Introduction
Ionizing radiation (IR)-induced apoptosis has been
known to play a primary role in the therapeutic effect of IR. Many
studies have addressed the mechanism of IR-induced apoptosis and
the most effective strategies to induce it in radiotherapy
(RT)-targeted cancer cells (1–4).
However, it has been reported that apoptosis may not be the
exclusive or even the primary mechanism underlying tumor regression
due to cancer therapy; many studies have indicated that premature
senescence also plays a crucial role in tumor regression as well as
cancer prevention (5–7). Cellular senescence is induced by a
variety of signals, such as telomere shortening, oncogene
activation, ROS and DNA damage (8,9).
Senescent cells undergo functional and morphological changes
including senescence-associated β-galactosidase (SA-β-Gal) activity
and large and flat morphology (10,11).
Previous studies showed that cancer cells may become prematurely
senescent rather than apoptotic in response to DNA damage depending
on cellular context, although the mechanism of this alternate
response is unclear. Senescent cells are found in pre-malignant
lesions in mice and humans, and induction of senescence prevents
malignant progression in certain mouse tumor models (12–14).
Premature senescence may be induced in cancer cells using lower
doses of a drug than required to induce apoptosis (15,16),
indicating that cancer therapies targeting cellular senescence may
reduce damage of normal tissues. However, despite the interest in
the clinical application of premature senescence to increase RT
efficacy, its importance in IR-exposed cells or tissues remains to
be further validated.
RT is an essential therapeutic modality for a wide
range of malignant tumors. Although 40–60% of cancer patients
receive RT, its clinical usage is hindered by the significant
morbidity of radiation-induced injury to surrounding normal tissue.
A major challenge in radiation oncology is minimizing the
detrimental effects of RT on normal tissue while maximizing its
tumoricidal effects (17,18). Fractionated radiation (FR) treatment
reduces the damage to the surrounding normal cells while
maintaining the probability of tumor control (19). In the clinical setting, the total IR
dose is carefully determined and is often fractionated to reduce
the injury to normal tissues. Due to differences in the repair of
sub-lethal damage in normal vs. tumor cells, 2 Gy fractional doses
are typically used in conventional FR (20,21).
It is not well known whether FR or any IR modality effectively
induces premature senescence in vivo, mainly due to the
shortage of reliable biomarkers of senescence as well as the
technical difficulty of validating permanent cell cycle arrest.
Although several novel markers of senescence have been identified
(15,22), their reliability, diagnostic and
prognostic values require further characterization.
In the present study, we assessed IR-induced cancer
cell senescence and senescence biomarkers in vitro and in
vivo, and compared the relative effectiveness of single
radiation (SR) vs. FR.
Materials and methods
Cell culture and animals
H460 and MCF7 cells were cultured as described by
Byun et al (15). Female
BALB/c athymic nude mice (Orient Co., Seongnam, Korea) were
maintained and studied as described by Byun et al (15).
Antibodies
Rabbit anti-eEF1A1 and anti-eEF1B2 antibodies were
purchased from Abcam (Cambridge, MA, USA). Rabbit anti-phospho-pRb
and mouse anti-p53 antibodies were purchased from Cell Signaling
Technology (Danvers, MA, USA) and Novocastra Inc. (Newcastle, UK),
respectively. Rabbit anti-p21, goat anti-cathepsin D, rabbit
anti-DcR2, mouse anti-DEC1, goat anti-β-actin, and horseradish
peroxidase-conjugated secondary antibodies were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA).
Irradiation
Cells were irradiated to γ-ray with 137Cs
γ-ray source (Atomic Energy of Canada Ltd., Mississauga, Canada) at
a dose rate of 3.2 Gy/min. Irradiation of cells was based on three
single irradiation doses (2, 6 and 12 Gy), while fractionated
irradiation of cells was conducted daily to 2 Gy three times with 4
h time intervals for single day or two consecutive days (3 × 2 Gy
or 6 × 2 Gy). Local regional irradiation of xenografted tumor was
performed under anesthesia using a 60Co source
irradiator (Theratron 780; Atomic Energy of Canada Ltd.) operating
at 1.3 Gy/min. When xenografted tumor volume reached 200–250
mm3, xenografted regions of mice were exposed to SR or
FR with the same procedures. Mice were sacrificed for further
experiments at 1 and 7 days after the last IR exposure.
Relative cell number, clonogenicity,
SA-β-Gal staining and western blotting
Analyses of relative cell number, clonogenicity,
SA-β-Gal activity and western blotting were performed as described
by Byun et al (15).
Xenograft mouse model
H460 lung cancer cells (5×106 cells) were
injected subcutaneously to the right side proximal hind legs of 6
week-old nude mice. Xenograft tumor growth was monitored every day
by measuring tumor width and length using a digital caliper. Tumor
volume was calculated by the following equation: Volume = 0.5 ×
Width2 × Length.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling assay
Apoptosis in tumor tissue was assayed using terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
kit (ApopTag® Peroxidase In Situ Apoptosis detection
kit; Millipore, Temecula, CA, USA) following the manufacturer’s
protocol. Sections were counterstained with hematoxylin. Negative
control sections were incubated with distilled water in the absence
of TdT.
Immunohistochemistry
Tumor tissues from xenografted mice were fixed and
embedded in paraffin. Immunohistochemistry was performed with the
Vectastain Elite ABC kit (Vector Laboratories Inc., Burlingame, CA,
USA) following the manufacturer’s protocol. Immunoreactive sites
were visualized by 3,3′-DAB. Subsequently, the slices were
counterstained by hematoxylin.
