Introduction
Cutaneous melanoma is an extremely aggressive skin
cancer with high metastatic potential. Indeed, melanoma
metastatisation to distant organs is the primary cause of human
skin cancer-related deaths. Although the majority of melanomas are
detected at an early stage (<1 mm Breslow thickness) and
surgically cured, melanomas diagnosed at later stages are
associated with poor survival rates, since they are refractory to
most of the available therapies (1). The molecular mechanisms associated
with the acquisition of a metastatic phenotype by melanoma cells
are not very well defined and, therefore, great effort is being
given to identify molecular determinants involved in the metastatic
switch that may either cause or contribute to the aggressiveness of
melanoma.
At the Markers and Tissue Resources for Melanoma
Meeting held in 2005 at Gaithersburg (MD, USA), a list of melanoma
biomarkers was generated (2). The
list included biomarkers associated with melanocytic tumour
progression, potential markers with prognostic significance and
putative therapeutic targets. These markers were established using
two criteria: differential expression during melanocytic tumour
progression and detection in routinely processed formalin-fixed
paraffin-embedded tissues by means of a reliable method. More
recently, a melanocytic tumour progression tissue microarray (TMA)
was developed in order to evaluate these candidate biomarkers by
immunohistochemical analysis (3).
The TMA includes samples from benign nevi, primary cutaneous
melanomas and melanoma metastases (lymph node and visceral
metastases). Nevertheless, TMA does not include cutaneous melanoma
metastases nor does it distinguish between the different types of
metastases.
The majority of human melanoma cell lines, derived
from primary or metastatic lesions, secrete the vascular
endothelial growth factor (VEGF)-A and express the receptors
VEGFR-1, VEGFR-2 and neuropilins (NRP) (4). The simultaneous presence of the growth
factor and its receptors in melanoma cells allows the establishment
of an autocrine loop, sustained by the interaction of VEGF-A mainly
with VEGFR-2, promoting the ability of melanoma cells to migrate
and invade the extracellular matrix (ECM) (5). We also demonstrated the existence in
cutaneous melanomas and lymph node metastases of a differential
VEGFR expression pattern (6,7), which
seems to confer distinct invasive properties to the different types
of metastases.
In an attempt to identify new players involved in
melanoma progression that may favour tumour spreading to distant
sites, we compared the gene expression profiles of two cell clones
derived from the human cutaneous metastatic melanoma cell line M14:
a highly invasive clone (M14C2/MK18) and a clone (M14C2/C4) with
low ability to invade the ECM. The highly invasive phenotype of
M14C2/MK18 cells was correlated with NRP-1 expression, activation
of a VEGF-A/VEGFR-2 autocrine loop and secretion of matrix
metalloprotease-2 (MMP-2), and resulted in a high in vivo
growth rate in a murine model. The results indicated that
platelet-derived growth factor (PDGF)-C upregulation and calpain-3
downregulation were directly involved in M14C2/MK18 melanoma cell
invasiveness.
Materials and methods
Reagents
Cell culture media and reagents were purchased from
Lonza (Basel, Switzerland); fetal bovine serum (FBS) was from
Euroclone (Pero, Italy), heparin, insulin/transferrin/selenium
supplement (ITS) and gelatin were from Sigma-Aldrich (St. Louis,
MO, USA) and fatty acid-free bovine serum albumin (BSA) was from
Roche (Mannheim, Germany). VEGF-A, PlGF and polyclonal goat
anti-VEGF-A antibodies used in ELISA (AF-293 and BAF-293) were from
R&D Systems (Abingdon, UK). Control goat and mouse IgGs were
from Sigma-Aldrich. The VEGFR tyrosine kinase inhibitor
4-[(4′-chloro-2′-fluoro)phenylamino]-6,7-dimethoxyquinazoline
(VEGFRin) (8) and the calpain
inhibitor N-Acetyl-Leu-Leu-Met (ALLM) (9) were from Merck (Darmstadt,
Germany).
Cell lines
The origin and culture conditions of M14C2/C4 and
M14C2/MK18 subclones, other human melanoma cell lines, melanocyte
and human umbilical vein endothelial cell (HUVEC) cultures, were
previously described (4,7).
RT-PCR analysis
Total cellular RNA from the different cell lines and
from HUVECs was prepared using a RNeasy Midi kit (Qiagen, Hilden,
Germany), following the manufacturer’s directions. Three micrograms
of total RNA/sample was subjected to reverse transcription by the
AMV enzyme (Roche) for 60 min at 42°C in 25 μl. Five microliters of
this cDNA preparation were used for each PCR amplification reaction
by 1 unit of Dynazyme II DNA polymerase (Finnzymes OY, Espoo,
Finland), utilizing the following primers and annealing conditions:
VEGFR-2 forward, 5′-CACAGGAAACCTGGAG AATCAGACGACAAG-3′ and reverse,
5′-TGGTCGACCATG ACGATGGACAAGTA-3′, which amplify a 402-bp fragment
(annealing for 1 min at 58°C); NRP-1 forward, 5′-ATGGAGAG
GGGGCTGCCG-3′ and reverse, 5′-CTATCGCGCTGTCG GTGTA-3′, which
amplify a 720-bp fragment (annealing for 30 sec at 52°C); PDGF-C
forward, 5′-GAGATGGCAGTTG GACTTAG-3′ and reverse,
5′-TCAGCCACTGCACTGCA CAG-3′, which amplify a 471-bp fragment
(annealing for 1 min at 60°C). The RNA integrity and the correct
reverse transcription of the samples were assessed by testing each
cDNA preparation for the amplification of the
glyceraldehyde-phosphate dehydrogenase (GAPDH) housekeeping gene:
forward, 5′-TCCCATCACCATCTTCCA-3′ and reverse primer, 5′-CAT
CACGCCACAGTTTCC-3′, which amplify a 380-bp fragment (annealing for
30 sec at 58°C).
Evaluation of VEGF-A secretion
Semi-confluent melanoma cell cultures were incubated
in 0.1% BSA/RPMI-1640 medium without FBS for 24 h. Culture
supernatants were then collected and concentrated at least 10-fold
in Centriplus concentrators (Amicon, Beverly, MA, USA). Cells were
detached from the flasks with a solution of 1 mM EDTA in PBS
(EDTA/PBS), and the total cell number/culture was recorded.
Quantification of the amount of VEGF-A in the concentrated
supernatants was performed using Maxisorp Nunc-Immuno™ plates
(Nunc, Roskilde, Denmark) coated with goat anti-VEGF-A IgGs, as
previously described (4).
Migration and invasion assays
The in vitro migration assay was performed
using Boyden chambers equipped with 8-μm pore diameter
polycarbonate filters (Nuclepore; Whatman, Inc., Clifton, NJ, USA),
coated with 5 μg/ml gelatin (10).
Briefly, melanoma cells were collected from continuous cultures,
washed, suspended in migration medium (1 μg/ml heparin/0.1% BSA in
RPMI-1640) and loaded (2×105 cells) into the upper
compartment of the Boyden chambers. Migration medium with or
without the chemotactic stimuli was added to the lower compartment
of the chambers. After incubation of the chambers at 37°C in a
CO2 incubator for 5 h, the filters were removed from the
chambers, the cells were fixed in ethanol and stained in 0.5%
crystal violet. The migrated cells, attached to the lower surface
of the filters, were counted under a microscope. Twelve
high-magnification microscopic fields (×200 magnification),
randomly selected on triplicate filters, were scored for each
experimental condition.
