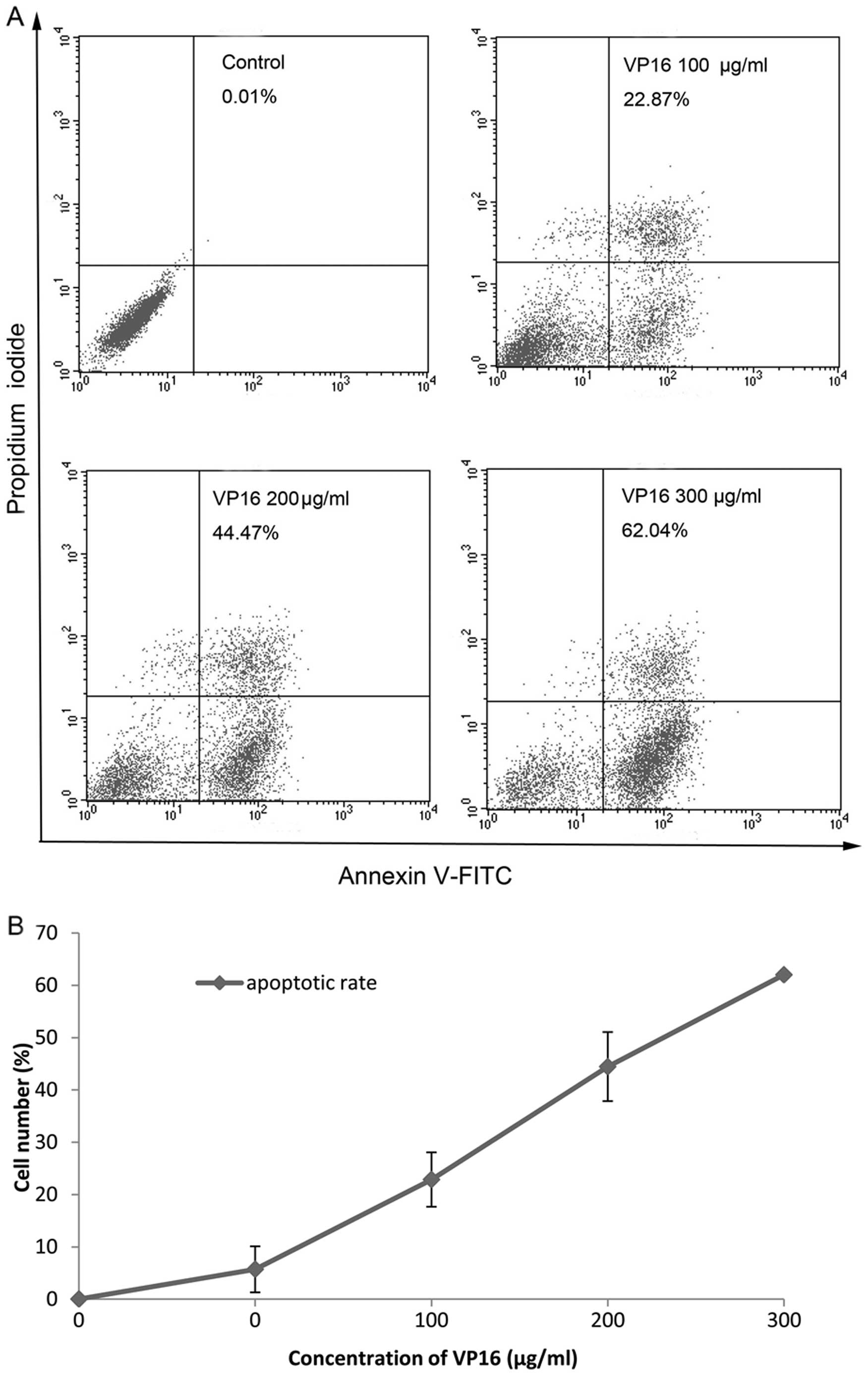
When we stained Panc-1 cells with Annexin V-FITC and
PI, the proportion of apoptotic cells was significantly increased
in a dose-dependent manner (Fig.
2).