Results
Effect of SR and FR on premature
senescence in carcinoma cell lines
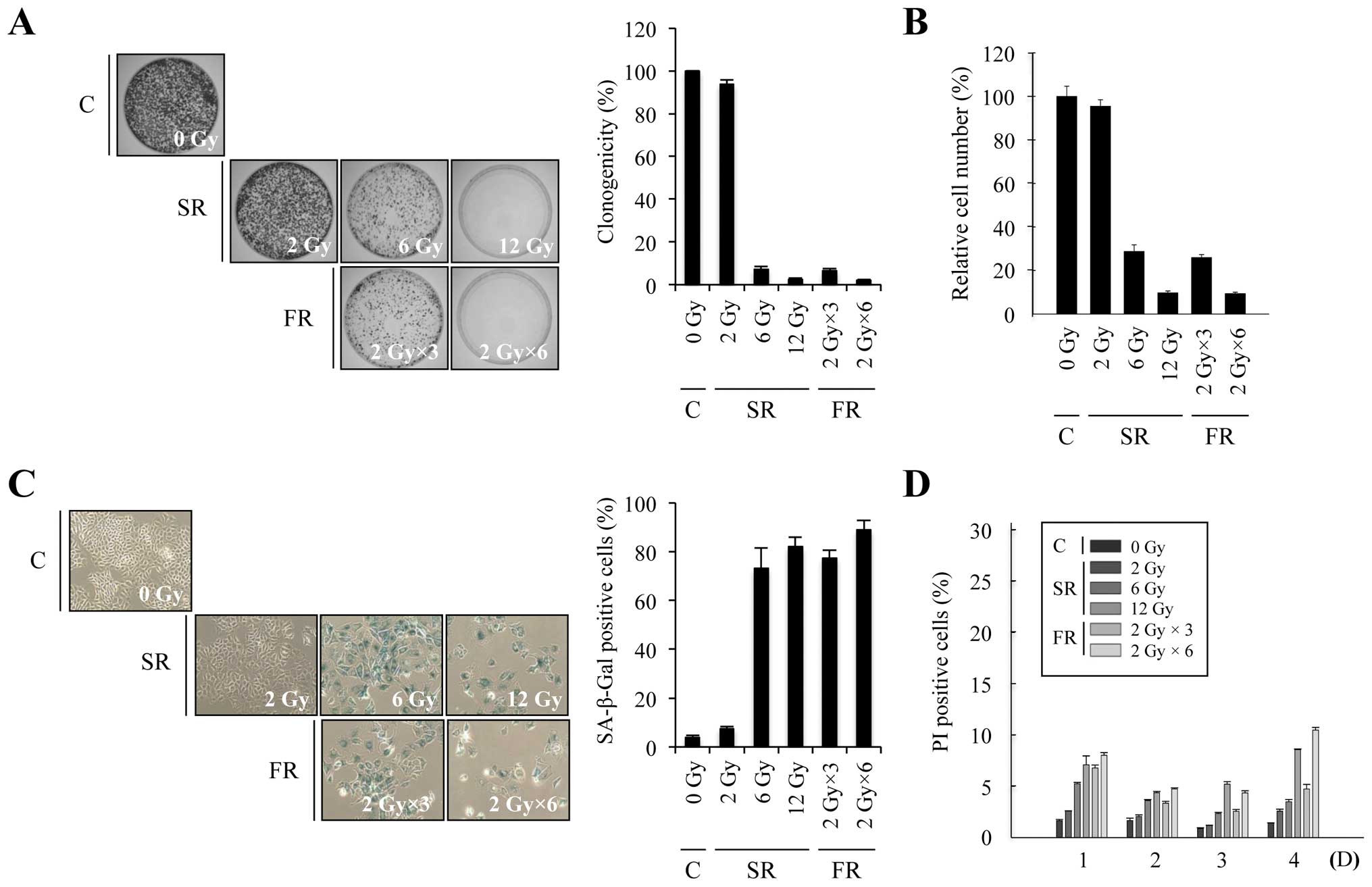
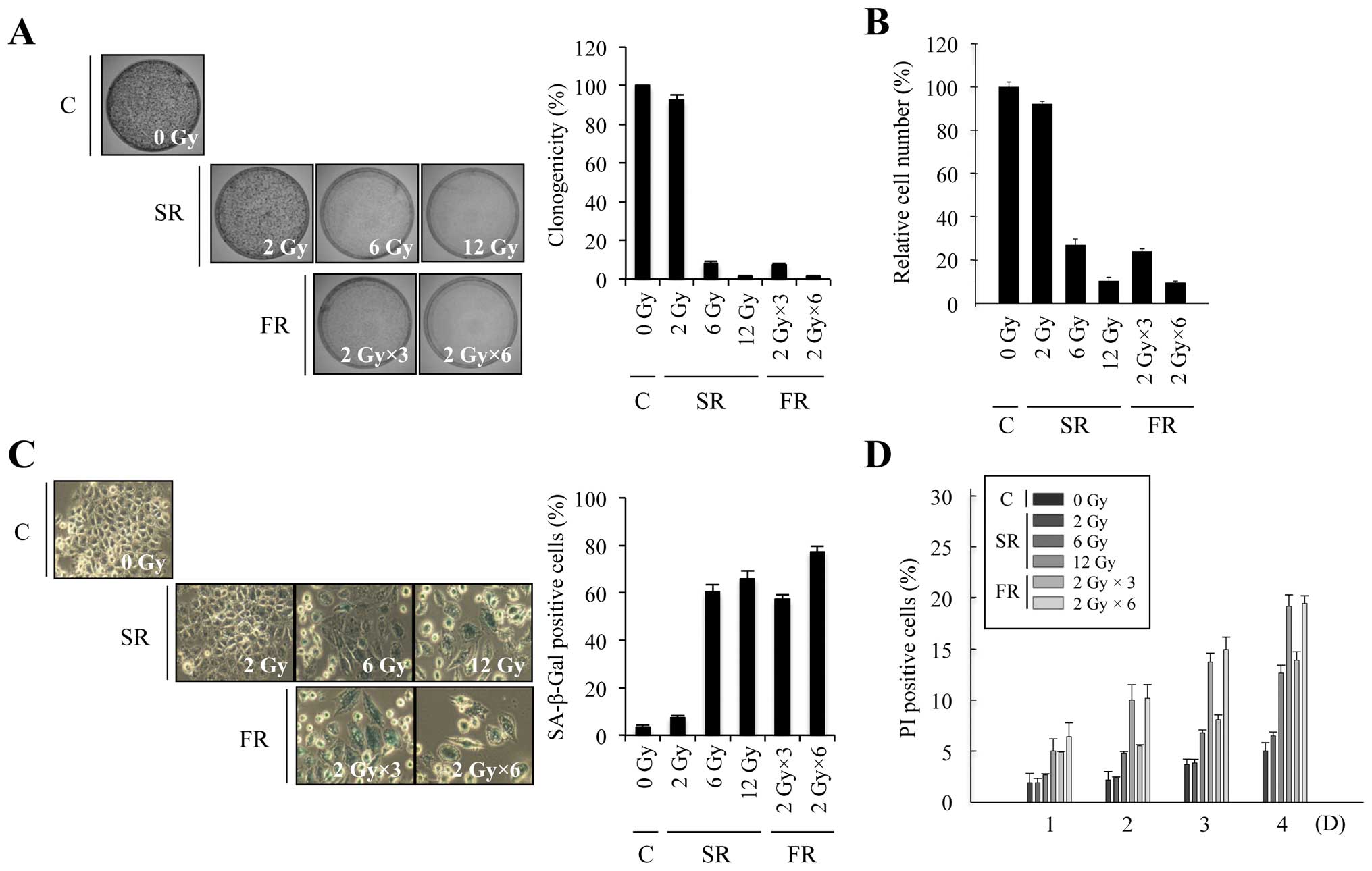
To investigate whether equivalent total doses of SR
or FR induced premature senescence, we exposed MCF7 and H460
carcinoma cells to a range of doses of SR (2, 6 or 12 Gy) or FR (3
× 2 Gy or 6 × 2 Gy). In both cell lines, 6 or 12 Gy SR
dose-dependently decreased clonogenicity and relative cell number,
increased SA-β-Gal activity and induced large and flat cell
morphology (Figs. 1A–C and 2A–C). The percentage of cells that
underwent cell death after SR exposure was up to ~20% in time- and
dose-dependent manners in both cell lines (Figs. 1D and 2D). These results indicate that either 6
or 12 Gy SR effectively induced cellular senescence rather than
cell death. Since RT is conventionally delivered in fractions of 2
Gy, we compared the effects of 6 or 12 Gy FR on MCF7 and H460 cells
(Figs. 1 and 2). The same total dose of FR resulted in
similar effects as SR on clonogenicity, relative cell number and
