Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide. Small cell lung cancer (SCLC) accounts for 13%
of lung cancer-related deaths and 12% of deaths from all other
types of cancer (1,2). SCLC is recognized for its
aggressiveness, displaying rapid growth throughout and early,
widespread metastasis (3). In
addition, chemotherapy does not consistently prolong the survival
of SCLC patients, especially following relapse, as demonstrated by
several previous studies (4). Aside
from the lack of efficacy of current treatments, the relatively few
molecules identified for precise targeting is a key contributing
factor to the low survival rate. Thus, understanding the molecular
underpinnings of SCLC, and revealing effective treatment options
for SCLC, are necessary to improve outcomes for SCLC patients.
Aurora kinase A (AURKA) is a centrosome-associated
serine/threonine kinase. AURKA participates in several crucial
mitotic events, including centrosome maturation and separation,
formation of the bipolar spindle, chromosome alignment and
segregation, and cytokinesis (5,6).
Performing an essential role as a facilitator in cell cycle
progression, AURKA is an important oncogene. The AURKA is located
in chromosome 20q13.2. This region is frequently amplified in a
variety of cancers. Also, AURKA overexpression is identified in
gastric, breast, ovarian, colorectal and esophageal tissues
(7). Overexpression of AURKA has
been shown to paralyze the G2/M checkpoint and spindle assembly
checkpoints, enabling rogue cells with defective spindles and
damaged DNA to enter mitosis and anaphase. These defects likely
contribute to genomic instability, and ultimately, carcinogenesis
(8,9). Centrosome amplification and
aneuploidy, two additional molecular drivers of genomic instability
and tumorigenesis, have also been associated with the
overexpression of AURKA (10,11).
In the last decade, RNA interference (RNAi) has
served as a powerful tool for precisely inhibiting selective gene
expression. The application of RNAi to medicine is a promising
treatment option for various diseases, especially cancer (12,13).
The introduction of nucleic acids such as non-coding, synthetic
small interfering RNAs (siRNAs) can modulate the expression of
target genes in a sequence-dependent manner. This occurs through
two mechanisms that siRNA incorporates into RNA-induced silencing
complexes (RISCs), and thereafter actively degrades and represses
the translation of corresponding messenger RNAs (mRNAs) (14,15).
There is little knowledge regarding the function of
AURKA in SCLC cells. In the present study, we knocked down AURKA
gene in two SCLC cell lines, H446 and H1688, and the apoptosis of
these cells increased through downregulation of Bcl-2 and
upregulation of Bax, which provides new insights into the roles of
AURKA in SCLC treatment.
Materials and methods
Cell culture
The human SCLC cell lines H446 and H1688 were
maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco,
Grand Island, NY, USA) supplemented with 10% fetal bovine serum
(FBS), 100 U/ml penicillin and 0.1 mg/ml streptomycin under
normoxic conditions (95% O2 and 5% CO2) at
37°C.
Cell growth inhibition assay
The effects of AURKA shRNA infection on the
proliferation of H446 and H1688 cells were analyzed by
methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay. The SCLC
cells were seeded into a 96-well plate at a concentration of
5×103 cells/well for several different time courses. A
total of 20 μl of MTT (Sigma, St. Louis, MO, USA) stock solution (5
mg/ml) was added to each well and the cultures were incubated at
37°C for an additional 4 h. This period of time was sufficient for
formazan crystals to precipitate. These were then dissolved by
adding 150 μl of dimethyl sulfoxide (DMSO) to each well. The
optical density was read by an enzyme-linked immunosorbent assay
(490 nm).
Recombination lentivirus generation
The sequence for human AURKA specific small
interfering RNA (siRNA) sequence is 5′-GAAAGCTCCACATCAATAA-3′; this
was designed with the sequence generator accessible on Invitrogen’s
website, using the AURKA sequence as a reference (GeneBank code:
NM_003600). The non-silencing (NS) sequence (5′-TTCTCC
GAACGTGTCACGT-3′) was used as a random, scrambled control. The
short hairpin RNA (shRNA) cassette against AURKA was:
5′-CCGGCAGAAAGCTCCACATCAATAAT
TCAAGAGATTATTGATGTGGAGCTTTCTGTTTTTG-3′, with two cohesive ends for
ligation into the pGCSIL-GFP vector. Pairs of complementary
oligonucleotides of these sequences were ligated into the
pGCSIL-GFP vector. The constructed lentiviral plasmid was hereafter
denoted as pGCSIL-GFP-sh AURKA. By co-transfection of
pGCSIL-GFP-shAURKA into pHelper 1.0 and pHelper 2.0 plasmids, AURKA
and lentivirus were re-generated in 293T cells. The final
lentiviral construct was referred to as LV-AURKA siRNA or
AURKA-siRNA. We generated lentiviruses that express non-silencing,
scrambled shRNA as a control, which we refer to as scr-siRNA. The
SCLC cells were transfected with the AURKA siRNA and scramble
siRNA, respectively.