In vitro ECM cell invasion was analysed in
Boyden chambers as described for the migration assay, but utilising
polycarbonate filters coated with 20 μg of the basement membrane
matrix Matrigel (BD Biosciences, Buccinasco, Italy) (5).
In a set of experiments, migration or invasion
assays were performed in the presence of VEGFRin, the calpain
inhibitor ALLM, antibodies against NRP-1 (H-286; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), PDGF-C (AF1560; R&D
Systems) or MMP-2 (clone 42-5D11; Merck) or the corresponding
control immunoglobulins (Igs). In the case of VEGFRin, the drug was
dissolved in DMSO at a stock concentration of 20 mM, and melanoma
cells were pre-incubated in the presence of 20 μM of the inhibitor
for 24 h at 37°C in a CO2 incubator. ALLM was also
dissolved in DMSO at a stock concentration of 50 mM, and cells were
pre-incubated in the presence of 15 or 30 μM of the inhibitor for
40 min at room temperature in a rotating wheel. The concentrations
of VEGFRin or ALLM tested had no effect on cell viability. The
final concentrations of DMSO used as solvent of the inhibitors did
not affect cell migration. Pretreatment with the antibodies was
performed for 45 min at room temperature in a rotating wheel. The
cells were then loaded in the Boyden chambers without removing the
antibody, ALLM or adding fresh VEGFRin.
Western blot analysis
Specific protein expression in the different cell
lines was analysed in semi-confluent cell cultures, growing in
6-well plates. Cells were washed with PBS, lysed and boiled in 400
μl of SDS sample buffer (50 mM Tris-HCl pH 6.8, 100 mM
dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol).
Secreted proteins were tested in cell culture supernatants,
obtained as indicated for the evaluation of VEGF-A secretion,
mixing the supernatants with an equal amount of 2X SDS sample
buffer before boiling. Proteins were then separated on
polyacrylamide gels and immunoblotting was performed (experimental
details provided upon request), incubating the membranes overnight
at 4°C with the following primary antibodies: rabbit polyclonal
antibody anti-β-tubulin (H-235, diluted to 0.2 μg/ml; Santa Cruz
Biotechnology, Inc.); mouse monoclonal antibody anti-NRP-1 (A12,
diluted to 1 μg/ml; Santa Cruz Biotechnology, Inc.); mouse
monoclonal antibody anti-MMP-2 (clone 42-5D11, diluted to 1 μg/ml)
and anti-TIMP-2 antibody (clone T2-101, diluted to 1 μg/ml; Merck);
and goat polyclonal antibody anti-PDGF-C (AF1560, diluted to 1
μg/ml; R&D Systems).
Microarray analysis of gene
expression
Gene expression profiling, utilizing
GeneChip® Human Genome U133A Arrays (Affymetrix, Inc.,
High Wycombe, UK), and data analysis, using the Prediction Analysis
of Microarrays (PAM) and the Significant Analysis of Microarrays
(SAM) tests, which allow identification of significant differences
between different groups, were performed as previously described
(11). The genes identified as
modulated in M14C2/MK18 cells corresponded to genes whose
transcripts were significantly up- or down-modulated according to
both PAM and SAM analyses.
In vivo tumour growth
Tumours were induced in 5 week-old male CD1 Nu/Nu
mice (10 mice/group; Charles River, Calco, Italy) by intramuscular
injection of 5×106 M14C2/C4 or M14C2/MK18 melanoma
cells. Melanoma growth was monitored by measuring tumour nodules
with callipers every 2–3 days, and the tumour volumes were
calculated according to the following formula: volume =
[(width)2 × length]/2. The animals were euthanised when
their tumours reached a volume of ~1,500 mm3. All
procedures involving mice and care were performed in compliance
with our institutional guidelines and with international directives
(Directive 2010/63/EU of the European Parliament and of the
Council; Guide for the Care and Use of Laboratory Animals, United
States National Research Council, 2011).
Evaluation of in vitro cell growth
Cells (5×104/well) were re-suspended in
complete medium, seeded in 24-well plates and allowed to grow at
37°C in a CO2 incubator. Selected wells were previously
coated with 0.5 ml of a 200 μg/ml Matrigel solution and incubated
overnight at 37°C. Evaluation of cell growth was performed by
detaching and counting the cells at the indicated times.
Results
Characterization of the M14C2/MK18 cell
line
We previously published the isolation of the
M14C2/MK18 cell subclone obtained by transfection of an expression
vector that contains the cDNA encoding for VEGFR-2 in a cell clone
isolated from the metastatic cutaneous melanoma cell line M14
(7). These cells also express NRP-1
and MMP-2 (Fig. 1A and E),
molecules that are involved in ECM invasion by melanoma cells.
Indeed, ECM invasion by M14C2/MK18 cells in the absence of a
stimulus was ~40-fold more efficient than that observed in control
cells (parental cells or cells from the M14C2/C4 clone, transfected
with the pcDNA3 empty vector) (Fig.
1B). Exposure to a stimulus not related to VEGF-A or NRP-1
(i.e., ITS, at 5 μg/ml), further increased ECM invasion by
M14C2/MK18 cells and stimulated invasion by parental and M14C2/C4
cells. Moreover, M14C2/MK18 cells, which do not express VEGFR-1,
showed a specific chemotactic response to VEGF-A mediated by
VEGFR-2 (Fig. 1C), as demonstrated
by its down-modulation after treatment with a VEGFR-2 inhibitor and
by the lack of response to PlGF, a VEGF-A-related growth factor
that does not bind to VEGFR-2 (Fig.
1D). M14C2/MK18 cells, already described as melanoma cells
expressing VEGFR-2 (7,12), showed an increased ability to
secrete VEGF-A (5.35±0.89 ng/106 cells) as compared with
M14C2/C4 cells or with the parental cell clone (0.46±0.05 and
0.63±0.06 ng/106 cells, respectively). High levels of
the fully processed form of MMP-2 were also found in the
supernatant of M14C2/MK18 cells, whereas M14C2/C4 cells showed
barely detectable amounts of this protein (Fig. 1E). By contrast, the tissue inhibitor
of metalloproteases (TIMP-2), used as a loading control, was
expressed at similar levels in the supernatants of both cell clones
(Fig. 1E). TIMP-2 is involved in
docking pro-MMP-2 to the cell surface, where the pro-enzyme is
activated by membrane-bound MMPs and by an additional TIMP-2
molecule (13,14). Therefore, the balance between the
amount of MMP-2 and TIMP-2 determines the extent of tissue
remodeling and ECM invasion. Indeed, blockage of MMP-2 proteolytic
activity with a specific antibody down-modulated the invasiveness
of M14C2/MK18 cells (Fig. 1F).
Altogether, these results demonstrated that M14C2/MK18 cells
displayed in vitro highly invasive characteristics.