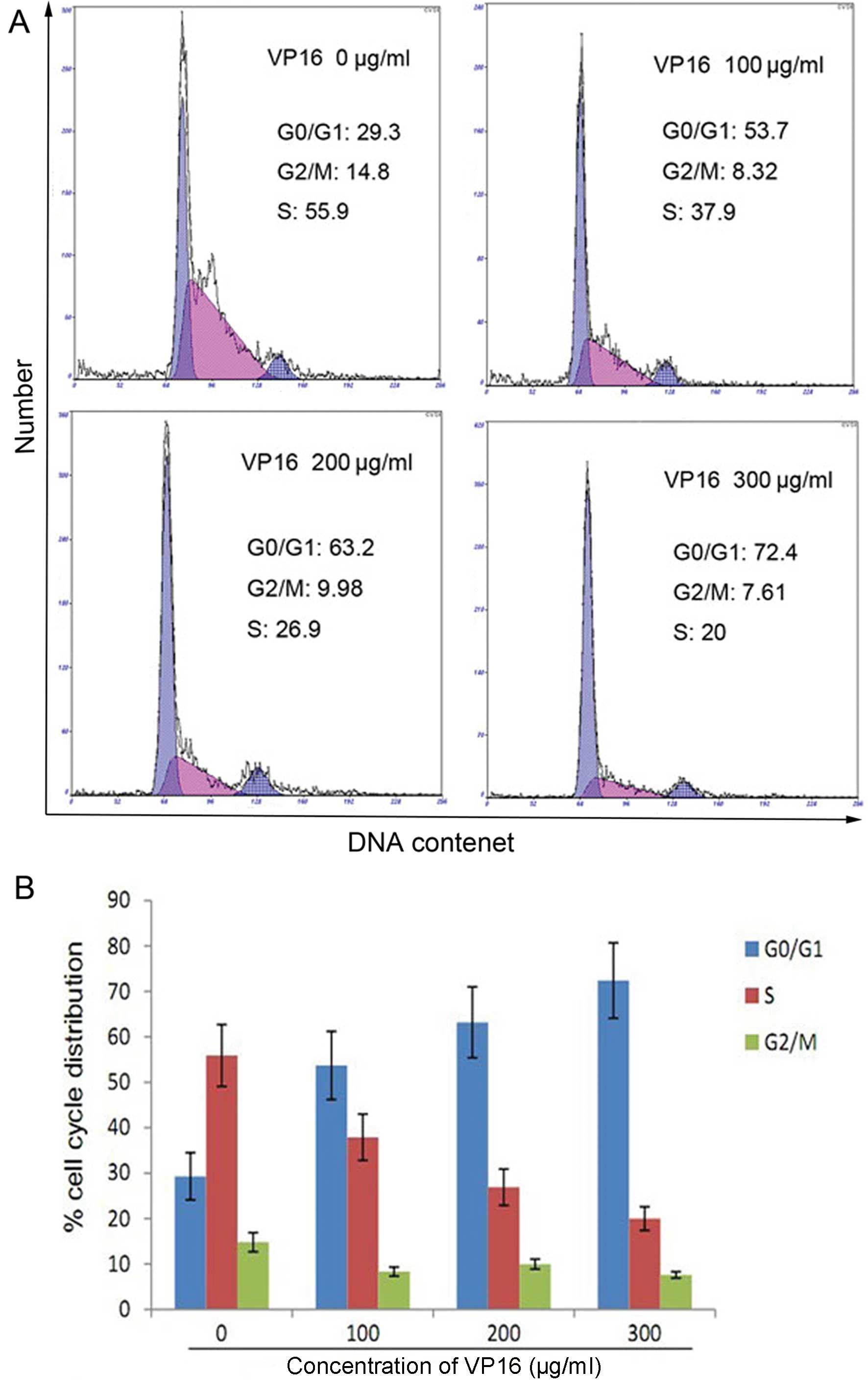
The effect of VP16 on cell cycle distribution
indicated that there was a higher number of cells in the G1 phase,
in higher concentration of VP16 compared with the control
(P<0.05) and, at the same time, the number of S phase cells was
significantly decreased (Fig.
3).
