Introduction
The estrogen receptors (ERs) ER-α and ER-β are
members of the nuclear hormone receptor superfamily of
ligand-activated transcription factors (1). The status of ER expression in human
breast tumors is an extremely important prognostic marker for
selecting the appropriate hormonal therapy (2,3).
Approximately 75% of breast cancers express ER and/or the
progesterone receptor (PR) and are treated with targeted
anti-estrogen therapy such as tamoxifen (2,4).
Tamoxifen is widely used for treating
hormone-dependent, ER/PR-positive breast cancer (5). It is effective for inducing the arrest
of tumor progression in 50% of patients with breast cancer
(6). However, although
anti-estrogen therapies targeting ER-α prevent disease recurrence
in patients with hormone-dependent breast cancer, de novo or
acquired resistance remains a major problem (7,8). To
date, many mechanisms have been suggested for the
tamoxifen-resistant model, yet the mechanisms are not fully
understood.
Protein kinase C (PKC) is a member of a family of
serine/threonine protein kinases and is involved in a wide variety
of fundamental physiological processes including cell proliferation
and apoptosis (9,10). Estrogen-treated breast cancer cells
such as MCF-7 and HCC38 show rapid increases in PKC activity
(11). PKC activity is
significantly elevated in malignant tumor tissues when compared
with that in normal human breast tissues (12). We reported that PKC-α mediates cell
invasion and migration by inducing matrix metalloproteinase (MMP)-1
and MMP-9 expression in breast cancer cells (13).
The aim of the present study was to investigate the
effect of PKC-α on the level of ER-α expression and the regulatory
mechanism of PKC-α-induced downregulation of ER-α in ER-positive
breast cancer cells.
Materials and methods
Reagents
4-Hydroxytamoxifen (4-OHT) was purchased from Sigma
(St. Louis, MO, USA). Fetal bovine serum (FBS), RPMI-1640 and
Dulbecco’s modified Eagle’s medium (DMEM) were purchased from
Thermo Scientific (Hemel Hempstead, UK). Penicillin (100 U/ml) and
100 mg/ml streptomycin were purchased from Life Technologies
(Rockville, MD, USA). Go6983 was purchased from Tocris (Ellisville,
MO, USA). SR11304 and mouse monoclonal anti-ER-α and anti-β-actin
antibodies were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). 12-O-Tetradecanoylphorbol-13-acetate (TPA)
was purchased from R&D Systems (Minneapolis, MN, USA). The
ECLplus reagents were from Amersham (Buckinghamshire, UK).
Cell culture and establishment of
tamoxifen-resistant (TAMR) MCF-7 breast cancer cells
MCF-7 breast cancer cells were grown in a humidified
atmosphere of 95% air and 5% CO2 at 37°C in DMEM
supplemented with 10% FBS, 2 mM glutamine, 100 IU/ml penicillin and
100 μg/ml streptomycin. T47D and ZR75-1 breast cancer cells were
cultured in RPMI-1640. The TAMR breast cancer cell line was kindly
provided by Professor Keun Wook Kang (Seoul National University,
Seoul, Korea). The TAMR cell line was established using a
previously reported methodology (14). Briefly, MCF-7 cells were washed with
PBS, and the culture medium was replaced with phenol red-free DMEM
containing 10% charcoal-stripped steroid-depleted FBS (HyClone,
Logan, UT, USA) and 0.1 mM 4-OHT. The cells were continuously
exposed to this treatment regimen for 2 weeks, and the 4-OHT
concentration was increased gradually up to 3 mM over a 9-month
period. Initially, cell growth rates were depressed. However, after
exposure to the medium for 9 months, the rate of cell growth
increased gradually, indicating the establishment of
tamoxifen-resistant cells.
Cell proliferation
Cell proliferation was measured using a
Countess® automated cell counter (Invitrogen, Carlsbad,
CA, USA). Cells were plated at 5×104/well in 6-well
plates. Tamoxifen-sensitive (TAMS) and TAMR cells were incubated in
phenol red-free DMEM containing 10% charcoal-stripped
steroid-depleted FBS with or without 3 mM 4-OHT for the indicated
times.
Western blotting
The cell lysates were used in the immunoblot
analysis for ER-α, PKC-α and β-actin. The proteins were boiled for
5 min in Laemmli sample buffer and then electrophoresed on 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. The
proteins were transferred to PVDF membranes, and the membranes were
blocked with 10% skim milk in TBS with 0.01% Tween-20 for 15 min.
The blots were incubated with anti-matrix metalloproteinase
(MMP)-1, PKC-α and β-actin antibodies (1:1,000 dilution) in 1%
TBS/T buffer (0.01% Tween-20 in TBS) at 4°C overnight. The blots
were washed three times, in TBS with 0.01% Tween-20, and they were
subsequently incubated with anti-rabbit peroxidase-conjugated
antibody (1:2,000 dilution) in TBS/T buffer. After a 1-h incubation
at room temperature, the blots were washed three times and enhanced
chemiluminescence reagents (Amersham Bioscience) were used for
development.
Real-time polymerase chain reaction
(RT-PCR)
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen), according to the manufacturer’s protocol.
