Introduction
Green tea is a popular type of tea worldwide.
Epidemiological investigations and experimental studies have shown
that green tea has chemopreventive effects for a wide range of
malignancies, including lung, stomach, breast, prostate,
pancreatic, ovarian, liver, colorectal and skin cancer (1–10).
Mechanistic studies indicate that EGCG, the major component of
green tea, appears to be the primary active ingredient responsible
for its biological effects. EGCG reportedly exhibits its cancer
preventive effects through regulation of multiple signaling
pathways, including suppression of various protein kinases
(1,11,12),
disruption of the activation of transcription factors, and
alteration of the expression of genes involved in cell
proliferation, angiogenesis and apoptosis, thereby imparting strong
cancer chemopreventive as well as therapeutic effects (13,14).
Epidermal growth factor receptor (EGFR) is a
transmembrane glycoprotein that possesses intrinsic receptor
tyrosine kinase activity. The EGFR signaling pathway is known to
play a major role in tumor genesis by regulating cell
proliferation, survival and metabolism (15). Upon ligand binding, activated EGFR
recruits, phosphorylates and activates a number of important
signaling molecules such as PLC-γ, Ras, PI-3K and JAK2 (16,17).
These EGFR downstream signaling cascades are conventional early
transient responses and mainly depend on EGFR tyrosine kinase
activity (18). Moreover, EGFR and
its ligands have repeatedly been observed in the nucleus (15,19,20).
Different stimuli such as the EGF ligand or ultraviolet radiation
may promote the nuclear translocation of EGFR. Nuclear EGFR
interacts with numerous transcription factors such as signal
transducer and activator of transcription 3 (STAT3), STAT5 and
transcription factor E2F1, and acts as a transcriptional factor to
regulate the expression of downstream target genes. Additionally,
other DNA-binding partners such as proliferating cell nuclear
antigen or DNA protein kinase (DNA-PK) are able to bind with
nuclear EGFR and induce PCNA stability or DNA repair (19,21–24).
These findings indicate that nuclear EGFR is involved in a number
of physiological and pathological processes, such as proliferation,
inflammation, metastasis, DNA repair and resistance to DNA-damaging
radiation or chemotherapy drugs (15).
Lung cancer is the leading cause of cancer-related
death in the world, and non-small cell lung carcinoma (NSCLC)
accounts for 80% of lung cancer cases (25–27).
Overexpression and hyperactivation of receptor tyrosine kinases
such as EGFR are known to be important in lung cancer
carcinogenesis, leading to the development of specific targeted
therapies (28,29). Specific receptor antagonists have
shown efficacy in clinical practice, yet tumors often become
resistant to these therapies due to an EGFR second mutation or
other tyrosine kinases such as c-Met overexpression, and the 5-year
survival for these patients remains poor at <15% (28). A major challenge in the treatment of
lung cancer is the identification of novel therapeutic targets or
the development of new anticancer agents that can supplement
current chemotherapy (30).
In the present study, we demonstrated that natural
compound EGCG markedly inhibited the growth of lung cancer by
directly targeting the EGFR signaling pathway. EGCG not only
decreased EGFR tyrosine kinase activity, but also suppressed
nuclear localization of EGFR and EGFR-mediated expression of
downstream target gene cyclin D1.
Materials and methods
Cell culture and transfection
All cell lines were obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA) and grown in a 37°C
incubator with 5% CO2 according to ATCC protocols. For
transfection experiments, Lipofectamine™ 2000 transfection reagent
(Invitrogen, Carlsbad, CA, USA) was used according to the
manufacturer’s instructions.
Reagents and antibodies
EGCG was obtained from Sigma (St. Louis, MO, USA).
Anti-EGFR, anti-p-EGFR (Tyr1068), anti-AKT (pan), anti-p-AKT
(Ser473), anti-ERK1/2, anti-p-ERK1/2 (Thr202/Tyr204), anti-S6,
anti-p-S6 (Ser235/236), anti-cyclin D1, anti-α-tubulin and
anti-laminB antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Anti-β-actin, anti-rabbit
IgG-HRP and anti-mouse IgG-HRP were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Anti-N-cadherin was purchased
from BD Biosciences (San Jose, CA, USA).
Western blotting
Cells were harvested by trypsinization and pelleted
by centrifugation. Cell pellets were lysed in Nonidet P-40 cell
lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Nonidet
P-40 and protease inhibitor mixture). Protein concentrations were
determined by the Bradford assay (Bio-Rad Laboratories, Hercules,
CA, USA). Proteins were separated by SDS-PAGE and electrically
transferred to polyvinylidene difluoride membranes (Millipore,
Billerica, MA, USA). After blocking in 5% non-fat dry milk in TBS,
the membranes were hybridized to specific primary antibodies
overnight at 4°C, washed 3 times with TBS-Tween-20, and then
incubated with secondary antibodies conjugated with horseradish
peroxidase for 1 h at room temperature. Next, the membranes were
washed 3 times in TBS-Tween-20 at room temperature. The protein
bands were visualized using ECL chemiluminescence reagents (Pierce
Chemical Co., Rockford, IL, USA) according to the manufacturer’s
protocol.
Lentiviral infection
Lentivirus plasmids (pLKO.1-shEGFR #1,
TRCN0000039633; pLKO.1-shEGFR #2, TRCN0000121068) were purchased
from Thermo Scientific (Huntsville, AL, USA). We co-transfected
pLKO.1-shEGFR with PSPAX2 and PMD2-G into 293T
cells. Viral supernatant fractions were collected and infected into
A549 lung cancer cells with 10 μg/ml Polybrene. After a 24-h
infection, the medium was replaced with fresh medium containing the
appropriate concentration of puromycin. Appropriate experiments
were conducted with these cells until the control cells (without
infection) completely died (usually 2–3 days) in the puromycin
medium.