cell death in both cell lines. Thus, our data demonstrate that
premature senescence is the major response of carcinoma cells to 6
or 12 Gy of IR, regardless of SR or FR.
Evaluation of senescence markers in SR-
or FR-exposed carcinoma cell lines
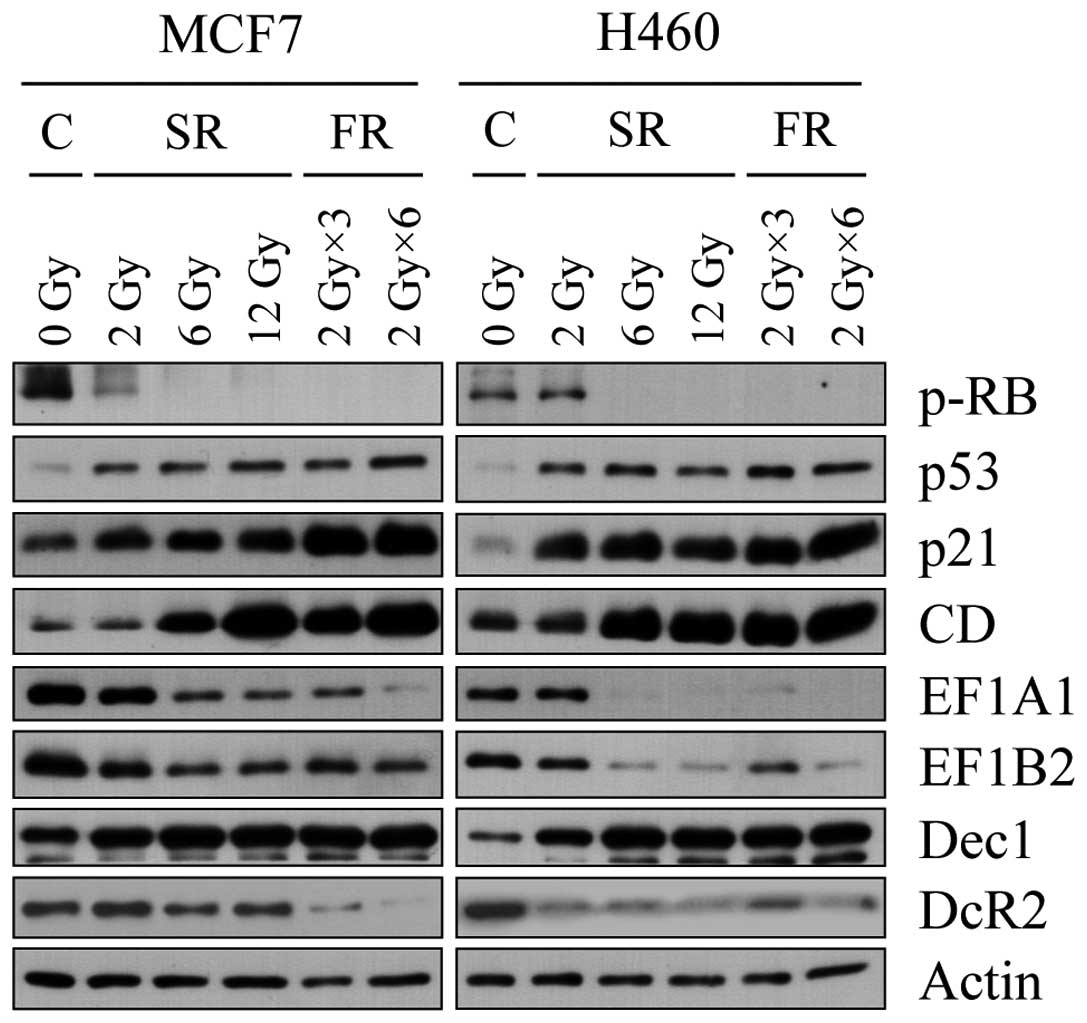
We reported CD, eEF1A1 and eEF1B2 and others have
reported DcR2 and Dec1 as senescence markers (15,22);
however, the reliability of these markers requires further
validation. We examined expression changes of these markers during
premature senescence in SR- or FR-exposed cells (Fig. 3). In either cell line, expression of
CD was increased and eEF1A1 and eEF1B2 were decreased by 6 or 12 Gy
of SR or FR. Notably, p53 and p21 accumulated not only in 6 or 12
Gy of SR- or FR-exposed cells, but also in 2 Gy of SR-exposed cells
which did not undergo either apoptosis or cellular senescence. The
changes in expression of CD, eEF1A1 and eEF1B2 closely correlated
with hypophosphorylation of pRb, a reliable indication of cell