Isolated RNA samples were then used for RT-PCR. Samples (1 μg total
RNA) were reverse-transcribed into cDNA in 20-μl reaction volumes
using a First-Strand cDNA Synthesis kit for RT-PCR, according to
the manufacturer’s instructions (MBI Fermentas, Hanover, MD,
USA).
Gene expression was quantified by real-time PCR
using a SensiMix SYBR kit (Bioline Ltd., London, UK) and 100 ng of
cDNA per reaction. The sequences of the primer sets used for this
analysis were: human ER-α (forward, 5′-CGC TAC TGT GCA GTG TGC
AAT-3′ and reverse, 5′-CCT CAC AGG ACC AGA CTC CAT AA-3′) and GAPDH
as an internal control (forward, 5′-ATT GTT GCC ATC AAT GAC CC-3′
and reverse, 5′-AGT AGA GGC AGG GAT GAT GT-3′). An annealing
temperature of 60°C was used for all primers. PCRs were performed
in a standard plate format with an ABI 7900HT real-time PCR
detection system. The raw threshold cycle (CT) value was
first normalized to the housekeeping gene for each sample to obtain
ΔCT. The normalized ΔCT was then calibrated
to the control cell samples to obtain ΔΔCT. All cDNA
samples were analyzed in three independent experiments.
PKC-α siRNA and myr-PKC-α
transfection
PKC-α siRNA was purchased from Bioneer Corp.
(Daejeon, Korea). Myr-PKC-α FLAG was a gift from Dr R.R. Hodges
(Addgene plasmid #10807) (15). We
found that the optimal siRNA knockdown and overexpression
conditions involved transfection of the MCF-7 breast cancer cells
at 80% confluence, and the cells were maintained in DMEM with 10%
FBS; Effectene® (Qiagen, Valencia, CA, USA) was used for
the transfections with PKC-α siRNA (25 and 50 nM, or as noted) or
Myr-PKC-α FLAG following the manufacturer’s protocols. Fresh
serum-free media with or without 20 nM TPA were added 24 h after
the 48-h transfection. The level of PKC-α protein expression was
analyzed by western blotting.
Statistical analysis
Statistical significance was determined using the
Student’s t-test. Results are presented as means ± standard errors.
All P-values are two-tailed, and differences were considered
significant at P<0.05.
Results
Inverse co-relationship between PKC-α and
ER-α in the TAMR cell line
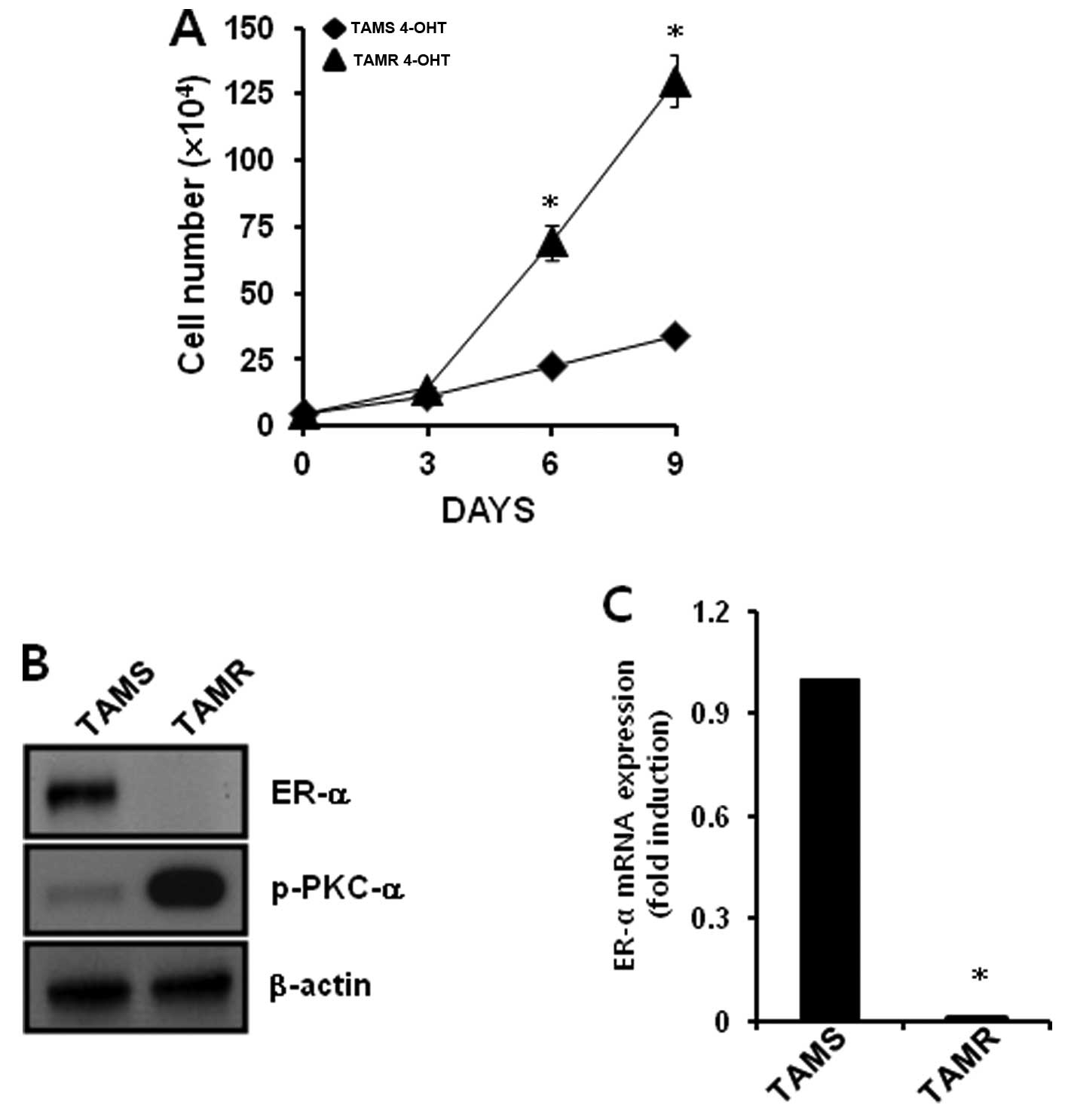
We chose TAMS and TAMR MCF-7 breast cancer cells to
verify the relationship between PKC-α and ER-α. We treated each
cell type with 3 mM 4-OHT for the indicated times. As shown in
Fig. 1A, the proliferation of TAMS
cells in response to 3 mM 4-OHT was significantly suppressed.