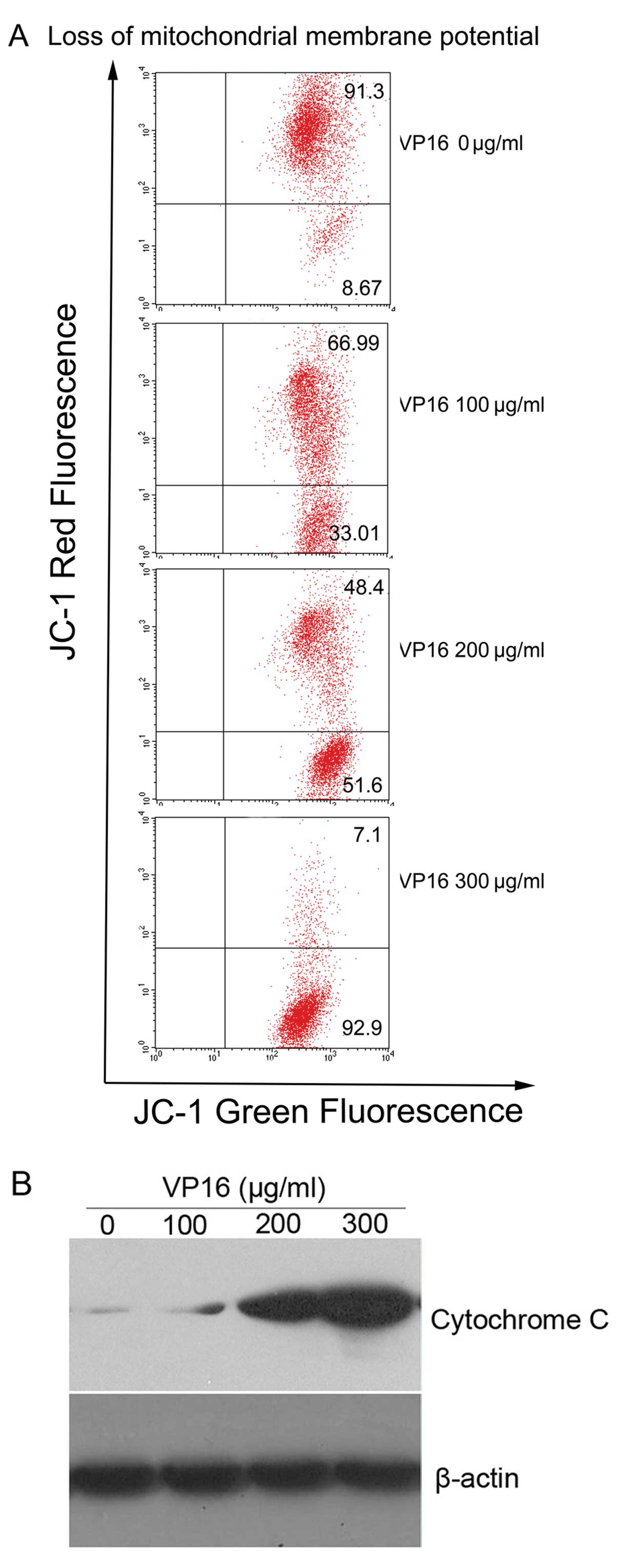
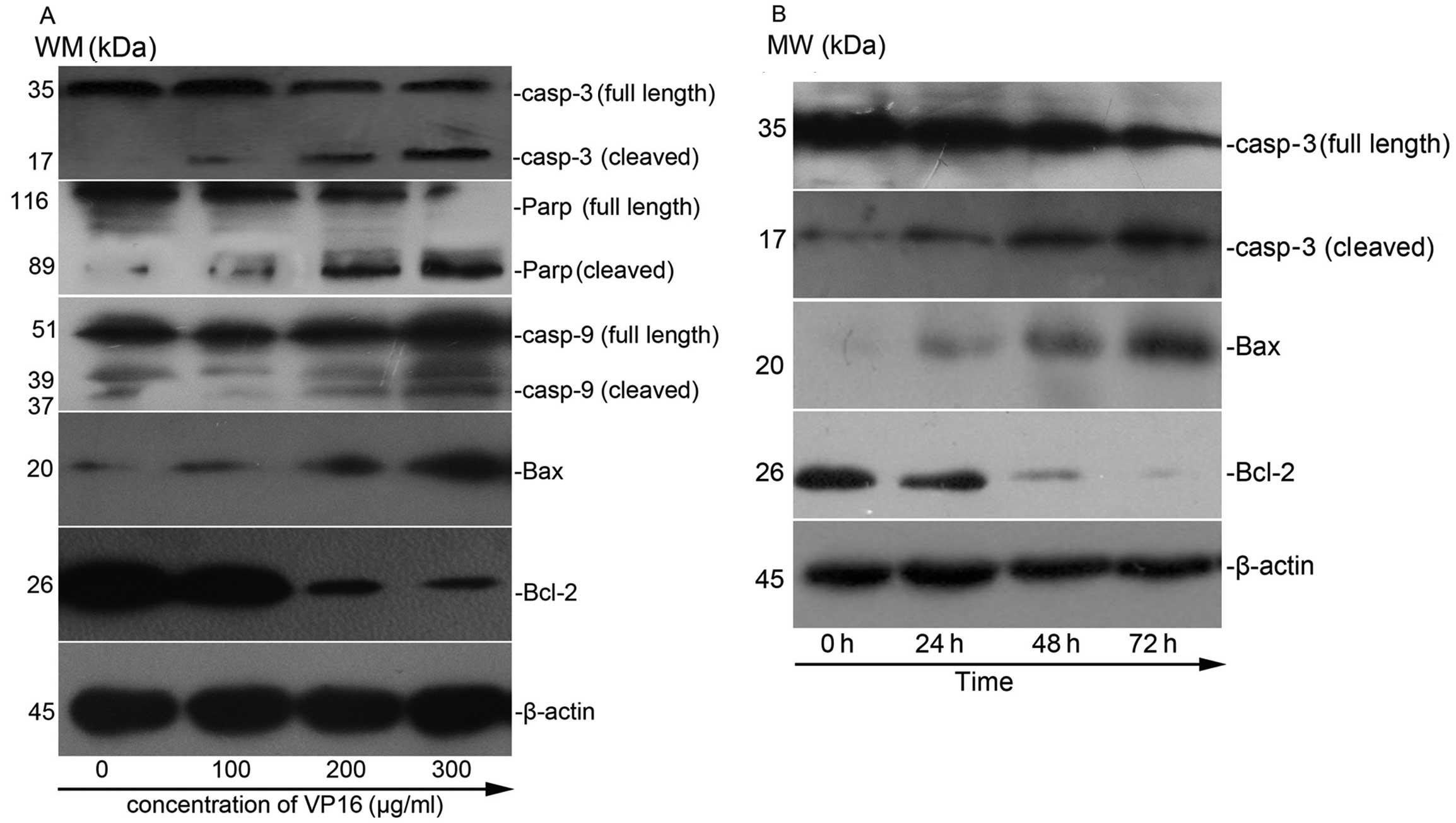
VP16 induces loss of Δψm and subsequently
enhances the release of cytochrome c in Panc-1 cells
VP16 in Panc-1 cells resulted in a dose-dependent
increase in the number of JC-1 dye-positive cells from 8.67 to
92.9%. Western blot analysis revealed that VP16 caused a
dose-dependent increase in the release of cytochrome c into
cytosol, which confirmed the disruption of the Δψm after VP16
treatment (Fig. 4).