cycle arrest. In contrast to the report of increased Dec1 and DcR2
expression in oncogene-induced prematurely senescent tumor cells
(13), after IR exposure in our
experiments, DcR2 expression decreased in both cell lines and Dec1
was evidently increased only in H460 cells. These data indicate
that IR exposure induced senescence effectively in either SR (6 or
12 Gy)- or FR (3 × 2 Gy or 6 × 2 Gy)-exposed H460 and MCF7 cells,
and we further validated CD, eEF1A1 and eEF1B2 as valuable markers
of cellular senescence.
Effect of SR and FR on apoptosis, cell
proliferation and premature senescence in tumor tissue of xenograft
mice
We next examined whether IR-induced premature
senescence was involved in inhibition of tumor growth in
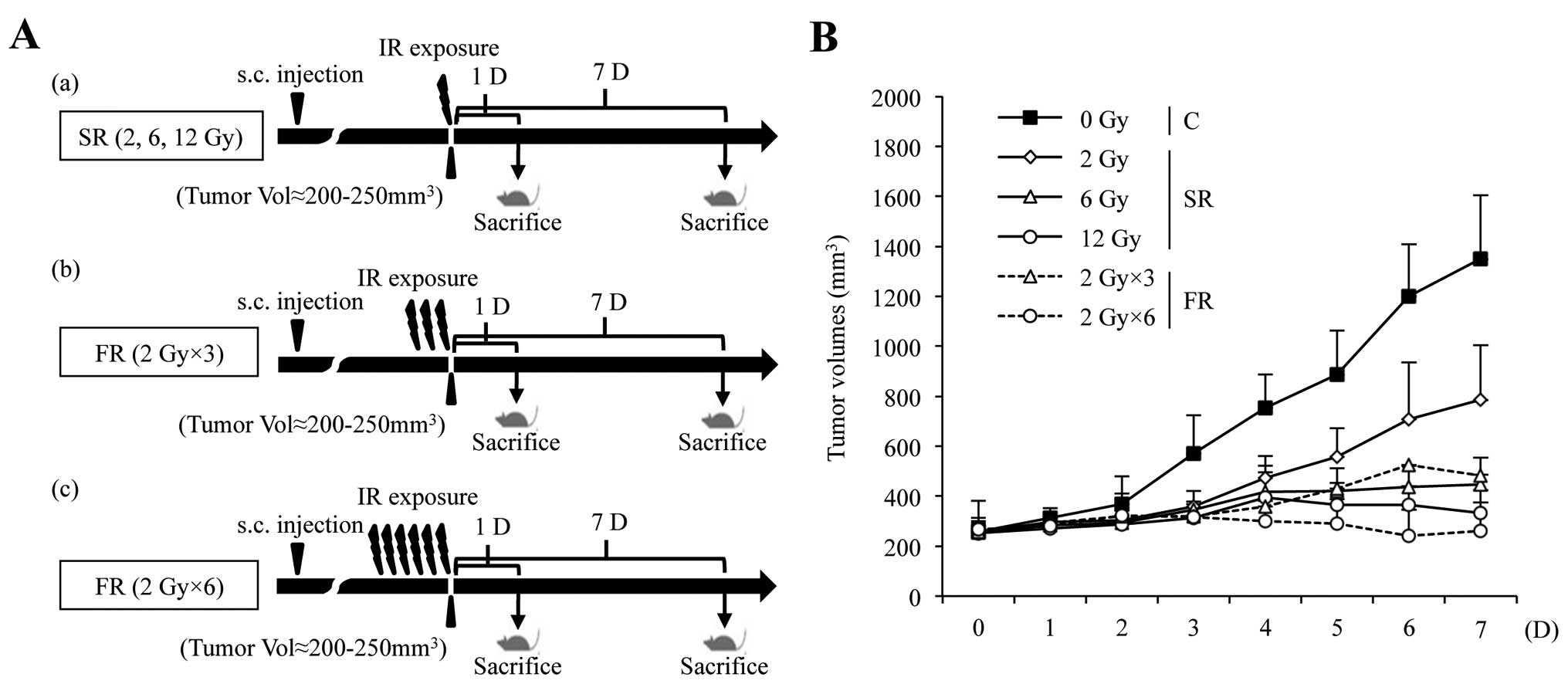
vivo. We developed a mouse xenograft model by subcutaneously
transplanting H460 cells in athymic nude mice and exposed mice to
either SR or FR (Fig. 4A). The
xenograft tumor volume was markedly reduced in a dose-dependent
manner following FR and SR exposure (Fig. 4B).