However, the proliferation of 3 mM 4-OHT-treated TAMR cells
increased in a time-dependent manner, and the gap in proliferation
was significantly different after 6 days (Fig. 1A). We also compared the expression
level of PKC-α and ER-α in TAMS and TAMR cells. The levels of ER-α
protein and mRNA expression were significantly decreased in the
TAMR cells when compared with the levels in TAMS cells (Fig. 1B and C). In contrast, PKC-α
phosphorylation was significantly increased in the TAMR cells
(Fig. 1B). Therefore, an inverse
correlation was noted between PKC-α and ER-α in the TAMR MCF-7
breast cancer cell line.
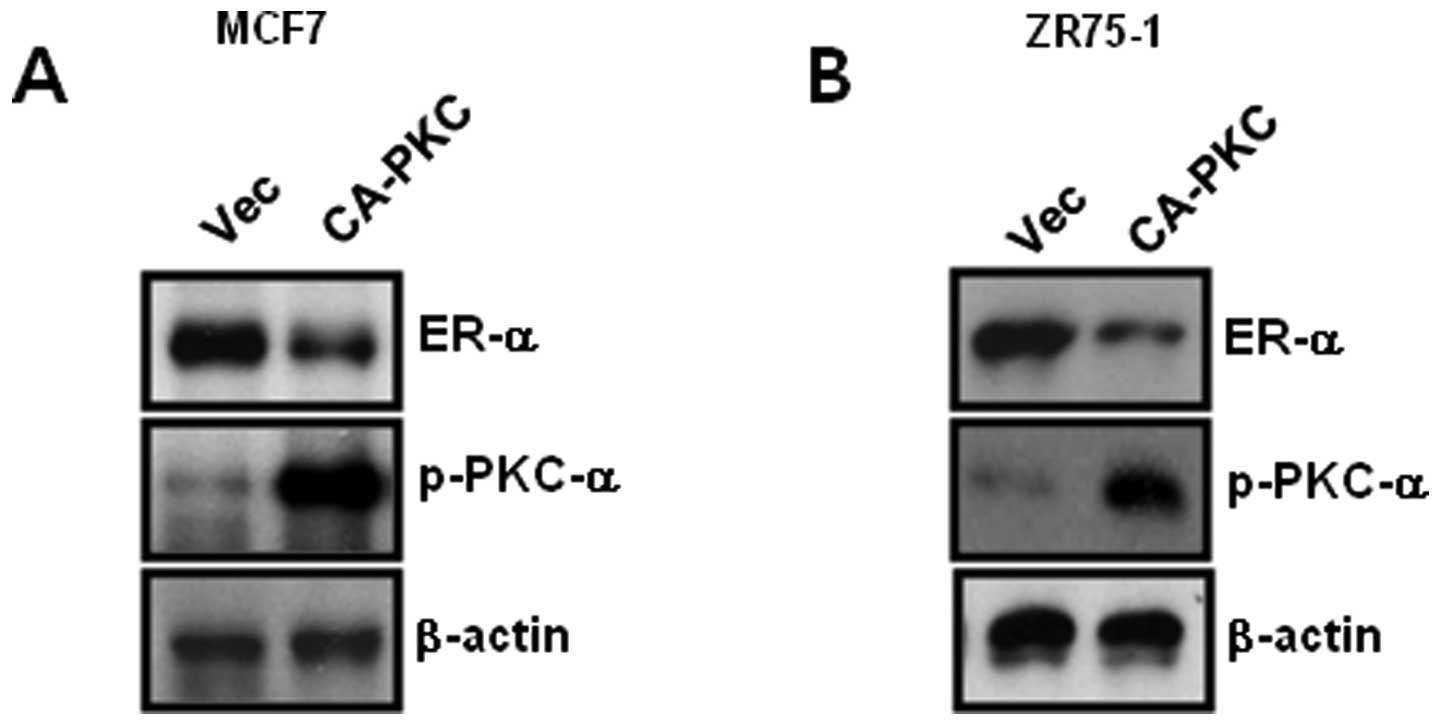
Overexpression of constitutively active
PKC-α (CA-PKC-α) decreases the level of ER-α expression in
ER-α-positive breast cancer cells
Based on Fig. 1, we
examined the direct relationship between PKC-α and ER-α in
ER-positive breast cancer cells. The cells were transfected with
CA-PKC-α for 48 h, and the cell lysates were harvested to detect
PKC-α and ER-α expression. The results showed that the level of
ER-α expression decreased due to CA-PKC-α overexpression in MCF-7
and ZR75-1 breast cancer cells (Fig. 2A
and B). Therefore, we demonstrated that the level of ER-α
expression was regulated through a PKC-α-dependent mechanism in
ER-positive breast cancer cells.