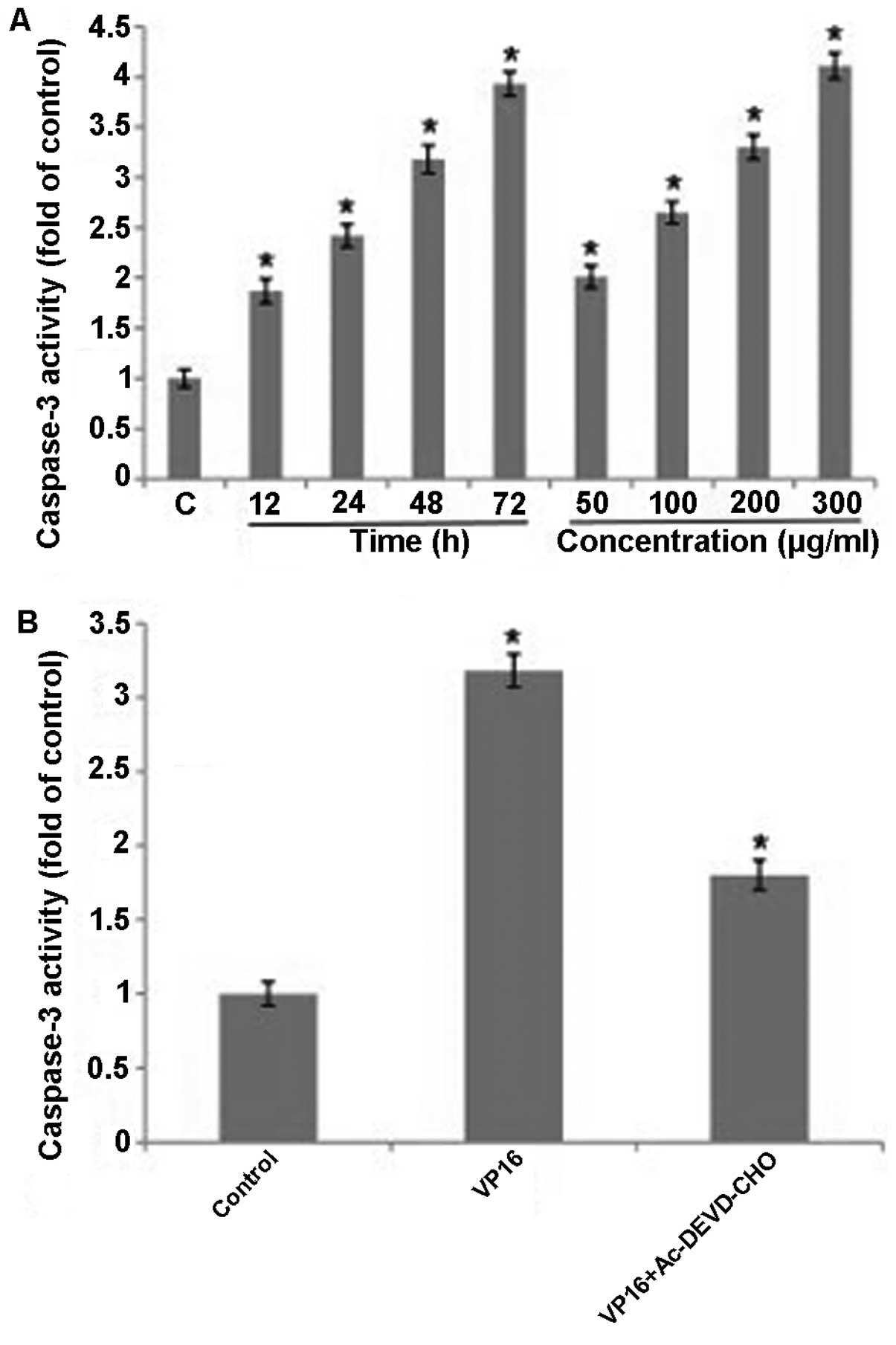
Involvement of caspase-3 activation
Caspases are a family of cysteine proteases that are
essentially involved in the apoptotic pathway. We investigated
whether caspase-3 was involved in apoptosis induced by VP16 in
Panc-1 cells. The data showed that activity of caspase-3 increased
significantly in a dose- and time-dependent manner in the cells
treated with VP16 (Fig. 5A). In
addition, activity of caspase-3 decreased in the combined treatment
with VP16 and caspase-3 inhibitor (Ac-DEVD-CHO) compared with the
VP16 treatment group (Fig. 5B). The
results demonstrated that VP16-induced apoptosis was dependent on
caspase-3.
Expression of apoptosis-related proteins
in VP16-treated cells
These data showed that treatment of VP16 in Panc-1
cells resulted in a dose- and time-dependent reduction in the
levels of the anti-apoptotic proteins and an increase in the level
of pro-apoptotic proteins (Fig.
6).