Subcellular proteome fractionation
The subcellular proteome fractions were prepared
using the ProteoExtract Subcellular Proteome Extraction kit
(Millipore) according to the manufacturer’s instructions.
Soft agar colony assay
To examine the anchorage-independent growth, lung
cancer cells were suspended (10,000 cells/ml) in 1 ml 0.3% agar
with Eagle’s basal medium containing 10% FBS, 1% antibiotics, and
different concentrations of EGCG (0, 10, 20 and 40 μmol/l) overlaid
into 6-well plates containing a 0.6% agar base. The cultures were
maintained in an incubator at 37°C with 5% CO2 for 1–2
weeks. The colonies were counted under a microscope with the
Image-Pro Plus software program (Media Cybernetics, Silver Spring,
MD, USA).
Flow cytometry
Flow cytometry was used to quantify cells in each
phase of the cell cycle. Cells (2×105) were seeded into
6-well plates and treated with various concentrations of EGCG for
24 h. The cells were harvested and washed with PBS twice and then
fixed in 70% ethanol overnight at 4°C. The cells were
counterstained in the dark with 50 μg/ml phosphatidyl inositol and
0.1% ribonuclease A (RNase A) in 400 μl PBS at 25°C for 30 min. The
stained cells were assayed and quantified by a FACSort flow
cytometer (BD Biosciences).
Statistical analysis
All statistical analyses were performed with SPSS
software (version 13.0). The experiments were performed in
triplicate. All quantitative data are expressed as mean values ±
standard deviation. The significant differences between two groups
were assessed by a 2-tailed Student’s t-test. A probability value
(P) <0.05 was considered to represent a statistically
significant difference.
Results
EGCG inhibits the anchorage-independent
growth of human lung cancer cells
In the present study, we first examined the effect
of EGCG on the anchorage-independent growth of human lung cancer
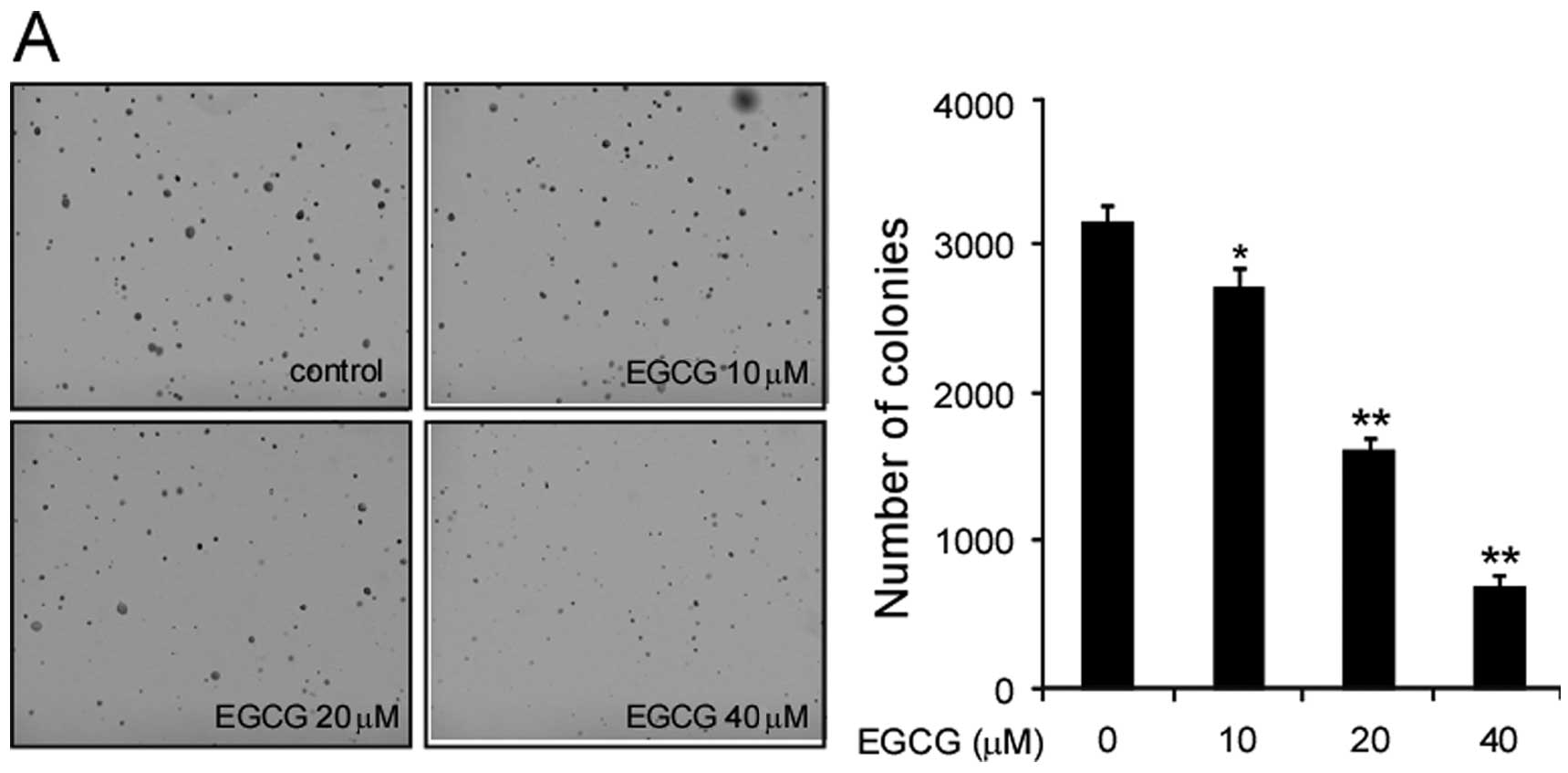
cell lines, A549, H1650 and H460. EGCG significantly inhibited A549
cell growth in soft agar in a dose-dependent manner (Fig. 1A). At 20 μM, EGCG substantially
inhibited the colony formation of A549 cells in soft agar, and the
inhibition rate reached 80% at the concentration of 40 μM.