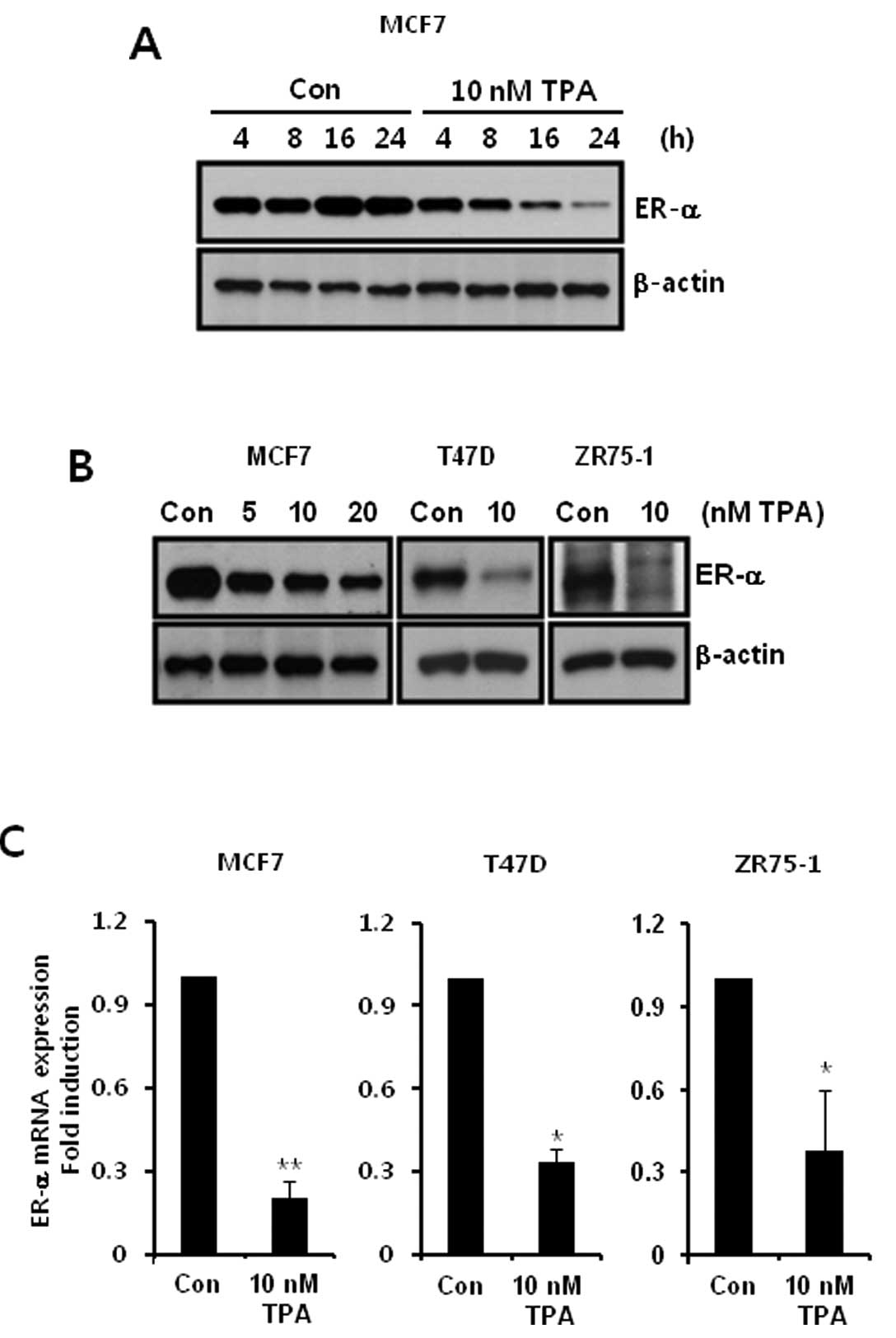
Expression of ER-α was decreased in
ER-α-positive breast cancer cells by TPA in a time- and
dose-dependent manner
Next, we investigated the effect of TPA on ER-α
expression. TPA is a natural molecule that is a well-known tumor
promoter and reversible activator of PKC (16). We treated MCF-7 cells with 10 nM TPA
for the indicated times. As shown in Fig. 3A, the level of ER-α protein
expression was decreased in a time-dependent manner following TPA
treatment. Furthermore, ER-α protein expression in response to TPA
was significantly decreased in T47D and ZR75-1 breast cancer cells
(Fig. 3B). In addition, we
confirmed the level of ER-α mRNA expression following TPA
treatment. The level of ER-α mRNA expression was also significantly
decreased to 0.2±0.06-fold (in MCF-7 cells), 0.34±0.04-fold (in
T47D cells) and 0.39±0.23-fold (in ZR75-1 cells) of the control
level following 10 nM TPA treatment, respectively (Fig. 3C). These results demonstrated that
TPA downregulates the expression of ER-α by activating PKC-α in
ER-positive breast cancer cells.