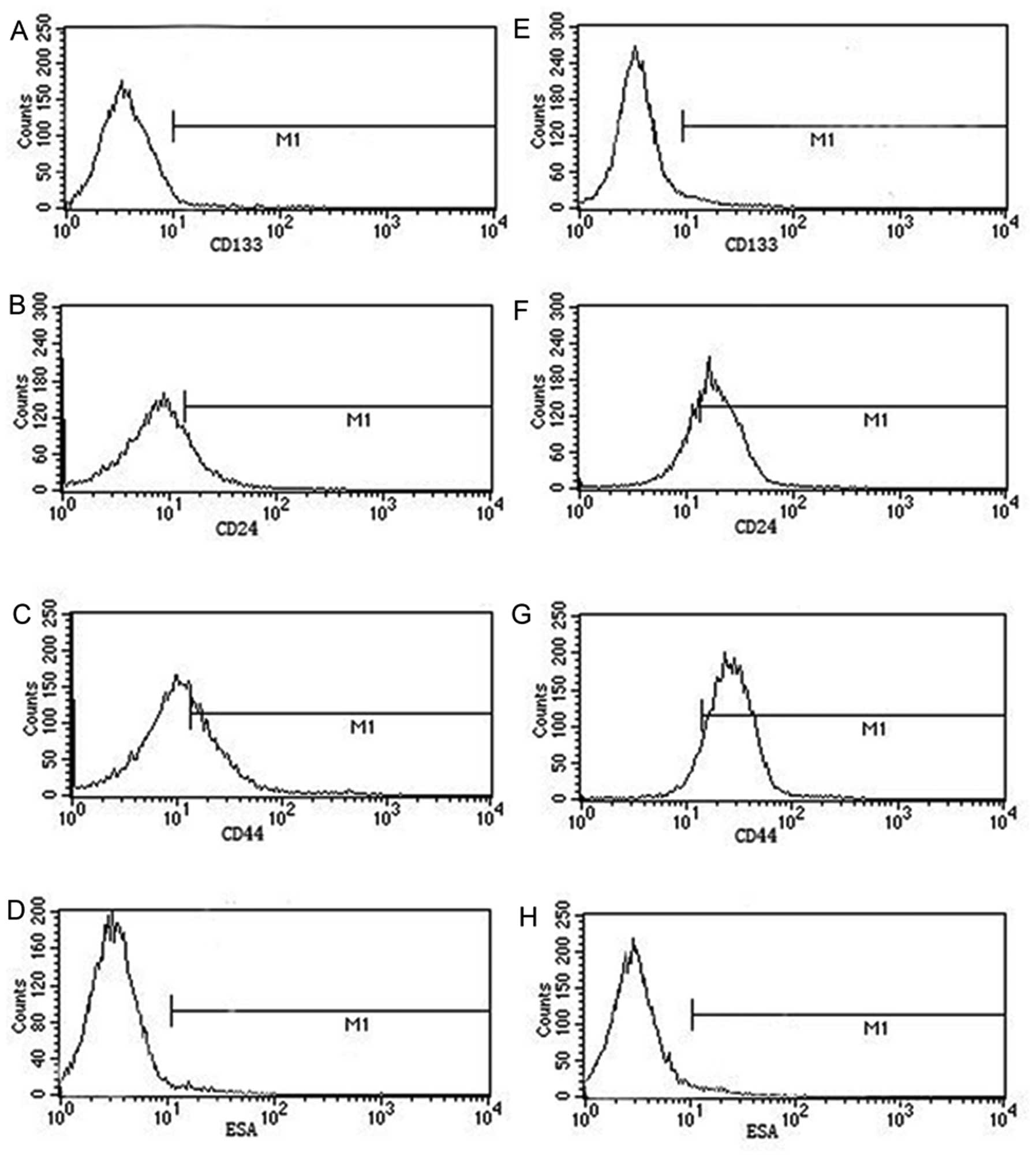
Cells treated with VP16 highly express
CD44, CD24, ESA and CD133
Results from the above studies and hypotheses of
CSCs, prompted us to study the change of CSC-related markers. The
data were: CD133, 0.31 vs. 1.54%; CD24, 19.51 vs. 69.47%; CD44,
21.55 vs. 93.23%; ESA, 1.51 vs. 8.38% (P<0.05, Fig. 7). The data demonstrated that most of
the cells killed by VP16 were non-CSCs and that CSCs derived from
Panc-1 cells played key role in chemotherapy resistance.