Meanwhile, we also investigated the effect of EGCG on the growth of
2 other lung cancer cell lines H1650 and H460. EGCG
dose-dependently inhibited the growth of these cell lines on soft
agar. At a concentration of 20 μM, the inhibition rate reached 60
and 30%, respectively (Fig. 1B and
C). EGCG dose-dependently inhibited the growth of lung cancer
cells in soft agar.
Short-term exposure to EGCG inhibits
EGF-induced EGFR phosphorylation and its downstream signaling
pathway
Previous studies have reported that the EGFR
signaling pathway is often deregulated in human lung cancer and
plays a dominant role in the proliferation and survival of cancer
cells. Therefore, we investigated the effect of EGCG on the EGFR
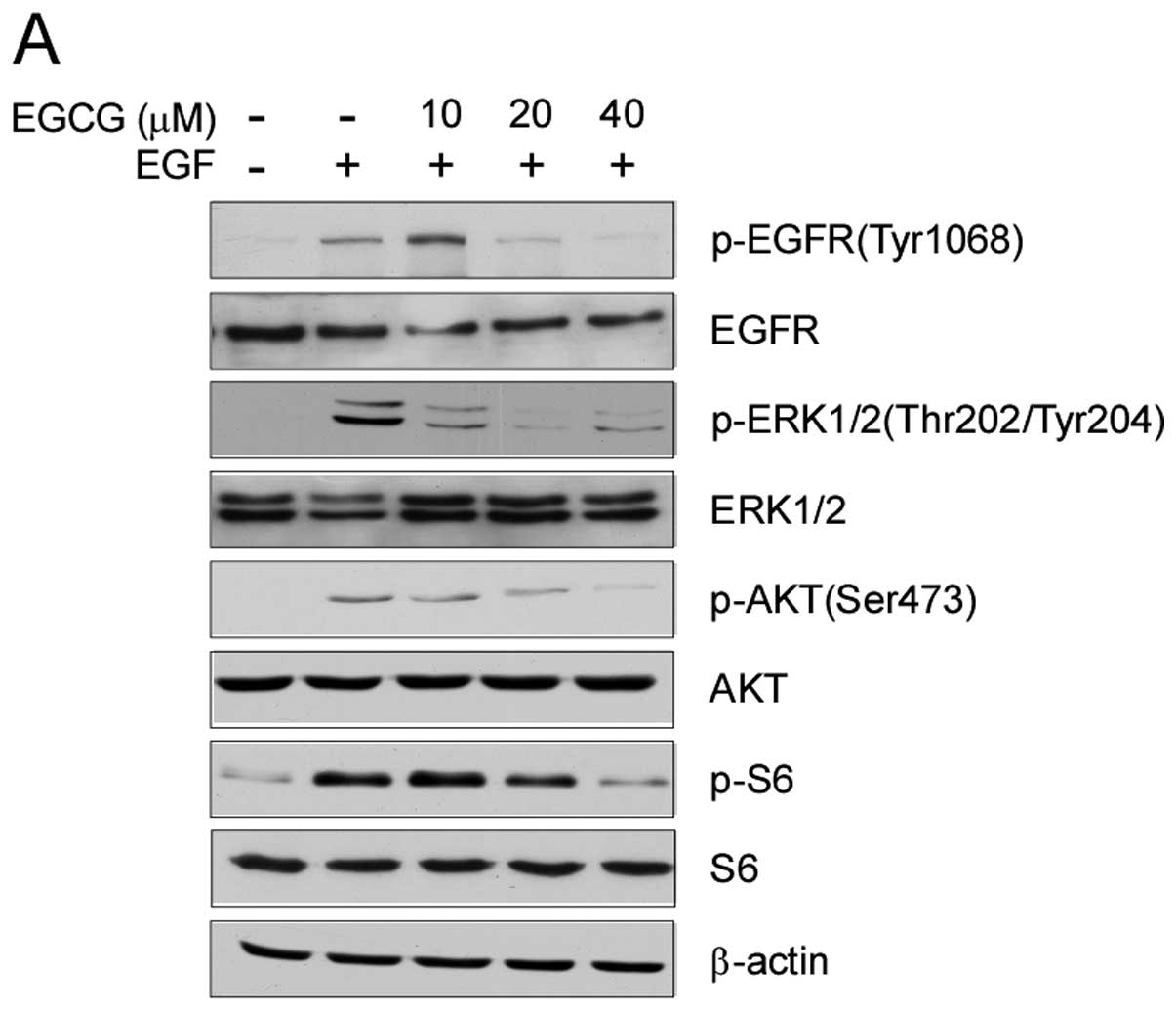
signaling pathway. Treatment of A549 cells with EGCG significantly
inhibited EGFR activation. Although a low concentration (10 μM) of
EGCG had little effect on EGFR phosphorylation, the EGF-induced
EGFR activation was inhibited >50% in the A549 cells in a
dose-dependent manner following treatment with 20 to 40 μM EGCG.