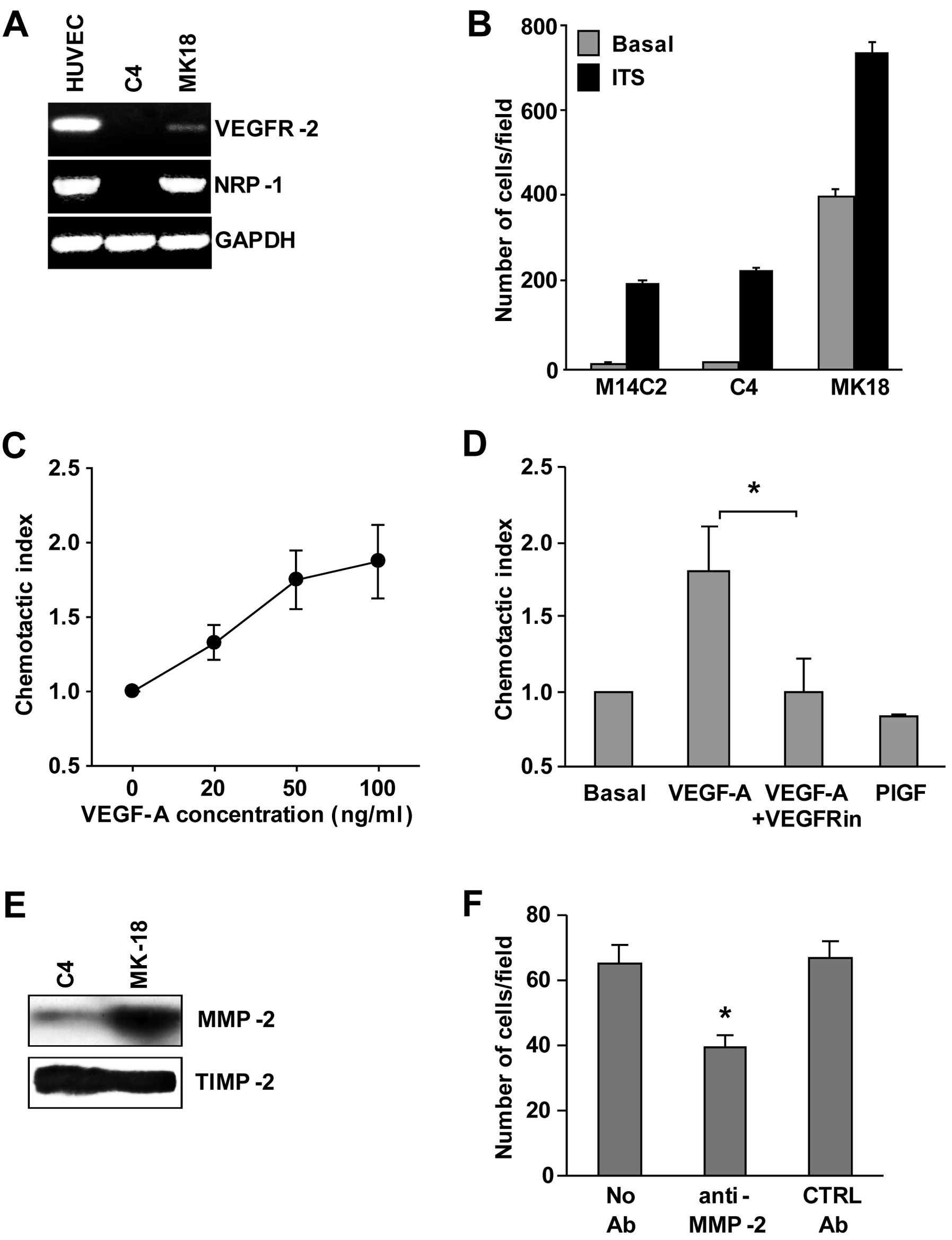 | Figure 1Characterization of M14C2/MK18 cells.
(A) RT-PCR analysis of VEGFR-2 and NRP-1 expression in M14C2/MK18
and M14C2/C4 cells. As a control for RNA integrity, analysis of the
GAPDH gene was also performed. HUVECs were included as positive
control. Results are representative of 1 out of 3 different
experiments with similar results. (B) ECM invasion by M14C2/MK18
and M14C2/C4 subclones and parental M14C2 cells was evaluated using
Boyden chambers equipped with Matrigel-coated filters, allowing the
cells to invade the Matrigel for 4 h, in response to migration
medium (basal invasion) or migration medium supplemented with ITS
(5 μg/ml). Each value represents the mean ± standard deviation (SD)
of the number of cells that had migrated per microscopic field from
three independent experiments. (C) Migration of M14C2/MK18 cells
was evaluated using Boyden chambers equipped with polycarbonate
gelatin-coated filters, allowing the cells to migrate for 5 h in
response to increasing concentrations of VEGF-A. Data are expressed
in terms of chemotactic index, calculated as the ratio between the
number of cells per microscopic field in the experimental condition
analysed and the number of cells per microscopic field in the basal
condition (i.e., in the absence of any stimulus). Each value
represents the mean ± SD of three independent experiments. (D) The
specificity of the chemotactic response induced by VEGF-A (30
ng/ml) in M14C2/MK18 cells was evaluated as described in panel C,
pre-incubating the cells (24 h at 37°C in a CO2
incubator) with culture medium in the absence or in the presence of
VEGFRin (20 μM). The response to VEGF-A was also compared with the
response to PlGF (50 ng/ml) that interacts only with VEGFR-1. Each
value represents the mean ± SD of three independent experiments.
*P<0.05, according to Student’s t-test analysis. (E)
Secretion of MMP-2 into the cell culture supernatant from
M14C2/MK18 and M14C2/C4 subclones was evaluated by western blot
analysis. Fifty microliters of concentrated cell supernatant,
corresponding to an equal number of cultured cells, was loaded in a
6–12% SDS-polyacrylamide gel gradient. As loading control, TIMP-2
protein secretion in the supernatant from both cell lines was also
evaluated. Results are representative of one out of three
independent experiments giving comparable results. (F) The
influence of anti-MMP-2 antibody treatment (3 μg/ml) on the ability
of M14C2/MK18 cells to spontaneously invade the ECM was analysed
using Boyden chambers equipped with Matrigel-coated filters,
allowing cells to invade for 2 h. As a negative control, a
non-specific antibody (mouse control IgG, CTRL Ab) was used. Each
value represents the mean ± SD of the number of cells that had
migrated per microscopic field from three independent experiments.