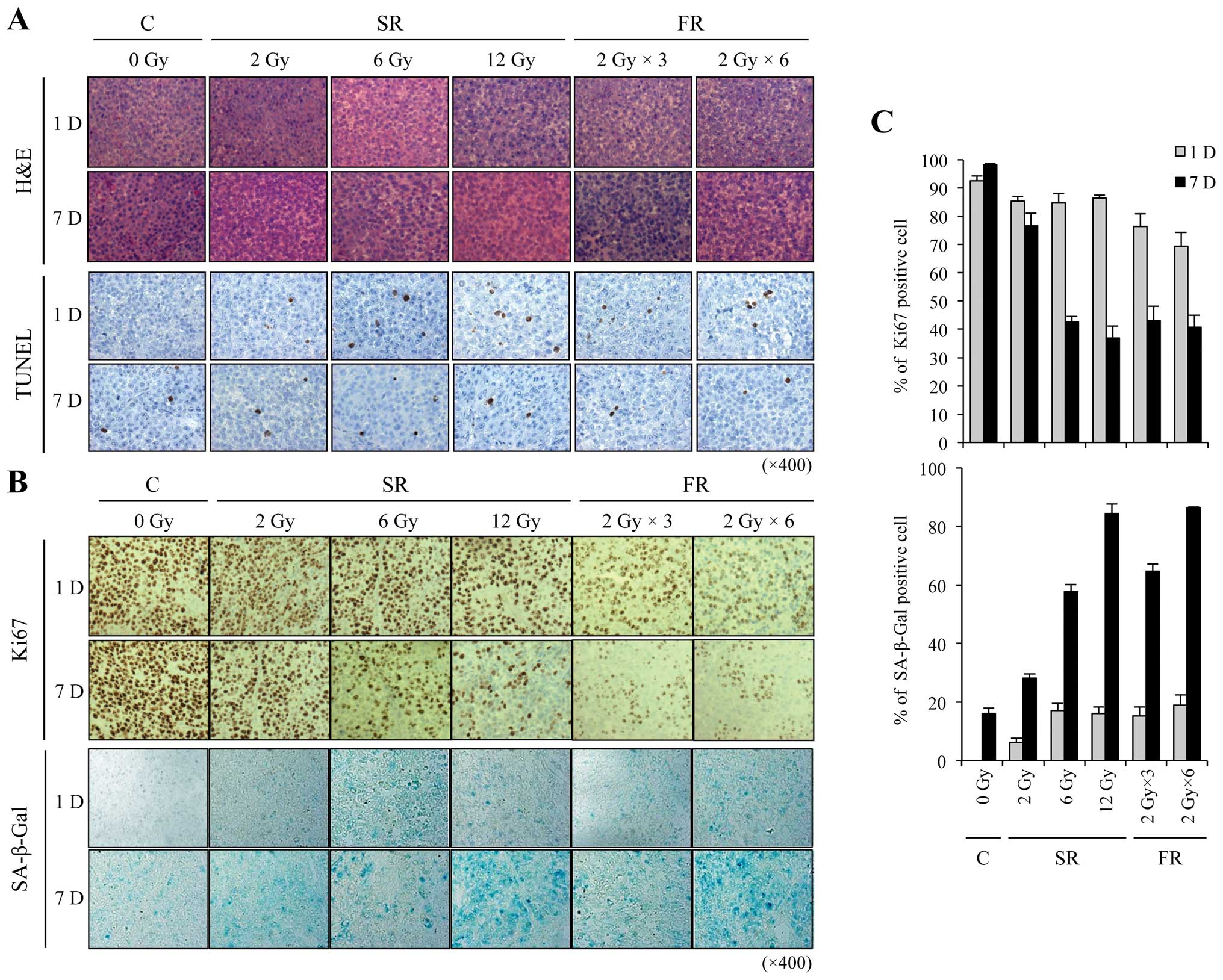
Xenograft tumors were processed for histology and
stained with hematoxylin and eosin (H&E) (Fig. 5A). To assess apoptosis, we performed
TUNEL assays and found no marked change in IR-induced apoptosis at
any IR dose or time examined (Fig.
5A). We next assessed cell proliferation and premature
senescence using Ki-67 immunostaining and SA-β-Gal staining,
respectively. Cellular proliferation was obviously decreased by
either 6 or 12 Gy SR or FR exposure (Fig. 5B). SA-β-Gal staining was strongly
apparent 7 days after exposure to 6 or 12 Gy of either SR or FR
(Fig. 5B). The Ki-67 and SA-β-Gal
labeling indices showed that the effects at 7 days were
dose-dependent (Fig. 5C). SR and FR
were equally effective at inducing SA-β-Gal positivity. These
results indicate that IR-induced inhibition of cell proliferation
may be related to the induction of premature senescence, and that
FR was as effective as SR at inducing premature senescence.