EGCG suppressed the activation of EGFR downstream kinases, such as
Akt, ERK1/2 and S6 in the A549 cells (Fig. 2A). To further confirm the effect of
EGCG on EGFR phosphorylation, we determined EGF-induced EGFR
phosphorylation at various time-points. Consistent with the above
results, EGCG suppressed EGFR activation as well as its downstream
kinases Akt and ERK1/2 at all time points (15, 30 and 60 min)
(Fig. 2B). As c-Fos is one of the
most important immediate early genes regulated by EGFR to initiate
downstream target gene transcription upon growth factor
stimulation, EGCG may regulate c-Fos expression. As expected,
together with the suppression of phosphorylation of EGFR,
EGF-induced c-Fos expression was inhibited by EGCG treatment
(Fig. 2B), suggesting that EGCG
decreases EGF-induced EGFR activation and signaling
transduction.
Long-term exposure to EGCG decreases the
expression levels of EGFR in human lung cancer cells
EGF-induced phosphorylation and activation of the
EGFR signaling pathway were inhibited after transient EGCG
treatment, indicating that EGCG dose-dependently inhibited EGFR
tyrosine kinase activity. Then, we investigated the effect of EGCG
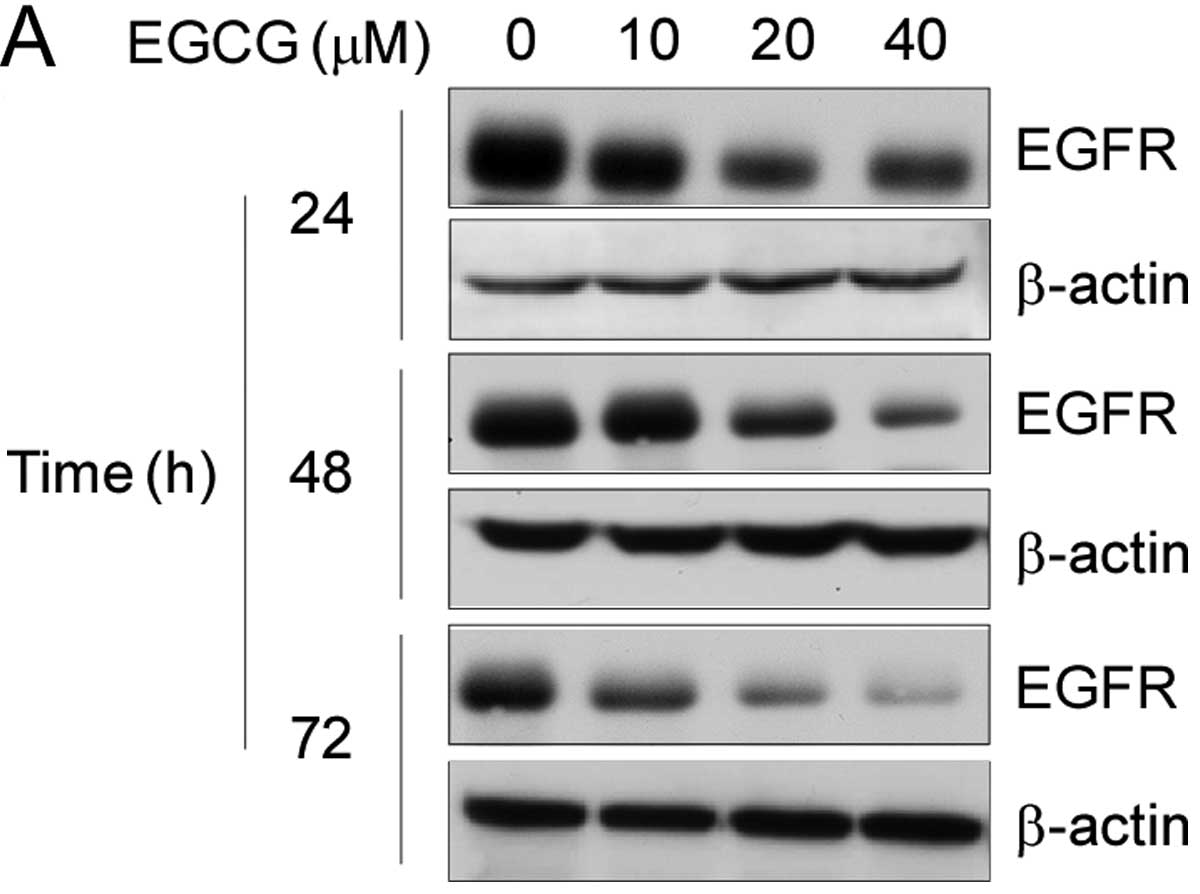
on EGFR expression after long-term exposure. As shown in Fig. 3A, long-term exposure to EGCG
significantly suppressed the expression of EGFR in a dose-dependent
and time-dependent manner. Following treatment with EGCG at 20 μM
for 48 h, the total EGFR protein level was markedly decreased.
Next, we purified the subcellular fractions to determine whether
EGCG treatment regulates EGFR expression both on the cellular
membrane and in the nucleus. As shown in Fig. 3B, in A549 lung cancer cells, the
membranous expression level of EGFR exhibited no obvious decrease
following treatment with 40 μM EGCG for 24 h; however, following a
72-h EGCG treatment, the EGFR membranous expression level was
significantly reduced. Moreover, EGCG had a high inhibitory effect
on nuclear EGFR expression, following treatment with 40 μM EGCG for
24 h (Fig. 3C). The expression of
EGFR was markedly decreased at 72 h, and the expression of EGFR was
almost completely suppressed, indicating that long-term EGCG
treatment inhibited the EGFR expression both on the cellular
membrane and in the nucleus.
Knockdown of EGFR reduces the sensitivity
of lung cancer cells to EGCG
We examined whether the knockdown of EGFR expression
influences the sensitivity of A549 lung cancer cells to EGCG.