TPA-induced downregulation of ER-α is
mediated by a PKC-α-dependent pathway but not by a
PI-3K/Akt-dependent pathway or p38-dependent pathway
To investigate the regulatory mechanisms of
TPA-induced downregulation of ER-α, we treated ER-positive breast
cancer cells with specific inhibitors such as the PKC inhibitor
Go6983, the PI-3K inhibitor LY294002, and the p38 inhibitor
SB203580. Our results showed that TPA-induced downregulation of
ER-α protein and mRNA expression was prevented by Go6983 in MCF-7
cells but not by LY294002 or SB203580 (Fig. 4A). The level of ER-α mRNA expression
was significantly decreased to 0.35±0.03-fold of the control level
following TPA treatment (Fig. 4A).
In contrast, TPA-induced downregulation of ER-α mRNA expression was
suppressed to 1.37±0.01-fold of the control level by 10 μM Go6983
treatment (Fig. 4A). Under the same
conditions, our results also showed that TPA-induced downregulation
of ER-α protein and mRNA expression was prevented by Go6983 in T47D
(Fig. 4B) and ZR75-1 (Fig. 4C) breast cancer cells. Therefore, we
demonstrated that TPA also regulates ER-α expression through a
PKC-dependent pathway in ER-positive breast cancer cells.
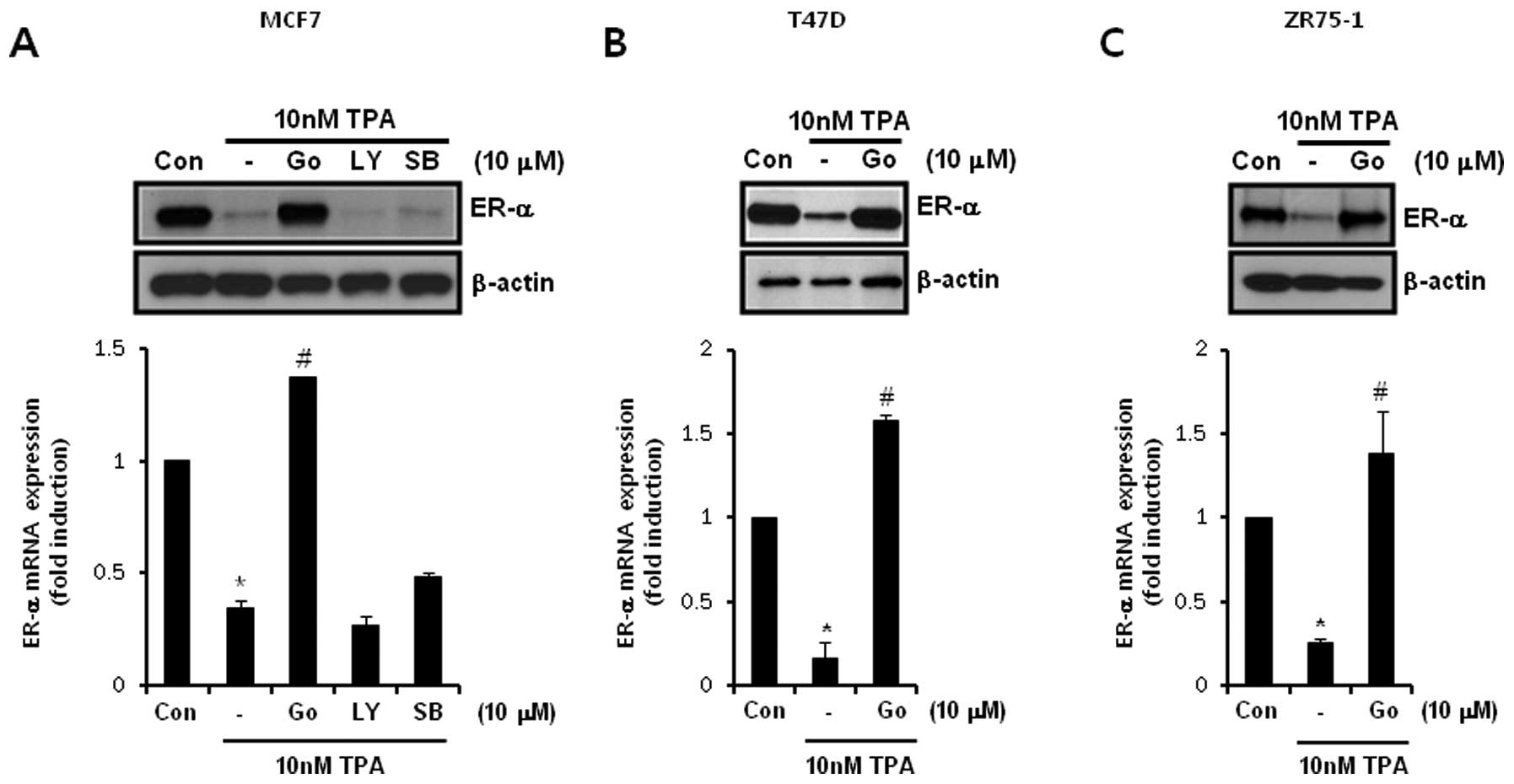 | Figure 4
12-O-Tetradecanoylphorbol-13-acetate (TPA)-induced
downregulation of estrogen receptor (ER)-α is prevented by Go6983
but not by LY294002 or SB203580. (A) After serum-starvation for 24
h, MCF-7 cells were pretreated with 10 μM Go6983 (Go), 10 μM
LY294002 (LY), and 10 μM SB203580 (SB), respectively, for 30 min