*P<0.05, according to Student’s t-test analysis,
comparing the invasion of cells treated with anti-MMP-2 antibodies
with that of cells untreated or treated with control
antibodies. |
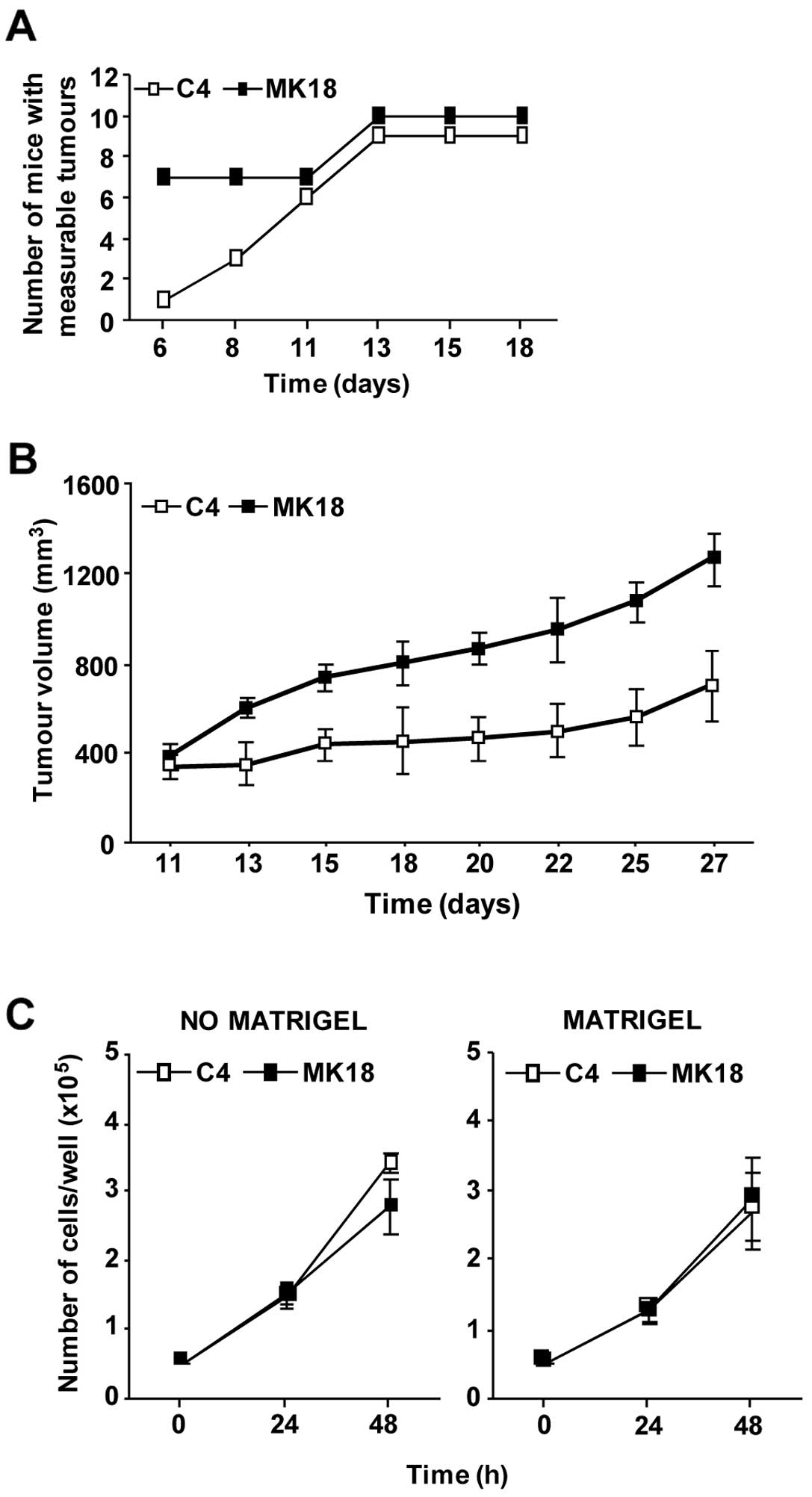
We next analysed whether the aggressive phenotype
showed by M14C2/MK18 cells in vitro was a characteristic
maintained by these cells when growing in an in vivo model.
Thus, cells were intramuscularly injected in CD1 Nu/Nu mice, and
the volume of the tumour nodules was monitored. Six days after
injection, 70% of the animals inoculated with M14C2/MK18 cells had
already developed measurable tumours, as compared to only 10% of
the mice injected with the M14C2/C4 cells (Fig. 2A). Only on day 11 after challenge
the number of mice showing measurable M14C2/C4 nodules was
identical to that of animals injected with M14C2/MK18 cells.
Moreover, from day 13 onward M14C2/MK18 tumour volume was
significantly higher with respect to that of control cells
(Fig. 2B). Notably, the augmented
in vivo growth of M14C2/MK18 cells was not due to an
elevated in vitro proliferation rate. In fact, M14C2/MK18
and M14C2/C4 cells showed comparable proliferation rates, even when
cells were allowed to grow on a basement membrane matrix (Fig. 2C). Therefore, the highly invasive
characteristics of M14C2/MK18 cells seemed to favour tumour
formation and growth also in vivo, likely as a result of the
interaction with the stromal compartment.
Differential gene expression in
M14C2/MK18 cells as compared with M14C2/C4 cells
In an attempt to identify the molecules responsible
for the elevated invasiveness of M14C2/MK18 cells, differential
gene expression analysis was performed in these cells and in the
M14C2/C4 control cells, utilizing GeneChip Human Genome Arrays. The
results showed a significant up-modulation of 18 genes (Table I) and down-modulation of 19 genes
(Table II) in the M14C2/MK18
cells. The up-modulated gene transcripts included genes encoding
for proteins involved in the regulation of the calcium metabolism
pathway [S100 calcium binding protein A2 (S100A2), stanniocalcin
(STC1), S100 calcium binding protein A3 (S100A3)],
cell-extracellular matrix interactions (COL5A2, LAMB3, PCOLCE2,
NRP-1) and PDGF-C, a cytokine involved in angiogenesis that
displays pleiotropic effects on multiple cellular targets.
Down-modulated molecules included gene products involved in
melanocytic differentiation (MLANA, DCT, RAB38), in lipid and
glucidic metabolism (APOE, ASAH1, GPM6B, GYG2) and CAPN3
(calpain-3), an endopeptidase endowed with numerous functions.
Notably, 11 out of 19 genes found to be down-modulated in the
M14C2/MK18 cells (genes highlighted in bold in Table II) are known to be under the
control of the microphthalmia-associated transcription factor
(MITF) (15).
 | Table IUpregulated genes in M14C2/MK18 cells
as compared with M14C2/C4 cells. |
Table I
Upregulated genes in M14C2/MK18 cells
as compared with M14C2/C4 cells.
| | | Fold-changeb | |
|---|
| | |
| |
|---|
| Probe | Symbol | Descriptiona | PAM | SAM | P-valuec |
|---|
| 218718_at | PDGF-C | Platelet-derived
growth factor-C | 2.38 | 132.27 | 0.0019 |
| 212298_at | NRP-1 | Neuropilin-1 | 2.33 | 103.09 | 0.0479 |
| 221730_at | COL5A2 | Collagen, type V,
α2 | 1.91 | 42.37 | 0.0331 |
| 205404_at | HSD11B1 | Hydroxysteroid
(11β) dehydrogenase 1 | 1.86 | 156.25 | 0.0003 |
| 204268_at | S100A2 | S100 calcium
binding protein A2 | 1.58 | 149.25 | 0.0012 |
| 207723_s_at | KLRC3 | Killer cell
lectin-like receptor subfamily C, member 3 | 1.57 | 12.31 | 0.0469 |
| 204597_x_at | STC1 | Stanniocalcin | 1.51 | 97.17 | 0.0196 |
| 201243_s_at | ATP1B1 | ATPase,
Na+/K+ transporting, β1 polypeptide | 1.48 | 10.13 | 0.0061 |
| 209270_at | LAMB3 | Laminin, β3 | 1.46 | 35.71 | 0.0023 |
| 202270_at | GBP1 | Guanylate binding
protein 1, interferon-inducible, 67 kDa | 1.45 | 3.74 | 0.0419 |
| 206027_at | S100A3 | S100 calcium
binding protein A3 | 1.44 | 20.02 | 0.0266 |
| 204205_at | APOBEC3G | Apolipoprotein B
mRNA editing enzyme, catalytic polypeptide-like 3G | 1.41 | 12.61 | 0.0181 |
| 206067_s_at | WT1 | Wilms tumour 1 | 1.41 | 15.57 | 0.0128 |
| 208025_s_at | HMGA2 | High mobility group
AT-hook 2 | 1.40 | 7.59 | 0.0007 |
| 212667_at | SPARC | Secreted protein,
acidic, cysteine-rich (osteonectin) | 1.39 | 9.20 | 0.0007 |
| 212097_at | CAV1 | Caveolin 1,
caveolae protein, 22 kDa | 1.33 | 12.56 | 0.0199 |
| 210510_at | NRP-1 | Neuropilin-1 | 1.28 | 10.91 | 0.0182 |
| 219295_at | PCOLCE2 | Procollagen
C-endopeptidase enhancer 2 | 1.28 | 6.39 | 0.0164 |
| 204279_at | PSMB9 | Proteasome
(prosome, macropain) subunit, β type, 9 | 1.23 | 4.54 | 0.0097 |
| Table IIDownregulated genes in M14C2/MK18
cells as compared with M14C2/C4 cells. |
Table II
Downregulated genes in M14C2/MK18
cells as compared with M14C2/C4 cells.