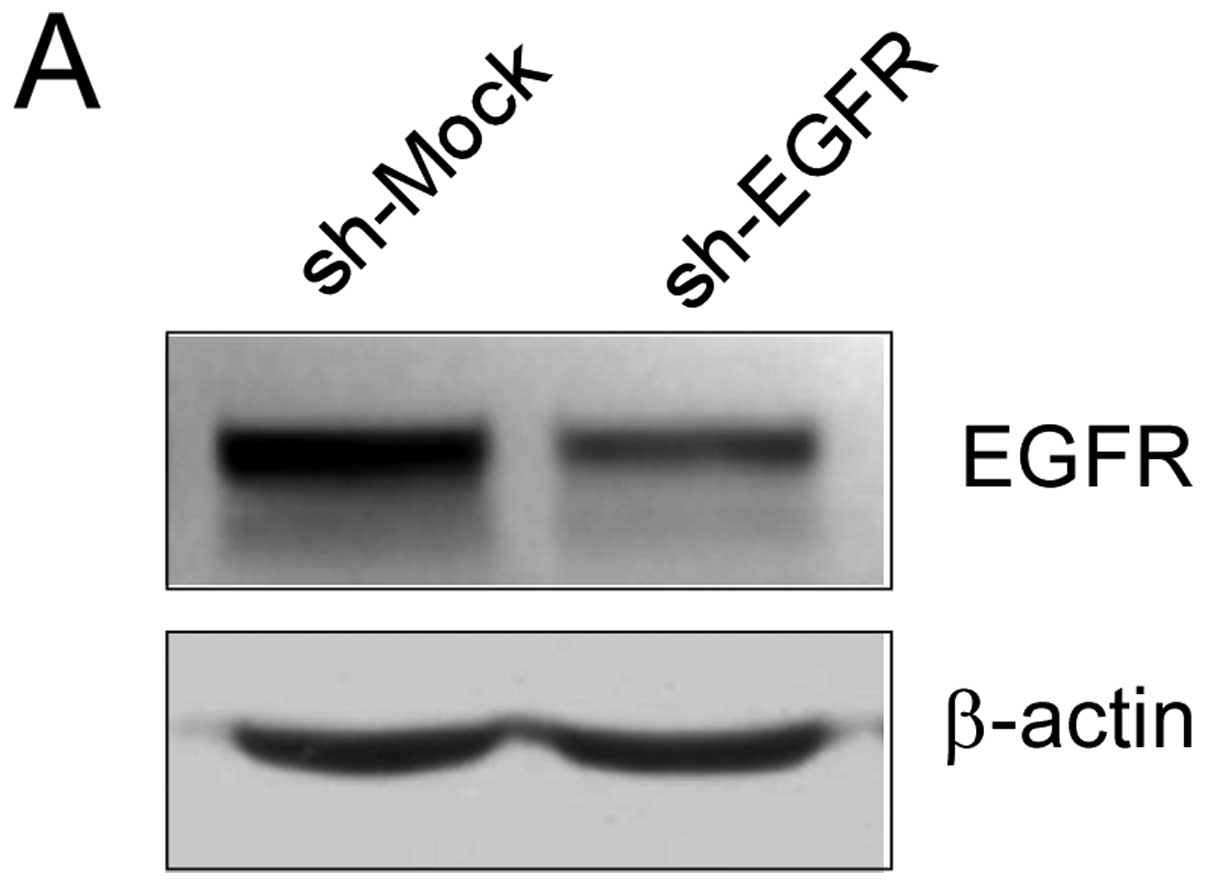
First, we determined the efficiency of shRNA knockdown, as well as
the effect of shRNA transfection on anchorage-independent growth.
The expression of EGFR was obviously decreased after shRNA
transfection (Fig. 4A). Moreover,
the growth of cells in soft agar decreased by >35% following
transfection when compared with the mock group (Fig. 4B). Next, A549 cells transfected with
sh-EGFR or sh-Mock control were treated with EGCG or vehicle and
subjected to a soft agar assay. EGCG (20 μM) inhibited the
anchorage-independent growth of A549 sh-Mock cells by ~50%. In
contrast, the inhibition was ~25% in A549 sh-EGFR cells, indicating
that A549 cells transfected with EGFR shRNA were resistant to EGCG
treatment (Fig. 4C). These results
imply that EGFR plays an important role in the sensitivity of A549
cells to the antiproliferative effect of EGCG.