and were then treated with 10 nM TPA for 24 h. ER-α protein and
mRNA expression was analyzed by western blotting and real-time
polymerase chain reaction analysis, respectively. (B and C) After
serum-starvation for 24 h, (B) T47D and (C) ZR75-1 cells were
pretreated with 10 μM (Go), respectively, for 30 min and then
treated with 10 nM TPA for 24 h. ER-α protein and mRNA expression
was analyzed by western blotting and real-time polymerase chain
reaction analysis, respectively. Results are representative of
three independent experiments. Values shown are means ± standard
errors. *P<0.05 vs. control, #P<0.05
vs. TPA-treated cells. Con, control. |
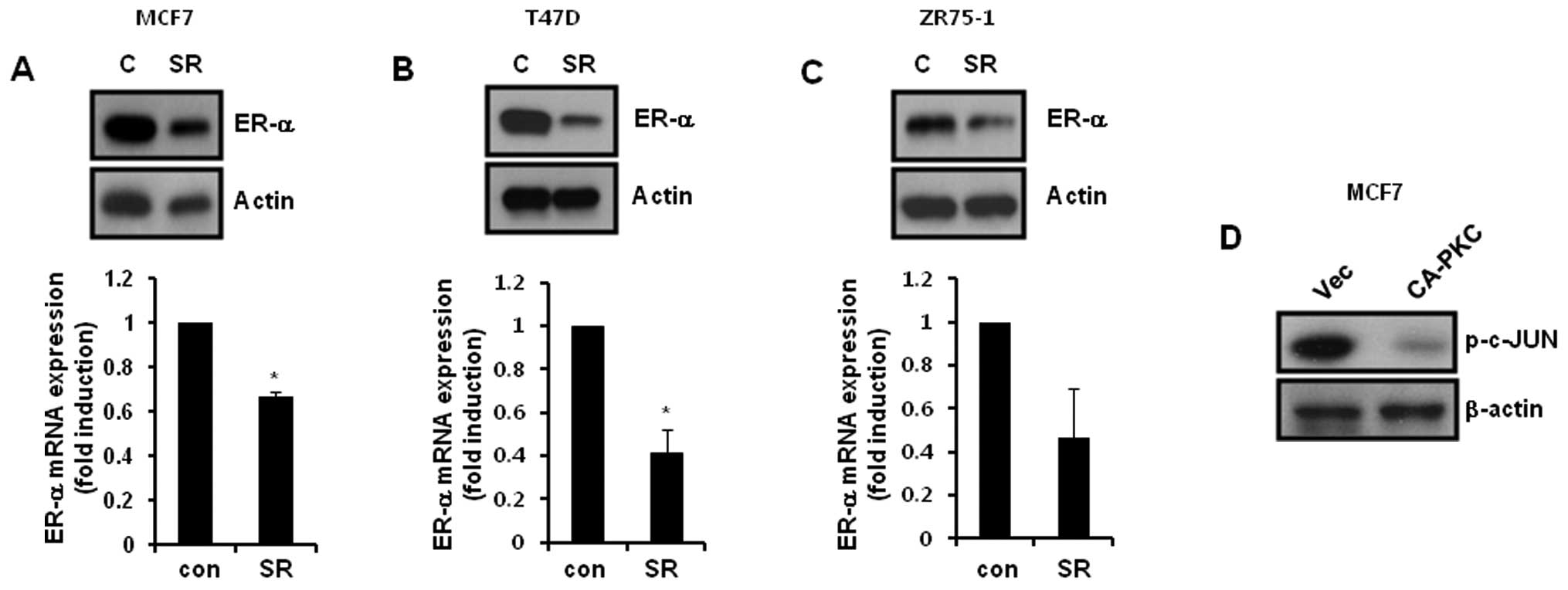
PKC-α regulates ER-α expression by
suppressing c-Jun activity in ER-α-positive breast cancer
cells
Finally, we investigated whether the activator
protein-1 (AP-1) transcriptional factor affects the level of ER-α
expression in ER-positive breast cancer cells. After treatment with
3 μM SR11302 for 24 h, the cells were harvested for detection of
ER-α mRNA and protein expression. Our results showed that the
levels of ER-α protein and mRNA expression was significantly
decreased following SR11302 treatment of ER-α-positive breast
cancer cells (Fig. 5A–C). In
addition, we examined the level of c-JUN phosphorylation in
CA-PKC-α-overexpressing MCF-7 cells. As shown in Fig. 5D, the level of c-JUN phosphorylation
was decreased due to CA-PKC-α overexpression. Therefore, we
demonstrated that PKC-α downregulated ER-α expression by
suppressing AP-1 activity in ER-α-positive breast cancer cells.