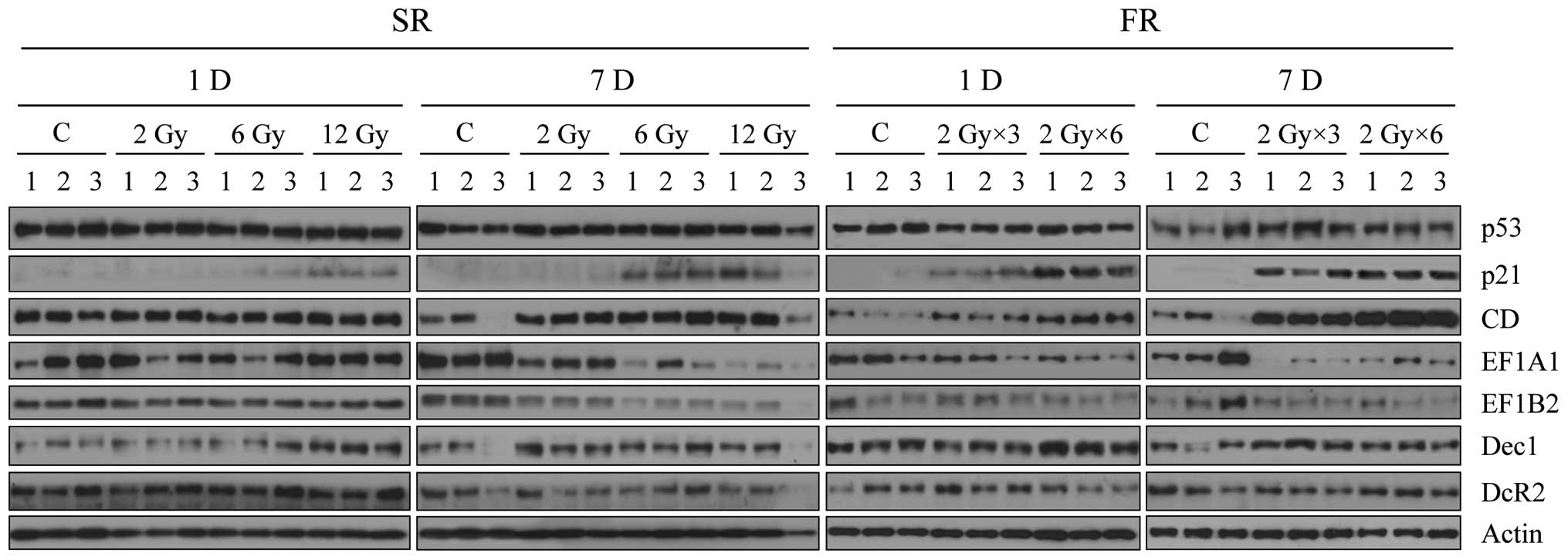
Finally, we analyzed expression of the senescence
bio-markers in the irradiated xenograft tumors (Fig. 6). Expression of p53 was not altered
by any dose of IR, in contrast to the in vitro result,
whereas p21 and CD expression increased after 6 or 12 Gy IR
exposure, particularly 7 days following FR. Expression of eEF1A1
and eEF1B2 were both decreased 7 days after exposure to 6 or 12 Gy
of SR or FR. The effects of FR on expression of p21, CD, eEF1A1 and
eEF1B2 were more pronounced than the effects of SR (Fig. 6). In contrast, SR or FR did not
alter expression of Dec1 and DcR2, indicating that these were not
markers for premature senescence in this context. Changes in
expression of CD, eEF1A1 and eEF1B2 in vivo (Fig. 6) were consistent with the in
vitro findings (Fig. 3) and are
further support that CD, eEF1A and eEF1B2 are promising senescence
biomarkers for clinical application.
Discussion
RT is based on eliminating the disease by depriving
the proliferative potential of the tumor cells. Frequently, the
desired effect of tumor treatment is achieved through the promotion
of apoptosis, particularly in solid tumors (23–25).
Although failure to undergo apoptosis in response to radiation may
be a mechanism of radiation resistance, it is gradually being
recognized that the loss of reproductive capacity in solid tumors
can occur through alternative pathways including mitotic
catastrophe, autophagic cell death and cellular senescence
(26,27). Haugstetter et al (28) showed that senescence index could
predict the treatment outcome in 30 metastatic colorectal cancer
patients. Our data showed that premature senescence rather than
apoptosis is the predominant response in vitro and in
vivo to either 6 or 12 Gy of SR or FR (Figs 1, 2
and 5). Apoptotic cells were rare
in vivo 1 or 7 days after SR or FR exposure (Fig. 5A). As the in vivo system may
be more clinically relevant than in vitro culture, this
suggests that apoptosis may not be the principal mechanism of
IR-mediated tumor regression in the clinical situation. Chang et
al (29) demonstrated that
induction of senescence-like phenotype seems to be common response
to IR under the conditions of minimal cytotoxicity, providing the
evidence that senescence may be an important determinant of cancer
treatment outcome. Of note, IR exposure is effective at reducing
tumor cell proliferation through multiple mechanisms, depending on
the complexity of the cellular context (25). We and others found that loss of PTEN
activity in either glioma cells or a mouse model of prostate cancer
results in widespread premature senescence rather than apoptosis
(12,25). Ruth and Roninson (30) reported that expression of
P-glycoprotein (Pgt) inhibits IR-induced apoptosis. However, this
effect of Pgt had no effect on radiation resistance since
inhibition of apoptosis is associated with a concurrent increase in
mitotic catastrophe and senescence in SR-damaged cells. Adjunctive
therapy of tyrosine kinase inhibitor or poly(ADP-ribose) polymerase
inhibitor with SR potentiates terminal growth arrest associated
with senescence in vitro and in vivo, suggesting that
tumor cell senescence is a mechanism for tumor targeting therapy in
combination with IR (31,32). DeMasters et al (33) demonstrated that vitamin D3 analog
delayed FR (5 × 2 Gy)-induced senescence arrest in breast cancer
cells. Based on these results, not only the genetic background of
the cancer patients but also the drug selection for combinational
treatment should be carefully considered when predicting the
primary mechanism underlying tumor regression after RT. Further
studies are needed to elucidate how widespread this phenomenon may
be and whether premature senescence practically contributes to the
treatment outcome in animal models and cancer patients.