| | | Fold-changeb | |
|---|
| | |
| |
|---|
| Probe | Symbold | Descriptiona | PAM | SAM | P-valuec |
|---|
| 214475_x_at | CAPN3 | Calpain-3,
(p94) | −2.21 | −123.90 | 0.0072 |
| 206426_at | MLANA | Melan-A | −1.93 | −76.24 | 0.0393 |
| 209167_at | GPM6B | Glycoprotein
M6B | −1.80 | −30.25 | 0.0373 |
| 216512_s_at | DCT | Dopachrome
tautomerase | −1.76 | −130.85 | 0.0287 |
| 210944_s_at | CAPN3 | Calpain-3,
(p94) | −1.68 | −92.29 | 0.0295 |
| 219412_at | RAB38 | RAB38, member RAS
oncogene family | −1.67 | −71.53 | 0.0011 |
| 211890_x_at | CAPN3 | Calpain-3,
(p94) | −1.58 | −69.13 | 0.0272 |
| 215695_s_at | GYG2 | Glycogenin 2 | −1.58 | −15.76 | 0.0445 |
| 203382_s_at | APOE | Apolipoprotein
E | −1.57 | −42.41 | 0.0016 |
| 203651_at | ZFYVE16 | Zinc finger, FYVE
domain containing 16 | −1.55 | −9.31 | 0.0132 |
| 216033_s_at | FYN | FYN oncogene
related to SRC, FGR, YES | −1.47 | −11.29 | 0.0285 |
| 209169_at | GPM6B | Glycoprotein
M6B | −1.41 | −30.25 | 0.0361 |
| 201534_s_at | UBL3 | Ubiquitin-like
3 | −1.39 | −3.82 | 0.0095 |
| 214028_x_at | TDRD3 | Tudor domain
containing 3 | −1.38 | −9.89 | 0.0003 |
| 213702_x_at | ASAH1 | N-acylsphingosine
amidohydrolase 1 | −1.38 | −20.27 | 0.0265 |
| 206864_s_at | HRK | Harakiri, BCL2
interacting protein (BH3 domain) | −1.32 | −8.67 | 0.0164 |
| 201362_at |
IVNS1ABP | Influenza virus
NS1A binding protein | −1.31 | −3.92 | 0.0387 |
| 209113_s_at | HMG20B | High-mobility group
20B | −1.28 | −11.18 | 0.0029 |
| 33304_at | ISG20 | Interferon
stimulated gene 20 kDa | −1.28 | −10.73 | 0.0141 |
| 202295_s_at | CTSH | Cathepsin H | −1.28 | −15.28 | 0.0048 |
| 209123_at | QDPR | Quinoid
dihydropteridine reductase | −1.25 | −7.46 | 0.0385 |
| 200924_s_at | SLC3A2 | Solute carrier
family 3 (activators of dibasic and neutral amino acid transport),
member 2 | −1.22 | −8.20 | 0.0155 |
Involvement of PDGF-C and calpain-3 in
melanoma cell invasiveness
The possibility that the up-modulation of PDGF-C may
play a role in the highly invasive properties of M14C2/MK18 cells
was analysed using the in vitro ECM invasion assay. Firstly,
western blot analysis confirmed the increased secretion of PDGF-C
into the culture supernatant of M14C2/MK18 cells (Fig. 3A). Two immunoreactive bands of ~45
and ~22 kDa were observed, corresponding to the pro-PDGF-C and to
the fully processed and active receptor binding form, respectively
(15). The expression of NRP-1
polypeptide, which we recently found to promote melanoma cell
invasion (16), was also confirmed
by immunoblot analysis (Fig. 3A).
Blockage of PDGF-C with a specific neutralizing antibody
down-modulated ECM invasion by M14C2/MK18 cells to the same extent
as NRP-1 inhibition (~35%). The simultaneous blockage of both
polypeptides resulted in an additive effect causing a decrease in
M14C2/MK18 cell invasiveness of ~70% (Fig. 3B).
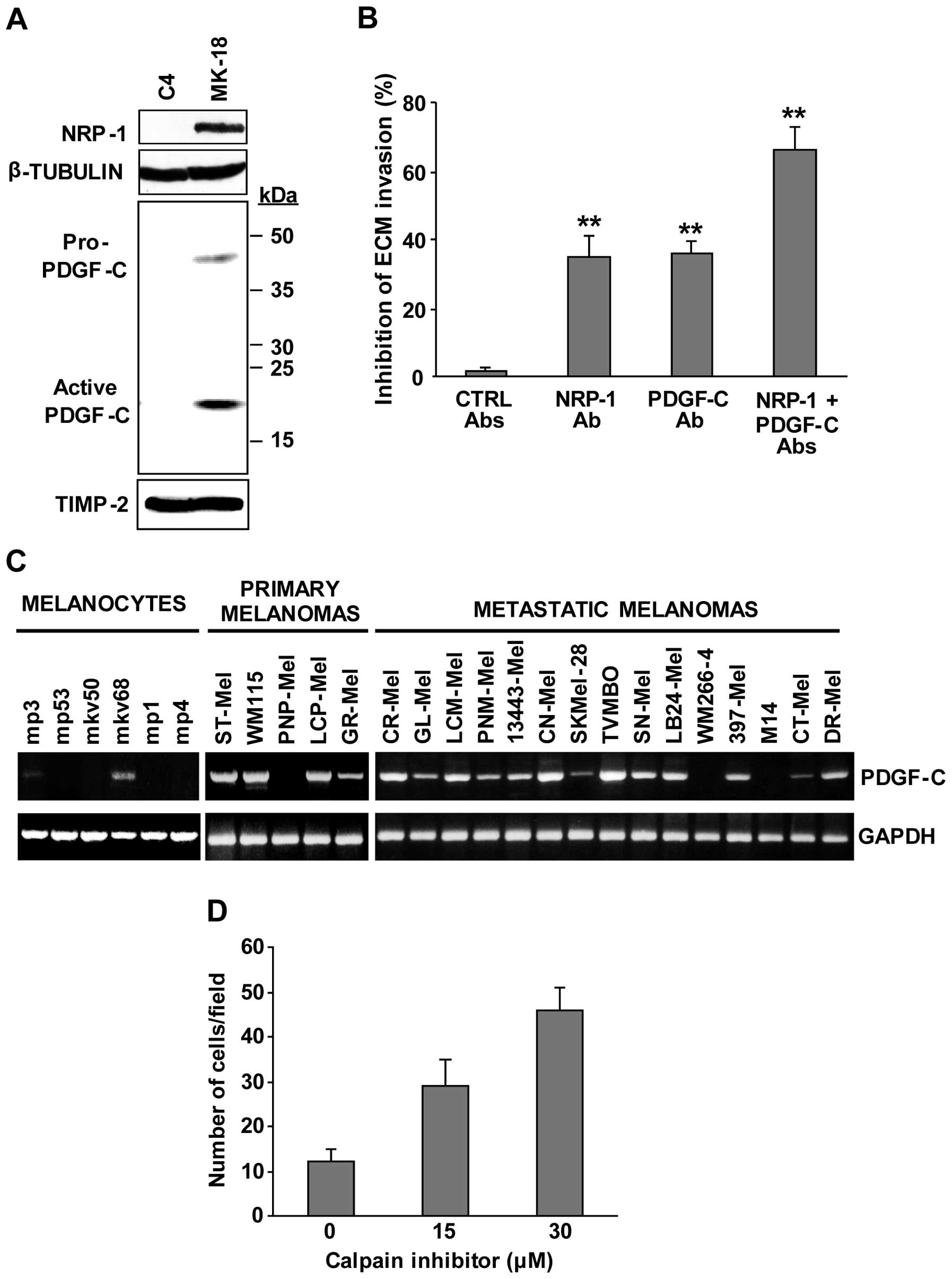 | Figure 3Involvement of PDGF-C and calpain-3
in the invasiveness of M14C2/MK18 cells. (A) The differential
expression of NRP-1 and secretion of PDGF-C polypeptides in
M14C2/C4 and M14C2/MK18 cells were evaluated by western blot
analysis. NRP-1 detection was analysed using 80 μg of proteins per
sample in a 7% SDS-polyacrylamide gel, and β-tubulin expression was
assessed as loading control. The bands corresponding to the
pro-PDGF-C and the fully processed, active PDGF-C forms were
detected by loading 50 μl of concentrated cell supernatants,
collected from an equal number of cultured cells, in a 6–12%
SDS-polyacrylamide gel gradient. TIMP-2 secretion was used as
loading control. Results are representative of one out of three
different experiments giving comparable results. (B) The influence
of antibodies that specifically block NRP-1 or PDGF-C on the
ability of M14C2/MK18 cells to spontaneously invade the ECM was
analysed using Boyden chambers equipped with Matrigel-coated
filters. Cells were allowed to invade Matrigel in response to
migration medium for 2 h. The assay was performed in the absence of
antibodies or in the presence of 2.5 μg/ml of antibodies anti-NRP-1
(H-286) and/or anti-PDGF-C (AF1560), or control antibodies (mouse
IgG + goat IgGs, CTRL Abs). Data are expressed as the percent
inhibition of ECM invasion calculated as compared with untreated
cells. Values represent the means ± SD from three independent
experiments. **P<0.01, according to Student’s t-test