Discussion
Although tamoxifen is widely used to treat all
stages of breast cancer, almost 50% of patients with breast cancer
fail to respond to tamoxifen and eventually acquire tamoxifen
resistance, leading to tumor progression and death (8,17). The
exact regulatory mechanism of tamoxifen resistance is not fully
understood. In the present study, we investigated whether the level
of ER-α expression is regulated by a PKC-α-dependent pathway in
ER-α-positive breast cancer cells.
PKC, a serine/threonine kinase, regulates
proliferation, differentiation and apoptosis in a variety of cells
including breast and ovarian cancer cells (18,19).
We previously reported that TPA-induced MMP-1 and MMP-9 expression
is mediated through a PKC-α-dependent pathway in breast cancer
cells (13). Upregulation of PKC-α
and PKC-ɛ is correlated with stimulation of human endometrial
cancer growth by tamoxifen (20).
Consistent with this report, the PKC-α phosphorylation level was
significantly augmented in the TAMR cell line compared with that in
the TAMS cell line. Overexpression of CA-PKC-α significantly
decreased the level of ER-α mRNA and protein expression in breast
cancer cells. Thus, we suggest that PKC-α expression or activity
may predict tamoxifen treatment failure.
Furthermore, the tumor promoter TPA is a natural
molecule and reversible activator of PKC (16). TPA suppressed ER expression in MCF-7
cells, similar to estrogen treatment and increased ER
phosphorylation (21). Elevated PKC
activity suppressed ER expression in breast cancer (22,23).
In addition, PKC-α levels may increase in patients with breast
cancer resulting in low or negative ER levels compared to those in
ER-positive patients (10,22,23).
PKC-α overexpression is associated with a more aggressive
neoplastic phenotype in MCF-7 breast cancer cells (24). Although we used treatment with TPA
to activate PKC in breast cancer cells, our results showed that
ER-α mRNA and protein expression decreased. Therefore, we suggest
that activation of PKC-α may trigger tamoxifen resistance by
downregulating ER-α in breast cancer cells.
AP-1 is a member of the Jun and/or Fos family. The
AP-1 complex regulates transcriptional activity of a variety of
genes including ER-α (25). ER-α
efficiently binds to c-Jun and JunB but does not directly bind to
any Fos family members (26). The
induction of AP-1 transcriptional activity requires TPA-induced
tumor promotion (27). Importantly,
our results showed that CA-PKC-α overexpression significantly
decreased c-Jun phosphorylation. In addition, the ER-α expression
level was suppressed by SR11302, an inhibitor of AP-1. Therefore,
we demonstrated that activation of PKC-α suppressed the level of
ER-α expression by inhibiting AP-1 activity in TAMR and ER-positive
breast cancer cells.
In conclusion, we demonstrated an inverse
relationship between PKC-α activity and the level of ER-α
expression in TAMR and ER-positive breast cancer cells. PKC-α
suppressed the level of ER-α expression by inhibiting c-Jun
phosphorylation in ER-positive breast cancer cells. Therefore, we
suggest that PKC-α may be a potential therapeutic target in TAMR
and ER-positive breast cancer.
Acknowledgements
The present study was supported by a grant from the
Korea Healthcare Technology R&D Project, Ministry for Health
and Welfare Affairs, Republic of Korea (A092255) and by a Samsung
Biomedical Research Institute grant (GL1B31311).
References
|
1
|
Osborne CK and Schiff R: Estrogen-receptor
biology: continuing progress and therapeutic implications. J Clin
Oncol. 23:1616–1622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osborne CK, Zhao H and Fuqua SA: Selective
estrogen receptor modulators: structure, function, and clinical
use. J Clin Oncol. 18:3172–3186. 2000.PubMed/NCBI
|
|
3
|
Harvey JM, Clark GM, Osborne CK and Allred
DC: Estrogen receptor status by immunohistochemistry is superior to
the ligand-binding assay for predicting response to adjuvant
endocrine therapy in breast cancer. J Clin Oncol. 17:1474–1481.