Molecular changes associated with cell cycle arrest,
such as changes in phospho-Rb, p53 and p21 in IR-exposed cells
occurred in similar patterns in SR- and FR-treated cells. However,
since these molecules change under the conditions of not only
permanent cell cycle arrest but also transient cell cycle arrest,
they are indicators of cell cycle arrest (15). Expression of CD, eEF1A1 and eEF1B2
was evidently altered in cells exposed to the same total doses of
SR or FR (6 or 12 Gy; Figs. 3 and
6) and were not altered in
transient cell cycle arrest (15).
CD, eEF1A1 and eEF1B2 expression correlated more closely with the
SA-β-Gal labeling index than the accumulation of p53 and p21 did
(Figs. 3 and 6), providing further support of our
previous report that these molecules are promising markers for the
detection of senescence (15).
Expression of CD, eEF1A1 and eEF1B2 in vivo was more
sensitive to FR than SR (Fig. 6),
indicating that conventional FR could be the preferred regimen for
inducing premature senescence. However, since we did not detect any
change in DcR2 in IR-induced senescent cells, neither in
vitro nor in vivo, DcR2 expression may depend upon the
specific cellular context and may not be a general marker for
cellular senescence. Similarly, although Dec1 expression increased
following SR or FR exposure in vitro, Dec1 expression did
not correlate with cellular senescence in vivo (Figs. 3 and 6). Although several recently identified
biomarkers, such as HP1α, PML, p16, p15 and 12C-FDG may
indicate cellular senescence, these need to be further evaluated
before their clinical application in vivo (13,24,27,28,34).
SA-β-Gal activity assay is the gold standard for senescence
detection but is not suitable for clinical use since it is an
enzymatic assay that requires fresh, frozen tissue sections
(24). Our results strongly suggest
that analysis of CD, eEF1A1 and eEF1B2 expression in combination
with p21, pRB and proliferation markers such as Ki-67 or
poly(ADP-ribose) polymerase, is a promising method to assess
senescence in vitro and in vivo.
In summary, we have shown that premature senescence
was the predominant pathway for IR-mediated tumor regression in
vitro and in vivo, in response to 6 and 12 Gy of either
SR or FR. Reliable assays for senescence may provide prognostic
value on IR-mediated tumor regression in clinically relevant
situations, and might be a more accurate prognostic factor than the
apoptotic index in the treatment of solid tumors. Moreover, our
data further support that CD, eF1A1 and eEF1B2 are promising
biomarkers for the detection of IR-induced premature senescence
in vivo. Our data may contribute to the clinical application
of premature senescence for the prediction of RT outcome.
Acknowledgements
The present study was supported by the Nuclear
Research and Development Program of the National Research
Foundation grant funded by the Korean government (MSIP)
(2012-M2B2B1-2012055637).
References
|
1
|
Nomura T, Kinuta M, Hongyo T, Nakajima H
and Hatanaka T: Programmed cell death in whole body and organ
systems by low dose radiation. J Radiat Res. 33:109–123. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hendry JH, Potten CS and Merritt A:
Apoptosis induced by high- and low-LET radiations. Radiat Environ
Biophys. 34:59–62. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Y, Liu L, Pazhanisamy SK, Li H, Meng
A and Zhou D: Total body irradiation causes residual bone marrow
injury by induction of persistent oxidative stress in murine
hematopoietic stem cells. Free Radic Biol Med. 48:348–356. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bruskov VI, Karp OE, Garmash SA, Shtarkman
IN, Chernikov AV and Gudkov SV: Prolongation of oxidative stress by
long-lived reactive protein species induced by X-ray radiation and
their genotoxic action. Free Radic Res. 46:1280–1290. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gewirtz DA, Holt SE and Elmore LW:
Accelerated senescence: an emerging role in tumor cell response to
chemotherapy and radiation. Biochem Pharmacol. 76:947–957. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Collado M and Serrano M: Senescence in
tumours: evidence from mice and humans. Nat Rev Cancer. 10:51–57.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nardella C, Clohessy JG, Alimonti A and
Pandolfi PP: Pro-senescence therapy for cancer treatment. Nat Rev
Cancer. 11:503–511. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ben-Porath I and Weinberg RA: The signals
and pathways activating cellular senescence. Int J Biochem Cell
Biol. 37:961–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohtani N, Mann DJ and Hara E: Cellular
senescence: its role in tumor suppression and aging. Cancer Sci.
100:792–797. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dimri GP, Lee X, Basile G, Acosta M, Scott
G, Roskelley C, et al: A biomarker that identifies senescent human
cells in culture and in aging skin in vivo. Proc Natl Acad
Sci USA. 92:9363–9367. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Debacq-Chainiaux F, Erusalimsky JD,
Campisi J and Toussaint O: Protocols to detect
senescence-associated beta-galactosidase (SA-βgal) activity, a
biomarker of senescent cells in culture and in vivo. Nat
Protoc. 4:1798–1806. 2009.PubMed/NCBI
|
|
12
|
Chen Z, Trotman LC, Shaffer D, Lin HK,
Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W,
Cordon-Cardo C and Pandolfi PP: Crucial role of p53-dependent
cellular senescence in suppression of Pten-deficient tumorigenesis.
Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Collado M, Gil J, Efeyan A, Guerra C,
Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM,
Barbacid M, Beach D and Serrano M: Tumour biology: senescence in
premalignant tumours. Nature. 436:6422005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dankort D, Filenova E, Collado M, Serrano
M, Jones K and McMahon M: A new mouse model to explore the
initiation, progression, and therapy of BRAFV600E-induced lung
tumors. Genes Dev. 21:379–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Byun HO, Han NK, Lee HJ, Kim KB, Ko YG,
Yoon G, Lee YS, Hong SI and Lee JS: Cathepsin D and eukaryotic
translation elongation factor 1 as promising markers of cellular
senescence. Cancer Res. 69:4638–4647. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Spallarossa P, Altieri P, Aloi C,
Garibaldi S, Barisione C, Ghigliotti G, Fugazza G, Barsotti A and
Brunelli C: Doxorubicin induces senescence or apoptosis in rat
neonatal cardiomyocytes by regulating the expression levels of the
telomere binding factors 1 and 2. Am J Physiol Heart Circ Physiol.
297:H2169–H2181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nübel T, Damrot J, Roos WP, Kaina B and
Fritz G: Lovastatin protects human endothelial cells from killing
by ionizing radiation without impairing induction and repair of DNA
double-strand breaks. Clin Cancer Res. 12:933–939. 2006.PubMed/NCBI
|
|
18
|
Rübe CE, Fricke A, Wendorf J, Stützel A,
Kühne M, Ong MF, Lipp P and Rübe C: Accumulation of DNA
double-strand breaks in normal tissues after fractionated
irradiation. Int J Radiat Oncol Biol Phys. 76:1206–1213.
2010.PubMed/NCBI
|
|
19
|
Kalderon N, Xu S, Koutcher JA and Fuks Z:
Fractionated radiation facilitates repair and functional motor
recovery after spinal cord transection in rat. Brain Res.
904:199–207. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dearnaley DP, Khoo VS, Norman AR, Meyer L,
Nahum A, Tait D, Yarnold J and Horwich A: Comparison of radiation
side-effects of conformal and conventional radiotherapy in prostate
cancer: a randomised trial. Lancet. 353:267–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Munshi A and Budrukkar A: Hypofractionated
radiation therapy in breast cancer: a revolutionary breakthrough or
a long way to go? J Clin Oncol. 25:458–459. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Collado M and Serrano M: The power and the
promise of oncogene-induced senescence markers. Nat Rev Cancer.
6:472–476. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suzuki M and Boothman DA: Stress-induced
premature senescence (SIPS) - influence of SIPS on radiotherapy. J
Radiat Res. 49:105–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JJ, Lee JH, Ko YG, Hong SI and Lee JS:
Prevention of premature senescence requires JNK regulation of Bcl-2
and reactive oxygen species. Oncogene. 29:561–575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN,
Lee BL and Lee JS: PTEN status switches cell fate between premature
senescence and apoptosis in glioma exposed to ionizing radiation.
Cell Death Differ. 18:666–677. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen WS, Yu YC, Lee YJ, Chen JH, Hsu HY
and Chiu SJ: Depletion of securin induces senescence after
irradiation and enhances radiosensitivity in human cancer cells
regardless of functional p53 expression. Int J Radiat Oncol Biol
Phys. 77:566–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jackson JG, Post SM and Lozano G:
Regulation of tissue- and stimulus-specific cell fate decisions by
p53 in vivo. J Pathol. 223:127–136. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Haugstetter AM, Loddenkemper C, Lenze D,
Gröne J, Standfuss C, Petersen I, Dörken B and Schmitt CA: Cellular
senescence predicts treatment outcome in metastasised colorectal
cancer. Br J Cancer. 103:505–509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang BD, Broude EV, Dokmanovic M, Zhu H,
Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K and Roninson IB: A
senescence-like phenotype distinguishes tumor cells that undergo
terminal proliferation arrest after exposure to anticancer agents.
Cancer Res. 59:3761–3767. 1999.
|
|
30
|
Ruth AC and Roninson IB: Effects of the
multidrug transporter P-glycoprotein on cellular responses to
ionizing radiation. Cancer Res. 60:2576–2578. 2000.PubMed/NCBI
|
|
31
|
Podtcheko A, Ohtsuru A, Namba H, Saenko V,
Starenki D, Palona I, Sedliarou I, Rogounovitch T and Yamashita S:
Inhibition of ABL tyrosine kinase potentiates radiation-induced
terminal growth arrest in anaplastic thyroid cancer cells. Radiat
Res. 165:35–42. 2006. View Article : Google Scholar
|
|
32
|
Efimova EV, Mauceri HJ, Golden DW, Labay
E, Bindokas VP, Darga TE, Chakraborty C, Barreto-Andrade JC,
Crawley C, Sutton HG, Kron SJ and Weichselbaum RR: Poly(ADP-ribose)
polymerase inhibitor induces accelerated senescence in irradiated
breast cancer cells and tumors. Cancer Res. 70:6277–6282. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
DeMasters GA, Gupta MS, Jones KR, Cabot M,
Wang H, Gennings C, Park M, Bratland A, Ree AH and Gewirtz DA:
Potentiation of cell killing by fractionated radiation and
suppression of proliferative recovery in MCF-7 breast tumor cells
by the Vitamin D3 analog EB 1089. J Steroid Biochem Mol Biol.
92:365–374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pearson M, Carbone R, Sebastiani C, Cioce
M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S,
Pandolfi PP and Pelicci PG: PML regulates p53 acetylation and
premature senescence induced by oncogenic Ras. Nature. 406:207–210.
2000. View Article : Google Scholar : PubMed/NCBI
|