Discussion
In the present study, we investigated the molecular
mechanisms of VP16-induced apoptosis in Panc-1 cells and the change
of markers of CSCs. VP16 inhibited the growth of cells in a dose-
and time-dependent manner. In addition, VP16 markedly induced
apoptosis in Panc-1 cells in the same manner, which was confirmed
by Annexin V and PI staining. Based on the above assays, we
observed a strong growth inhibitory effect of VP16 in Panc-1 cells;
we then determined the possible mechanism of anti-proliferative
activity of VP16. Our data demonstrated that treatment with VP16 in
Panc-1 cells induced G1 phase arrest of cell cycle, indicating that
one of the mechanisms was inhibition of cell cycle progression.
Loss of mitochondrial membrane potential was suicidal to cells as
they became bioenergetically deficient (30) and that led to release of cytochrome
c into the cytosol. As the level of cytochrome c
increased in the cytosol, it interacted with Apaf-1 and ATP formed
a complex with pro-caspase-9, leading to activation of caspases-9
and -3 which led to the cleavage of PARP (19,20).
Mitochondria depolarization, considered an
irreversible step in the apoptosis process, could trigger a cascade
of caspases. The mitochondrion was a major subcellular compartment
where the Bcl-2 family members interacted with each other or
exerted function independently (21).
The Bcl-2 family of proteins include proteins that
could either inhibit (Bcl-2, Bcl-xL, etc.) or induce (Bax, Bak,
Bad, etc.) apoptosis. The anti-apoptotic proteins prevented
cytochrome c release by forming heterodimer complexes with
pro-apoptotic Bcl-2 family proteins (22).
The pro-apoptotic Bcl-2 proteins including Bax and
Bak facilitate release of apoptogenic molecules from mitochondria
to the cytosol and accelerate apoptotic cell death (23–25).
To evaluate the role of caspases, the master
executioner during apoptotic cell death, in VP16-induced cell
death, the activities of caspase-3 were assessed using the
caspase-3 colorimetric assay kits. The data suggested that
caspase-3 inhibitor could significantly inhibit the activities of
caspase-3 in the VP16-induced cell death, indicating that apoptosis
in VP16-treated Panc-1 cells occurred in a caspase-dependent
manner.
VP16 induces apoptosis in various cancer cell lines
and performs antitumor activity (3–5). In
the present study, we observed a strong inhibitory effect of VP16
on Panc-1 cells in vitro, indicating chemotherapy drugs
could kill cancer cells. However, pancreatic cancer patients
continue to have a dismal prognosis with an average overall median
survival of 4–6 months. The overall 5-year survival is <5%
(1). The preliminary data suggested
that tumor-cell repopulation describes the continuing proliferation
of surviving tumor stem cells that can occur during a course of
radiotherapy or chemotherapy (26–29).
These findings led to our investigation of the role
of CSCs during cytotoxic cancer therapy. Our results suggested that
CSCs were more resistant to VP16 and most CSCs could escape
cytotoxic cancer therapy. The relationship between CSCs and
repopulation during the process of radiotherapy and chemotherapy
will be our next research project.
Acknowledgements
The authors thank the participants for taking part
in the study and for the laboratory assistance.
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2007. CA Cancer J Clin. 57:43–66. 2007. View Article : Google Scholar
|
|
2
|
Niederhuber JE, Brennan MF and Menck HR:
The National Cancer Data Base report on pancreatic cancer. Cancer.
76:1671–1677. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Thomas A, Murray T, et al: Cancer
statistics, 2002. CA Cancer J Clin. 52:23–47. 2002. View Article : Google Scholar
|
|
4
|
Bjerkvig R, Tysnes BB, Aboody KS, et al:
Opinion: the origin of the cancer stem cell: current controversies
and new insights. Nat Rev Cancer. 5:899–904. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baldwin EL and Osheroff N: Etoposide,
topoisomerase II and cancer. Curr Med Chem Anticancer Agents.
5:363–372. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meresse P, Dechaux E, Monneret C, et al:
Etoposide: discovery and medicinal chemistry. Curr Med Chem.