analysis, comparing the invasion of cells treated with anti-NRP-1
or anti-PDGF-C antibodies with that of cells treated with control
antibodies, or comparing the invasion of cells treated with both
antibodies with that of cells treated with only anti-NRP-1 or
anti-PDGF-C antibodies. (C) RT-PCR analysis of PDGF-C expression
was performed in a series of human melanocyte primary cultures and
in human melanoma cell lines derived from primary or metastatic
melanomas. As a control of RNA integrity, analysis of GAPDH gene
was performed. Results are representative of one out of three
different experiments giving comparable results. (D) The influence
of the treatment with the calpain inhibitor ALLM (15 and 30 μM) on
the ability of M14C2/C4 cells to spontaneously invade the ECM was
analysed using Boyden chambers equipped with Matrigel-coated
filters and allowing the cells to invade for 3 h. Samples of
untreated cells (calpain inhibitor concentration 0) were incubated
with the same final concentration of DMSO, used as drug solvent,
present in the sample treated with 30 μM of the inhibitor. Each
value represents the mean ± SD of the number of cells
migrated/microscopic field. |
We then investigated the expression of PDGF-C using
RT-PCR analysis in a series of human melanoma cell lines and
melanocytic cultures. The results indicated that most of the cell
lines under study (17 out of 20), derived from primary or
metastatic melanomas, expressed the PDGF-C transcript, while only 2
out of the 6 melanocytic primary cultures showed low PDGF-C levels
(Fig. 3C). This finding suggests
that PDGF-C expression may be required for melanoma development and
progression.
In order to investigate the possibility that
calpain-3 down-modulation, evidenced by microarray analysis, might
contribute to the melanoma aggressive phenotype, M14C2/C4 cells
were treated with a specific calpain inhibitor (ALLM) and analysed
for their ability to invade the ECM. Interestingly, ALLM treatment
significantly stimulated ECM invasion by these cells in a
concentration-dependent manner (Fig.
3D).
These data demonstrated that PDGF-C upregulation and
calpain-3 downregulation are both involved in the increase of
M14C2/MK18 cell aggressiveness.
Discussion
A previously isolated highly invasive cell clone
(M14C2/MK18 cells) derived from a metastatic cutaneous melanoma
lesion was characterised for the expression of several important
determinants of melanoma aggressiveness: the activation of a
VEGF-A/VEGFR-2 autocrine loop, NRP-1 expression and secretion of
fully active MMP-2. These cells displayed rapid engraftment and an
elevated growth rate in an in vivo murine model. With an
intent to identify further molecules and mechanisms responsible for
their elevated aggressiveness, the gene expression profile of
M14C2/MK18 cells was analysed and compared to that of a control
cell clone, derived from the same original melanoma, but with lower
aggressiveness in vitro and in vivo.
The results of the microarray analysis showed a
strong up-modulation of NRP-1 (known to be expressed at high levels
in these cells) but also of PDGF-C expression. PDGF-C is a member
of the PDGF family of growth factors secreted as a latent
homodimeric form and consisting of an N-terminal CUB (Clr/Cls,
urchin endothelial growth factor-like protein and bone morphogenic
protein 1) domain followed by a linker sequence and a C-terminal
growth factor domain (GFD) (17).
The CUB domain must be proteolytically removed to permit GFD to
bind and activate PDGFRs (principally PDGFRα homodimers, but PDGFRβ
can also be activated via PDGFRαβ heterodimerisation) (17–19).
In the adult, the angiogenic capabilities of PDGF-C are comparable
to those of VEGF in different model systems (18,20)
and are linked to its effects on fibroblasts, endothelial
progenitor cells, bone marrow cells and mature vascular cells by
promoting their recruitment, proliferation, differentiation and
migration (21–23).
In breast cancer, increased PDGF-C expression
correlates with lymph node metastases, higher proliferative
potential, and lower rates of 7-year disease-free survival
(23). A role for PDGF-C in Ewing
sarcoma has also been suggested by the observation that cell lines
derived from this tumour type overexpress this cytokine, which
promotes anchorage-independent growth (25,26).