1999.PubMed/NCBI
|
|
4
|
Herynk MH and Fuqua SA: Estrogen receptors
in resistance to hormone therapy. Adv Exp Med Biol. 608:130–143.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Milano A, Dal Lago L, Sotiriou C, Piccart
M and Cardoso F: What clinicians need to know about antioestrogen
resistance in breast cancer therapy. Eur J Cancer. 42:2692–2705.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ali S and Coombes RC: Endocrine-responsive
breast cancer and strategies for combating resistance. Nat Rev
Cancer. 2:101–112. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kurebayashi J: Endocrine-resistant breast
cancer: underlying mechanisms and strategies for overcoming
resistance. Breast Cancer. 10:112–119. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiff R, Massarweh S, Shou J and Osborne
CK: Breast cancer endocrine resistance: how growth factor signaling
and estrogen receptor coregulators modulate response. Clin Cancer
Res. 9:447S–454S. 2003.PubMed/NCBI
|
|
9
|
Dekker LV and Parker PJ: Protein kinase C
- a question of specificity. Trends Biochem Sci. 19:73–77. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fabbro D, Kung W, Roos W, Regazzi R and
Eppenberger U: Epidermal growth factor binding and protein kinase C
activities in human breast cancer cell lines: possible quantitative
relationship. Cancer Res. 46:2720–2725. 1986.
|
|
11
|
Boyan BD, Sylvia VL, Frambach T, Lohmann
CH, Dietl J, Dean DD and Schwartz Z: Estrogen-dependent rapid
activation of protein kinase C in estrogen receptor-positive MCF-7
breast cancer cells and estrogen receptor-negative HCC38 cells is
membrane-mediated and inhibited by tamoxifen. Endocrinology.
144:1812–1824. 2003. View Article : Google Scholar
|
|
12
|
Gordge PC, Hulme MJ, Clegg RA and Miller
WR: Elevation of protein kinase A and protein kinase C activities
in malignant as compared with normal human breast tissue. Eur J
Cancer. 32A:2120–2126. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim S, Han J, Lee SK, Choi MY, Kim J, Lee
J, Jung SP, Kim JS, Kim JH, Choe JH, Lee JE and Nam SJ: Berberine
suppresses the TPA-induced MMP-1 and MMP-9 expressions through the
inhibition of PKC-α in breast cancer cells. J Surg Res.
176:e21–e29. 2012.PubMed/NCBI
|
|
14
|
Knowlden JM, Hutcheson IR, Jones HE,
Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE and Nicholson
RI: Elevated levels of epidermal growth factor receptor/c-erbB2
heterodimers mediate an autocrine growth regulatory pathway in
tamoxifen-resistant MCF-7 cells. Endocrinology. 144:1032–1044.
2003. View Article : Google Scholar
|
|
15
|
Hodges RR, Kazlauskas A, Toker A and Dartt
DA: Effect of overexpression of protein kinase C alpha on rat
lacrimal gland protein secretion. Adv Exp Med Biol. 506:237–241.
2002.PubMed/NCBI
|
|
16
|
Kikkawa U, Takai Y, Tanaka Y, Miyake R and
Nishizuka Y: Protein kinase C as a possible receptor protein of
tumor-promoting phorbol esters. J Biol Chem. 258:11442–11445.
1983.PubMed/NCBI
|
|
17
|
Osborne CK: Tamoxifen in the treatment of
breast cancer. N Engl J Med. 339:1609–1618. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Newton AC: Protein kinase C: structure,
function, and regulation. J Biol Chem. 270:28495–28498. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gupta AK, Galoforo SS, Berns CM, Martinez
AA, Corry PM, Guan KL and Lee YJ: Elevated levels of ERK2 in human
breast carcinoma MCF-7 cells transfected with protein kinase C
alpha. Cell Prolif. 29:655–663. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tonetti DA, O’Regan R, Tanjore S, England
G and Jordan VC: Antiestrogen stimulated human endometrial cancer
growth: laboratory and clinical considerations. J Steroid Biochem
Mol Biol. 65:181–189. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McInerney EM, Weis KE, Sun J, Mosselman S
and Katzenellenbogen BS: Transcription activation by the human
estrogen receptor subtype β (ERβ) studied with ERβ and ERα receptor
chimeras. Endocrinology. 139:4513–4522. 1998.
|
|
22
|
Basu A: The potential of protein kinase C
as a target for anticancer treatment. Pharmacol Ther. 59:257–280.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Blobe GC, Obeid LM and Hannun YA:
Regulation of protein kinase C and role in cancer biology. Cancer
Metastasis Rev. 13:411–431. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ways DK, Kukoly CA, deVente J, Hooker JL,
Bryant WO, Posekany KJ, Fletcher DJ, Cook PP and Parker PJ: MCF-7
breast cancer cells transfected with protein kinase C-alpha exhibit
altered expression of other protein kinase C isoforms and display a
more aggressive neoplastic phenotype. J Clin Invest. 95:1906–1915.
1995. View Article : Google Scholar
|
|
25
|
Kushner PJ, Agard DA, Greene GL, Scanlan
TS, Shiau AK, Uht RM and Webb P: Estrogen receptor pathways to
AP-1. J Steroid Biochem Mol Biol. 74:311–317. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jonat C, Rahmsdorf HJ, Park KK, Cato AC,
Gebel S, Ponta H and Herrlich P: Antitumor promotion and
antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by
glucocorticoid hormone. Cell. 62:1189–1204. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Behrens A, Jochum W, Sibilia M and Wagner
EF: Oncogenic transformation by ras and fos is mediated by c-Jun
N-terminal phosphorylation. Oncogene. 19:2657–2663. 2000.
View Article : Google Scholar : PubMed/NCBI
|