11:2443–2466. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Zhang J, Wang H, et al: miRNA-135a
promotes breast cancer cell migration and invasion by targeting
HOXA10. BMC Cancer. 12:1112012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yingkun N, Lvsong Z and Huimin Y: Shikonin
inhibits the proliferation and induces the apoptosis of human HepG2
cells. Can J Physiol Pharmacol. 88:1138–1146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuo SY, Hsieh TJ, Wang YD, et al:
Cytotoxic constituents from the leaves of Cinnamomum
subavenium. Chem Pharm Bull. 56:97–101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Madan E, Prasad S, Roy P, et al:
Regulation of apoptosis by resveratrol through JAK/STAT and
mitochondria mediated pathway in human epidermoid carcinoma A431
cells. Biochem Biophys Res Commun. 377:1232–1237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu X, Wu N, Xia P, et al: Correlation
between low-level expression of the tumor suppressor gene TAp73 and
the chemoresistance of human glioma stem cells. Cancer Chemother
Pharmacol. 69:1205–1212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yen YP, Tsai KS, Chen YW, et al: Arsenic
induces apoptosis in myoblasts through a reactive oxygen
species-induced endoplasmic reticulum stress and mitochondrial
dysfunction pathway. Arch Toxicol. 86:923–933. 2012. View Article : Google Scholar
|
|
13
|
Cha JD and Kim JY: Essential oil from
Cryptomeria japonica induces apoptosis in human oral
epidermoid carcinoma cells via mitochondrial stress and activation
of caspases. Molecules. 17:3890–3901. 2012.PubMed/NCBI
|
|
14
|
Reya T, Morrison SJ, Clarke MF, et al:
Stem cells, cancer, and cancer stem cells. Nature. 414:105–111.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Collins AT, Berry PA, Hyde C, et al:
Prospective identification of tumorigenic prostate cancer stem
cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li C, Heidt DG, Dalerba P, et al:
Identification of pancreatic cancer stem cells. Cancer Res.
67:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fukuda K, Saikawa Y, Ohashi M, et al:
Tumor initiating potential of side population cells in human
gastric cancer. Int J Oncol. 34:1201–1207. 2009.PubMed/NCBI
|
|
19
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wolf BB and Green DR: Suicidal tendencies:
apoptotic cell death by caspase family proteinases. J Biol Chem.
274:20049–20052. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kluck RM, Bossy-Wetzel E, Green DR, et al:
The release of cytochrome c from mitochondria: a primary site for
Bcl-2 regulation of apoptosis. Science. 275:1132–1136. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huppertz B, Kadyrov M and Kingdom JC:
Apoptosis and its role in the trophoblast. Am J Obstet Gynecol.
195:29–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsujimoto Y and Shimizu S: Bcl-2 family:
life-or-death switch. FEBS Lett. 466:6–10. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hockenbery D, Nuñez G, Milliman C, et al:
Bcl-2 is an inner mitochondrial membrane protein that blocks
programmed cell death. Nature. 348:334–336. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qiu B, Wang Y, Tao J, et al: Expression
and correlation of Bcl-2 with pathological grades in human glioma
stem cells. Oncol Rep. 28:155–160. 2012.PubMed/NCBI
|
|
26
|
Davis AJ, Chapman W, Hedley DW, et al:
Assessment of tumor cell repopulation after chemotherapy for
advanced ovarian cancer: pilot study. Cytometry A. 51:1–6. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stephens TC and Peacock JH: Tumour volume
response, initial cell kill and cellular repopulation in B16
melanoma treated with cyclophosphamide and
1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea. Br J Cancer.
36:313–321. 1977.PubMed/NCBI
|
|
28
|
Wu L and Tannock IF: Repopulation in
murine breast tumours during and after sequential treatments with
cyclophosphamide and 5-fluorouracil. Cancer Res. 63:2134–2138.
2003.PubMed/NCBI
|
|
29
|
Wang L, Huang X, Zheng X, et al:
Enrichment of prostate cancer stem-like cells from human prostate
cancer cell lines by culture in serum-free medium and
chemoradiotherapy. Int J Biol Sci. 9:472–479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin F, Zhao L, Guo YJ, et al: Influence of
Etoposide on anti-apoptotic and multidrug resistance-associated
protein genes in CD133 positive U251 glioblastoma stem-like cells.
Brain Res. 1336:103–111. 2010. View Article : Google Scholar : PubMed/NCBI
|