PDGF-C autocrine signalling has also been suggested to be involved
in the initiation and progression of brain tumours such as
glioblastoma and medulloblastoma (27,28)
and in the chemoresistance of oral squamous cell carcinoma
(29). PDGF-C has been correlated
with melanoma progression as a paracrine factor that favours
melanoma dissemination by stimulating fibroblast reactivity and
fibrosis, and by inducing angiogenesis through its effect on the
fibroblasts and the endothelium surrounding the tumour (23). Herein, we show, for the first time,
that PDGF-C can also support melanoma cell invasiveness by
autocrine activation and suggest that this mechanism may be
frequently active in melanoma cells, since we also found that
PDGF-C mRNA is widely expressed in melanoma cell lines. The
production of the ~22 kDa fully active form of PDGF-C also requires
the presence of proteases that process this cytokine. Therefore,
further studies are necessary to identify the melanoma cell lines
that actually produce the fully processed form of PDGF-C. In
addition, PDGF-C expression may be responsible for tumour
resistance to anti-angiogenic therapies targeting VEGF. In fact,
levels of this cytokine are elevated in melanomas resistant to
anti-VEGF therapy (30,31), likely due to the role of PDGF-C in
tumour-induced vessel maturation and stabilisation (32). Therefore, the data herein shown
suggest that blocking of PDGF-C may synergise with anti-VEGF-A
therapies in combating tumour angiogenesis and tumour invasion.
On the other hand, we also found that the gene which
showed the most important down-modulation in M14C2/MK18 cells was
CAPN3, encoding for calpain-3. Calpains are a group of
calcium-dependent thiol-proteases that respond to Ca2+
signals by cleaving specific proteins, frequently components of
signalling cascades, irreversibly modifying their function.
Calpain-3 is a 94-kDa enzyme that is structurally similar to
calpain-1 and -2, but has an additional N-terminal sequence of
20–30 amino acids. The function of calpains is regulated by
phosphorylation, intracellular Na+ concentration,
calpastatin and their subcellular localization (33). In this context, three of the genes
found to be highly upregulated in M14C2/MK18 cells are involved in
the control of intracellular calcium concentration (STC1, S100A2
and S100A3). Their increased expression may cause a decrease in
calcium levels in melanoma cells. Moreover, the elevated expression
of Na+/K+ transporting ATPase β1 polypeptide
(ATP1B1) would likely result in reduced intracellular
Na+ levels. Therefore, the upregulation of these four
genes in M14C2/MK18 cells may contribute to the down-modulation, at
a post-translational level, of calpain-3 activity that is
maintained by a marked decrease in the CAPN3 transcript.
Calpains are involved in numerous cell functions,
and consequently they are key regulators of several important
pathways (34). For example, they
have a role in the remodelling of cytoskeletal attachments to the
plasma membrane during cell fusion and cell motility, in the
proteolytic modification of molecules involved in signal
transduction pathways, in the degradation of enzymes controlling
cell cycle progression or in the regulation of gene expression and
substrate degradation in various apoptotic pathways. A study in
human biopsies showed that the expression of calpain-3 is
significantly downregulated in the most aggressive melanomas
compared with benign nevi, suggesting that its downregulation may
contribute to melanoma progression (35). Consistent with this hypothesis, we
found that treatment of M14C2/C4 cells with a calpain inhibitor
significantly triggered ECM invasion by these cells, otherwise
unable to migrate across the basement matrix layer.
Notably, 11 out of the 19 genes down-modulated in
M14C2/MK18 cells are known to be under the control of the
micro-phthalmia-associated transcription factor (MITF). MITF
encodes for basic helix-loop-helix-leucine-zipper transcription
factors, among which the M-isoform is specifically expressed in
melanocytes (36) and plays a key
role in melanocyte development, survival and differentiation
(37–42). When the levels of MITF polypeptide
expression were evaluated during melanoma progression, results
indicated an intense nuclear staining for the MITF polypeptide in
83% of nevi and 56% of primary melanomas, but only 23% of
metastases (3). MITF is subjected
to various post-translational modifications (43), including phosphorylation at Ser73,
which targets MITF for ubiquitin-dependent proteolysis (44) and increases its interaction with the
repressor protein inhibitor of activated STAT3 (PIAS3). PIAS3
mediates MITF sumoylation, down-modulating its transcriptional
activity (45). Moreover, PIAS3 is
a substrate of calpain that negatively regulates its sumoylase
activity (46). Therefore, it can
be hypothesised that the MITF post-translational downregulation
consequent to the decrease in calpain-3 may be a determinant of
M14C2/MK18 cell invasiveness. Moreover, inhibition of the calpain-3
substrate PIAS3 may contribute to maintain a low aggressive
phenotype in melanoma cells, by preserving MITF activity.
In conclusion, we identified several molecules,
including PDGF-C and calpain-3, that may be involved in the
acquisition of an aggressive phenotype that transforms low invasive
lesions into highly invasive tumours, suggesting the possibility
that these proteins or their downstream effectors may represent
molecular targets for more effective therapies against malignant
melanoma.
Acknowledgements
The authors would like to thank Daniele Bartoloni
for the graphics and the Italian Ministry of Health for the
financial support.
References
|
1
|
Berwick M, Erdei E and Hay J: Melanoma
epidemiology and public health. Dermatol Clin. 27:205–214. 2009.
View Article : Google Scholar
|
|
2
|
Becker D, Mihm MC, Hewitt SM, Sondak VK,
Fountain JW and Thurin M: Markers and tissue resources for
melanoma: meeting report. Cancer Res. 66:10652–10657.
2006.PubMed/NCBI
|
|
3
|
Nazarian RM, Prieto VG, Elder DE and
Duncan LM: Melanoma biomarker expression in melanocytic tumor
progression: a tissue microarray study. J Cutan Pathol. 37:41–47.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lacal PM, Failla CM, Pagani E, et al:
Human melanoma cells secrete and respond to placenta growth factor
and vascular endothelial growth factor. J Invest Dermatol.
115:1000–1007. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lacal PM, Ruffini F, Pagani E and D’Atri
S: An autocrine loop directed by the vascular endothelial growth
factor promotes invasiveness of human melanoma cells. Int J Oncol.
27:1625–1632. 2005.PubMed/NCBI
|
|
6
|
Schietroma C, Cianfarani F, Lacal PM, et
al: Vascular endothelial growth factor-C expression correlates with
lymph node localization of human melanoma metastases. Cancer.
98:789–797. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ruffini F, Failla CM, Orecchia A, et al:
Expression of the soluble vascular endothelial growth factor
receptor-1 in cutaneous melanoma: role in tumour progression. Br J
Dermatol. 164:1061–1070. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hennequin LF, Thomas AP, Johnstone C, et
al: Design and structure-activity relationship of a new class of
potent VEGF receptor tyrosine kinase inhibitors. J Med Chem.
42:5369–5389. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sasaki T, Kishi M, Saito M, et al:
Inhibitory effect of di- and tripeptidyl aldehydes on calpains and
cathepsins. J Enzyme Inhib. 3:195–201. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lacal PM, Morea V, Ruffini F, et al:
Inhibition of endothelial cell migration and angiogenesis by a
vascular endothelial growth factor receptor-1 derived peptide. Eur
J Cancer. 44:1914–1921. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lacal PM, Tentori L, Muzi A, et al:
Pharmacological inhibition of poly(ADP-ribose) polymerase activity
down-regulates the expression of syndecan-4 and Id-1 in endothelial
cells. Int J Oncol. 34:861–872. 2009.PubMed/NCBI
|
|
12
|
Ruffini F, D’Atri S and Lacal PM:
Neuropilin-1 expression promotes invasiveness of melanoma cells
through vascular endothelial growth factor receptor-2 dependent and
independent mechanisms. Int J Oncol. 43:297–306. 2013.
|
|
13
|
Stetler-Stevenson WG: Matrix
metalloproteinases in angiogenesis: a moving target for therapeutic
intervention. J Clin Invest. 103:1237–1241. 1999.PubMed/NCBI
|
|
14
|
Wang Z, Juttermann R and Soloway PD:
TIMP-2 is required for efficient activation of proMMP-2 in vivo. J
Biol Chem. 275:26411–26415. 2000. View Article : Google Scholar
|
|
15
|
Hoek KS, Schlegel NC, Eichhoff OM, et al:
Novel MITF targets identified using a two-step DNA microarray
strategy. Pigment Cell Melanoma Res. 21:665–676. 2008. View Article : Google Scholar
|
|
16
|
Fredriksson L, Ehnman M, Fieber C and
Eriksson U: Structural requirements for activation of latent
platelet-derived growth factor CC by tissue plasminogen activator.