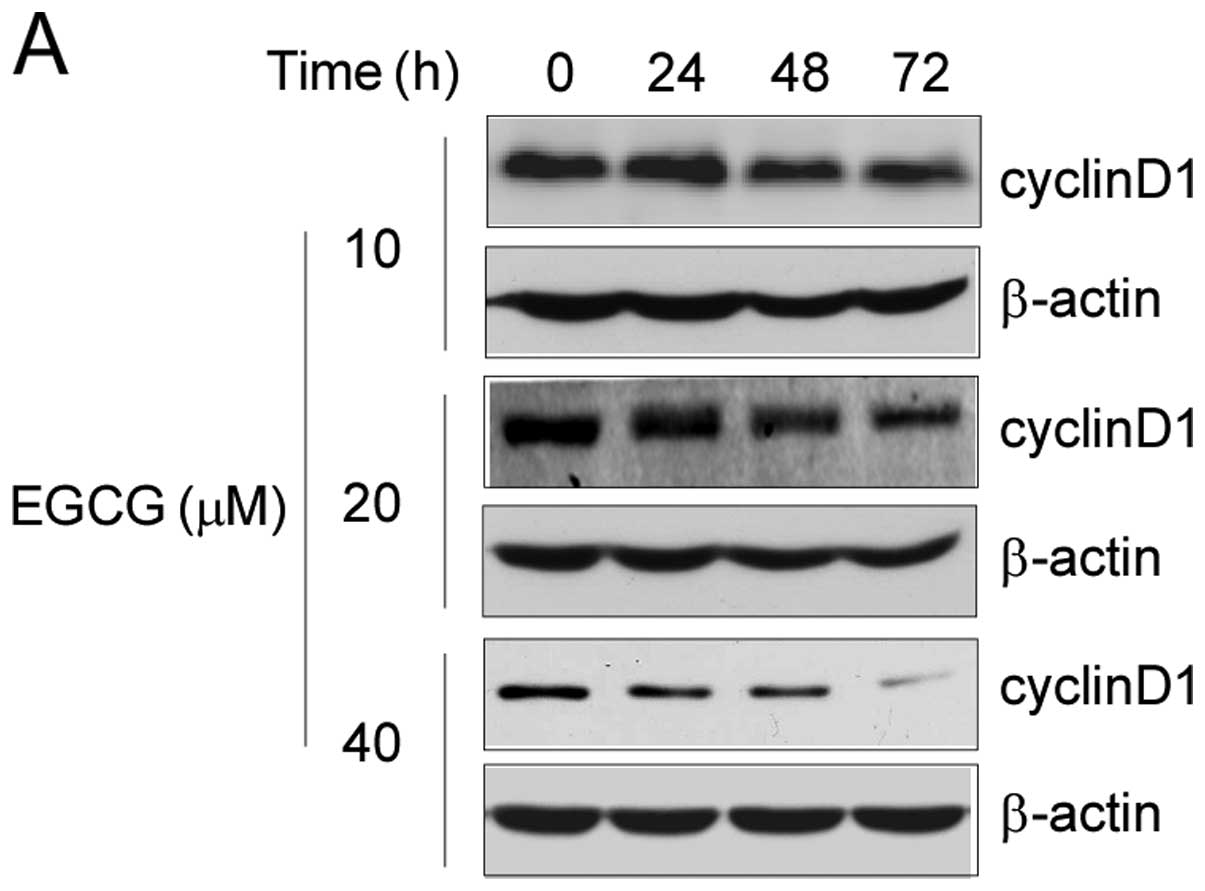
EGCG treatment suppresses cyclin D1
expression and induces cell cycle G0/G1
arrest
Previous studies demonstrated that cyclin D1 is an
important target gene for nuclear EGFR and is involved in cell
cycle regulation (31,32). Based on the above results, we
further investigated the effect of EGCG on cyclin D1 expression as
well as on cell cycle progression. Long-term treatment of EGCG
resulted in downregulation of cyclin D1 expression in A549 lung
cancer cells. Following treatment with 20 μM EGCG for 48 h, the
expression of cyclin D1 protein was substantially suppressed
(Fig. 5A). Together with the
inhibition of cyclin D1, EGCG treatment was accompanied by cell
cycle arrest at the G0/G1 phase in a
concentration-dependent manner. As shown in Fig. 5B, the percentage of cells in the
G0/G1 phase was increased to ~25% following
treatment with 40 μM EGCG. These data indicate that EGCG exerts its
antitumor activity via inhibition of EGFR transactivation ability
and regulation of expression of its target genes.
Discussion
The natural compound EGCG is the major polyphenol
component in green tea. EGCG is a highly active and promising
therapeutic and chemopreventive agent. However, the underlying
mechanism for its anticancer activity has not yet been elucidated.
The present study identified EGFR as a direct target of EGCG in
human lung cancer.
EGFR regulates important tumorigenic processes,
including proliferation, apoptosis, angiogenesis and invasion, and
along with its ligands, it is frequently overexpressed or
hyperactivated during the development and progression of human lung
cancer (28,33). Previous studies have revealed that
EGCG inhibits the activation of the EGFR and downstream signaling
pathways in several types of cancer cells (34–37).
Moreover, EGCG suppresses growth factor receptor signaling in human
non-small cell lung cancer cells and potentiates the
antiproliferative activity of c-Met and EGFR inhibitors (38). In the present study, we first
examined the antitumor effects of EGCG in several human lung cancer
cell lines. EGCG inhibited the anchorage-independent growth of
human lung cancer cells in a dose-dependent manner (Fig. 1). Western blotting demonstrated that
the EGFR signaling pathway appeared to play an important role in
EGCG-mediated lung cancer cell growth inhibition. EGF-induced EGFR
activation was markedly decreased by EGCG pre-treatment (Fig. 2). Our findings corroborated previous
studies which demonstrated that the effect of EGCG against lung
cancer may depend on immediate EGFR activity suppression.
Receptor downregulation is the most prominent
regulator of EGFR signal attenuation and involves the
internalization and subsequent degradation of the activated
receptor in lysosomes. Previous reports demonstrated that
EGCG-induced EGFR signaling reduction was partly regulated by
p38-mediated EGFR phosphorylation and internalization in colon
cancer (39,40). We then assessed EGFR expression
after long-term EGCG exposure in A549 cells. EGCG not only
decreased the total EGFR protein levels (Fig. 3A), but also inhibited the EGFR
expression on the cell membrane (Fig.
3B). More recent compelling evidence further indicates that
EGFR function depends on its subcellular location. EGFR can be
shuttled into the cell nucleus and the mitochondrion upon ligand
binding, radiation, EGFR-targeted therapy and other stimuli.
Nuclear EGFR serves as a transcriptional regulator, tyrosine kinase
and mediator of other physiological processes (15). In the present study, we demonstrated
for the first time that EGCG markedly inhibited EGFR nuclear
localization in a dose- and time-dependent manner (Fig. 3C). Based on these results, we
hypothesized that EGCG attenuates EGFR transcription activity.
Cyclin D1 is one of the well-known downstream target genes of EGFR.
We determined the effect of EGCG on cyclin D1 expression by western
blot analysis. Consistent with our hypothesis, the results clearly
showed that EGCG potently suppressed cyclin D1 expression in a
concentration-dependent manner (Fig.
5A). Additionally, EGCG-mediated cyclin D1 inhibition was
accompanied by cell cycle arrest at the G0/G1
phase (Fig. 5B). Moreover,
knockdown of EGFR expression suppressed A549 cell colony formation
in soft agar and decreased the sensitivity of A549 cells to EGCG
treatment (Fig. 4). These data
provide strong evidence that long-term EGCG exposure markedly
downregulates EGFR protein expression as well as its
transactivation.
Taken together, EGFR, as a potential and important
target of EGCG, offers useful evidence for the rational use and
combination treatment of EGCG in lung cancer therapy. We cannot
exclude the possibility that EGCG treatment, in addition to
decreasing EGFR kinase activity and protein expression, may have
antitumor activity by affecting other pathways. Numerous
preclinical studies and clinical trials of EGCG are still ongoing
in different countries (1), and
more research must be conducted to provide further valuable
evidence to guide the clinical use of EGCG in lung cancer
prevention and therapy.
Acknowledgements
The present study was supported by the Nature
Scientific Foundation of China (grant no. 81371690), the Nature
Scientific Foundation of Hunan Province (grant no. 08JJ6010), the
Research Program of the Science and Technology Department of Hunan
Province (grant no. 2012FJ4076) and the Research Program of the
Science and Technology Department of Hunan Province (grant no.
2012TT2011). We would like to thank Professor Jing Zhou for his
excellent English language editing of the manuscript.
Abbreviations:
|
EGCG
|
epigallocatechin gallate
|
|
EGFR
|
epidermal growth factor receptor
|
References
|
1
|
Kanwar J, Taskeen M, Mohammad I, Huo C,
Chan TH and Dou QP: Recent advances on tea polyphenols. Front
Biosci (Elite Ed). 4:111–131. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jin L, Li C, Xu Y, Wang L, Liu J, Wang D,
et al: Epigallocatechin gallate promotes p53 accumulation and
activity via the inhibition of MDM2-mediated p53 ubiquitination in
human lung cancer cells. Oncol Rep. 29:1983–1990. 2013.PubMed/NCBI
|
|
3
|
Park JS, Khoi PN, Joo YE, Lee YH, Lang SA,
Stoeltzing O, et al: EGCG inhibits recepteur d’origine nantais
expression by suppressing Egr-1 in gastric cancer cells. Int J
Oncol. 42:1120–1126. 2013.
|
|
4
|
Yiannakopoulou EC: Effect of green tea
catechins on breast carcinogenesis: a systematic review of in-vitro
and in-vivo experimental studies. Eur J Cancer Prev. Aug
9–2013.(Epub ahead of print).
|
|
5
|
Hagen RM, Chedea VS, Mintoff CP, Bowler E,
Morse HR and Ladomery MR: Epigallocatechin-3-gallate promotes
apoptosis and expression of the caspase 9a splice variant in PC3
prostate cancer cells. Int J Oncol. 43:194–200. 2013.PubMed/NCBI
|
|
6
|
Kim SO and Kim MR: (-)-Epigallocatechin
3-gallate inhibits invasion by inducing the expression of Raf
kinase inhibitor protein in AsPC1 human pancreatic adenocarcinoma
cells through the modulation of histone deacetylase activity. Int J
Oncol. 42:349–358. 2013.
|
|
7
|
Chen H, Landen CN, Li Y, Alvarez RD and
Tollefsbol TO: Enhancement of cisplatin-mediated apoptosis in
ovarian cancer cells through potentiating G2/M arrest and p21
upregulation by combinatorial epigallocatechin gallate and
sulforaphane. J Oncol. 2013:8729572013. View Article : Google Scholar
|
|
8
|
Kochi T, Shimizu M, Terakura D, Baba A,
Ohno T, Kubota M, et al: Non-alcoholic steatohepatitis and
preneoplastic lesions develop in the liver of obese and
hypertensive rats: suppressing effects of EGCG on the development
of liver lesions. Cancer Lett. 42:60–69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogawa K, Hara T, Shimizu M, Nagano J, Ohno
T, Hoshi M, et al: (-)-Epigallocatechin gallate inhibits the
expression of indoleamine 2,3-dioxygenase in human colorectal
cancer cells. Oncol Lett. 4:546–550. 2012.PubMed/NCBI
|
|
10
|
Chiou YS, Sang S, Cheng KH, Ho CT, Wang YJ
and Pan MH: Peracetylated (-)-epigallocatechin-3-gallate (AcEGCG)
potently prevents skin carcinogenesis by suppressing the
PKD1-dependent signaling pathway in CD34+ skin stem
cells and skin tumors. Carcinogenesis. 34:1315–1322. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shimizu M, Adachi S, Masuda M, Kozawa O
and Moriwaki H: Cancer chemoprevention with green tea catechins by
targeting receptor tyrosine kinases. Mol Nutr Food Res. 55:832–843.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujiki H and Suganuma M: Green tea: an
effective synergist with anticancer drugs for tertiary cancer
prevention. Cancer Lett. 324:119–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh BN, Shankar S and Srivastava RK:
Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms,
perspectives and clinical applications. Biochem Pharmacol.