J Biol Chem. 280:26856–26862. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li X, Pontén A, Aase K, et al: PDGF-C is a
new protease-activated ligand for the PDGF alpha-receptor. Nat Cell
Biol. 2:302–309. 2000. View
Article : Google Scholar
|
|
18
|
Gilbertson DG, Duff ME, West JW, et al:
Platelet-derived growth factor C (PDGF-C), a novel growth factor
that binds to PDGF alpha and beta receptor. J Biol Chem.
276:27406–27414. 2001.PubMed/NCBI
|
|
19
|
Fredriksson L, Li H, Fieber C, Li X and
Eriksson U: Tissue plasminogen activator is a potent activator of
PDGF-CC. EMBO J. 23:3793–3802. 2004.
|
|
20
|
Cao R, Bråkenhielm E, Li X, et al:
Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF
family, involves activation of PDGFR-αα and -αβ receptors. FASEB J.
16:1575–1583. 2002.PubMed/NCBI
|
|
21
|
Dimmeler S: Platelet-derived growth factor
CC - a clinically useful angiogenic factor at last? N Engl J Med.
352:1815–1816. 2005.PubMed/NCBI
|
|
22
|
Li X, Tjwa M, Moons L, et al:
Revascularization of ischemic tissues by PDGF-CC via effects on
endothelial cells and their progenitors. J Clin Invest.
115:118–127. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anderberg C, Li H, Fredriksson L, et al:
Paracrine signaling by platelet-derived growth factor-CC promotes
tumor growth by recruitment of cancer associated fibroblasts.
Cancer Res. 69:369–378. 2009.PubMed/NCBI
|
|
24
|
Hurst NJ Jr, Najy AJ, Ustach CV, Movilla L
and Kim HR: Platelet-derived growth factor-C (PDGF-C) activation by
serine proteases: implications for breast cancer progression.
Biochem J. 441:909–918. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zwerner JP and May WA: PDGF-C is an
EWS/FLI induced transforming growth factor in Ewing family tumors.
Oncogene. 20:626–633. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zwerner JP and May WA: Dominant negative
PDGF-C inhibits growth of Ewing family tumor cell lines. Oncogene.
21:3847–3854. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Andrae J, Molander C, Smits A, Funa K and
Nistér M: Platelet-derived growth factor-B and -C and active
alpha-receptors in medulloblastoma cells. Biochem Biophys Res
Commun. 296:604–611. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lokker NA, Sullivan CM, Hollenbach SJ,
Israel MA and Giese NA: Platelet-derived growth factor (PDGF)
autocrine signaling regulates survival and mitogenic pathways in
glioblastoma cells: evidence that the novel PDGF-C and PDGF-D
ligands may play a role in the development of brain tumours. Cancer
Res. 62:3729–3735. 2002.
|
|
29
|
Yamano Y, Uzawa K, Saito K, et al:
Identification of cisplatin-resistance related genes in head and
neck squamous cell carcinoma. Int J Cancer. 126:437–449. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Crawford Y, Kasman I, Yu L, et al: PDGF-C
mediates the angiogenic and tumorigenic properties of fibroblasts
associated with tumors refractory to anti-VEGF treatment. Cancer
Cell. 15:21–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li X, Kumar A, Zhang F, Lee C, Li Y, Tang
Z and Arjuna P: VEGF-independent angiogenic pathways induced by
PDGF-C. Oncotarget. 1:309–314. 2010.PubMed/NCBI
|
|
32
|
di Tomaso E, London N, Fuja D, et al:
PDGF-C induces maturation of blood vessels in a model of
glioblastoma and attenuates the response to anti-VEGF treatment.
PLoS One. 4:e51232009.PubMed/NCBI
|
|
33
|
Ono Y, Ojima K, Torii F, et al: Skeletal
muscle-specific calpain is an intracellular
Na+-dependent protease. J Biol Chem. 285:22986–22998.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Storr SJ, Carragher NO, Frame MC, Parr T
and Martin SG: The calpain system and cancer. Nat Rev Cancer.
11:364–374. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moretti D, Del Bello B, Cosci E, Biagioli
M, Miracco C and Maellaro E: Novel variants of muscle calpain 3
identified in human melanoma cells: cisplatin-induced changes in
vitro and differential expression in melanocytic lesions.
Carcinogenesis. 30:960–967. 2009. View Article : Google Scholar
|
|
36
|
Hodgkinson CA, Moore KJ, Nakayama A,
Steingrímsson E, Copeland NG, Jenkins NA and Arnheiter H: Mutations
at the mouse microphthalmia locus are associated with defects in a
gene encoding a novel basic-helix-loop-helix-zipper protein. Cell.
74:395–404. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vachtenheim J, Novotna H and Ghanem G:
Transcriptional repression of the microphthalmia gene in melanoma
cells correlates with the unresponsiveness of target genes to
ectopic microphthalmia-associated transcription factor. J Invest
Dermatol. 117:1505–1511. 2001. View Article : Google Scholar
|
|
38
|
McGill GG, Horstmann M, Widlund HR, et al:
Bcl2 regulation by the melanocyte master regulator Mitf modulates
lineage survival and melanoma cell viability. Cell. 109:707–718.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Du J, Widlund HR, Horstmann MA, et al:
Critical role of CDK2 for melanoma growth linked to its
melanocyte-specific transcriptional regulation by MITF. Cancer
Cell. 6:565–576. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Steingrimsson E, Copeland NG and Jenkins
NA: Melanocytes and the microphthalmia transcription factor
network. Annu Rev Genet. 38:365–411. 2004. View Article : Google Scholar
|
|
41
|
Carreira S, Goodall J, Aksan I, et al:
Mitf cooperates with Rb1 and activates p21Cip1
expression to regulate cell cycle progression. Nature. 433:764–769.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Loercher AE, Tank EM, Delston RB and
Harbour JW: MITF links differentiation with cell cycle arrest in
melanocytes by transcriptional activation of INK4A. J Cell Biol.
168:35–40. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Levy C, Khaled M and Fisher DE: MITF:
master regulator of melanocyte development and melanoma oncogene.
Trends Mol Med. 12:406–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu M, Hemesath TJ, Takemoto CM, et al:
c-Kit triggers dual phosphorylations, which couple activation and
degradation of the essential melanocyte factor Mi. Genes Dev.
14:301–312. 2000.PubMed/NCBI
|
|
45
|
Levy C, Sonnenblick A and Razin E: Role
played by microphthalmia transcription factor phosphorylation and
its Zip domain in its transcriptional inhibition by PIAS3. Mol Cell
Biol. 23:9073–9080. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
De Morrée A, Hulsik DL, Impagliazzo A, et
al: Calpain 3 is a rapid-action, unidirectional proteolytic switch
central to muscle remodelling. PLoS One. 5:e119402010.PubMed/NCBI
|