82:1807–1821. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han W and Lo HW: Landscape of EGFR
signaling network in human cancers: biology and therapeutic
response in relation to receptor subcellular locations. Cancer
Lett. 318:124–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tebbutt N, Pedersen MW and Johns TG:
Targeting the ERBB family in cancer: couples therapy. Nat Rev
Cancer. 13:663–673. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kolch W and Pitt A: Functional proteomics
to dissect tyrosine kinase signalling pathways in cancer. Nat Rev
Cancer. 10:618–629. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Avraham R and Yarden Y: Feedback
regulation of EGFR signalling: decision making by early and delayed
loops. Nat Rev Mol Cell Biol. 12:104–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marti U, Burwen SJ, Wells A, Barker ME,
Huling S, Feren AM, et al: Localization of epidermal growth factor
receptor in hepatocyte nuclei. Hepatology. 13:15–20. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brand TM, Iida M, Luthar N, Starr MM,
Huppert EJ and Wheeler DL: Nuclear EGFR as a molecular target in
cancer. Radiother Oncol. 108:370–377. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia
W, Wei Y, et al: Nuclear interaction of EGFR and STAT3 in the
activation of the iNOS/NO pathway. Cancer Cell. 7:575–589. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y
and Hung MC: Co-regulation of B-Myb expression by E2F1 and EGF
receptor. Mol Carcinog. 45:10–17. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hung LY, Tseng JT, Lee YC, Xia W, Wang YN,
Wu ML, et al: Nuclear epidermal growth factor receptor (EGFR)
interacts with signal transducer and activator of transcription 5
(STAT5) in activating Aurora-A gene expression. Nucleic Acids Res.
36:4337–4351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dittmann K, Mayer C, Fehrenbacher B,
Schaller M, Raju U, Milas L, et al: Radiation-induced epidermal
growth factor receptor nuclear import is linked to activation of
DNA-dependent protein kinase. J Biol Chem. 280:31182–31189. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
26
|
Katlic MR, Facktor MA, Berry SA, McKinley
KE, Bothe A Jr and Steele GD Jr: ProvenCare lung cancer: a
multi-institutional improvement collaborative. CA Cancer J Clin.
61:382–396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramalingam SS, Owonikoko TK and Khuri FR:
Lung cancer: new biological insights and recent therapeutic
advances. CA Cancer J Clin. 61:91–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herbst RS, Fukuoka M and Baselga J:
Gefitinib - a novel targeted approach to treating cancer. Nat Rev
Cancer. 4:956–965. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Janku F, Stewart DJ and Kurzrock R:
Targeted therapy in non-small-cell lung cancer - is it becoming a
reality? Nat Rev Clin Oncol. 7:401–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tao Y, Song X, Deng X, Xie D, Lee LM, Liu
Y, et al: Nuclear accumulation of epidermal growth factor receptor
and acceleration of G1/S stage by Epstein-Barr-encoded oncoprotein
latent membrane protein 1. Exp Cell Res. 303:240–251. 2005.
View Article : Google Scholar
|
|
32
|
Shi Y, Tao Y, Jiang Y, Xu Y, Yan B, Chen
X, et al: Nuclear epidermal growth factor receptor interacts with
transcriptional intermediary factor 2 to activate cyclin D1 gene
expression triggered by the oncoprotein latent membrane protein 1.
Carcinogenesis. 33:1468–1478. 2012. View Article : Google Scholar
|
|
33
|
Sato M, Shames DS, Gazdar AF and Minna JD:
A translational view of the molecular pathogenesis of lung cancer.
J Thorac Oncol. 2:327–343. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shimizu M, Shirakami Y and Moriwaki H:
Targeting receptor tyrosine kinases for chemoprevention by green
tea catechin, EGCG. Int J Mol Sci. 9:1034–1049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masuda M, Suzui M and Weinstein IB:
Effects of epigallocatechin-3-gallate on growth, epidermal growth
factor receptor signaling pathways, gene expression, and
chemosensitivity in human head and neck squamous cell carcinoma
cell lines. Clin Cancer Res. 7:4220–4229. 2001.PubMed/NCBI
|
|
36
|
Hou Z, Sang S, You H, Lee MJ, Hong J, Chin
KV, et al: Mechanism of action of (-)-epigallocatechin-3-gallate:
auto-oxidation-dependent inactivation of epidermal growth factor
receptor and direct effects on growth inhibition in human
esophageal cancer KYSE 150 cells. Cancer Res. 65:8049–8056.
2005.
|
|
37
|
Adachi S, Nagao T, Ingolfsson HI, Maxfield
FR, Andersen OS, Kopelovich L, et al: The inhibitory effect of
(-)-epigallocatechin gallate on activation of the epidermal growth
factor receptor is associated with altered lipid order in HT29
colon cancer cells. Cancer Res. 67:6493–6501. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Milligan SA, Burke P, Coleman DT, Bigelow
RL, Steffan JJ, Carroll JL, et al: The green tea polyphenol EGCG
potentiates the antiproliferative activity of c-Met and epidermal
growth factor receptor inhibitors in non-small cell lung cancer
cells. Clin Cancer Res. 15:4885–4894. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Adachi S, Shimizu M, Shirakami Y, Yamauchi
J, Natsume H, Matsushima-Nishiwaki R, et al: (-)-Epigallocatechin
gallate downregulates EGF receptor via phosphorylation at
Ser1046/1047 by p38 MAPK in colon cancer cells. Carcinogenesis.
30:1544–1552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arteaga CL: Epidermal growth factor
receptor dependence in human tumors: more than just expression?
Oncologist. 7(Suppl 4): S31–S39. 2002. View Article : Google Scholar : PubMed/NCBI
|