Lentivirus infection
Cells were incubated with lentivirus in a small
volume of serum-free DMEM at 37°C for 4 h. DMEM containing 10% FBS
was then added and cells were placed in the incubator for an
additional period of time, as indicated, for the remainder of the
experiment. Green fluorescent protein (GFP) showed that the
infection efficiency in SCLC cells was ~90% at a multiplicity of
infection (MOI) of 30, no viral cellular toxicity was noted at this
concentration of lentivirus. Thus, the following experiments were
performed using this concentration to achieve MOI of 30, except
where indicated otherwise.
Real-time PCR analysis
The mRNA of AURKA in SCLC cells was analyzed by
real-time PCR. RNA was initially extracted from lentivirus-infected
SCLC cells 5 days post-infection using TRIzol (Invitrogen). Reverse
transcription was performed using a Promega M-MLV cDNA synthesis
kit according to the manufacturer’s instructions. Real-time qPCR
analysis was performed using the SYBR-Green Master Mix kit and the
DNA Engine Opticon™ System (MJ Research, Waltham, MA, USA). The
expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was
used as an internal control. The forward and reverse primers of
AURKA were: 5′-GCC CTG TCT TAC TGT CAT TCG-3′ and 5′-AGG TCT CTT
GGT ATG TGT TTG C-3′, respectively. The forward and reverse primers
of GAPDH were: 5′-TGA CTT CAA CAG CGA CAC CCA-3′ and 5′-CAC CCT GTT
GCT GTA GCC AAA-3′, respectively. The relative gene expression
levels were calculated using the 2−ΔΔCT algorithm.
Colony-forming assay
A soft agar colony formation assay was performed to
assess the anchorage-independent growth ability of SCLC cells as a
characteristic of in vitro tumorigenicity. The infected SCLC
cells were counted and inoculated in 6-well plates at a density of
500 cells/well. Following incubation for 14 days, the SCLC cells
were immobilized by 4% paraformaldehyde and stained using Giemsa
dye for 20 min. Then, the cells were rinsed with distilled water
and colonies were counted and photographed under a microscope.
Flow cytometry analysis
The SCLC cells were briefly seeded and infected with
virus in stock medium. The stock medium was then removed and
replaced with a serum-free medium for 48 h incubation. After the
time period, adherent cells were harvested by trypsinization and
suspended in 0.5 ml of 70% alcohol. They were briefly subjected to
4°C conditions for 30 min. The cells were then incubated with 50
μg/ml of propidium iodide (PI; Sigma) for 1 h in the dark. A total
of 1.0×106 SCLC cells were washed twice with ice-cold
PBS and incubated for 15 min in 100 μl staining buffer that
included 5 μl APC labeled Annexin V. Fluorescence-activated cell
sorting (FACS) analysis for Annexin V staining was performed by
flow cytometer. The DNA content of stained nuclei was analyzed by a
flow cytometer using MultiCycle DNA cell cycle analyzed software to
determine the percentage of cells for each phase of the cell cycle.
Each experiment was performed in triplicate.
Western blot analysis
The SCLC cell pellets were lysed with protein
extraction solution and incubated at −20°C for 20 min. The cell
lysates were then centrifuged at 12,000 × g for 5 min and total
proteins were collected.
After protein quantization using a Coomassie
brilliant blue assay, 50 μg of protein was separated by 10%
SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF)
membrane. The membrane was blocked with 5% skim milk and proteins
were detected with primary antibodies against Aurora-A, Bcl-2, Bax,
CDK1 and GAPDH. The primary antibody was detected with horseradish
peroxidase-conjugated anti-mouse for 1 h at 25°C. The horseradish
peroxidase-conjugated secondary antibody was visualized with
enhanced chemiluminescence (ECL; Millipore, Bedford, MA, USA).
Protein band intensities were determined using the video imaging
CMIASWIN system (Bio-Rad, Hercules, CA, USA). GAPDH was once more
used as an internal positive control.
Statistical analysis
Data are represented as a means ± SD of results from
the three independent experiments with similar patterns.
Statistical significance of difference between AURKA-siRNA and
scr-siRNA groups was determined by Student’s t-test and one-way
ANOVA using GraphPad Prism 5.0 software. For all experiments,
p<0.05 was considered to indicate a statistically significant
difference.
Results
Construction of lentivirus vector
mediating RNAi targeting of AURKA (LV-AURKA siRNA) and its effects
on AURKA expression
To elucidate the role of AURKA in SCLC, we first
constructed lentivirus vectors to deliver either AURKA-specific
siRNA or non-specific scramble-siRNA. H446 and H1688 SCLC cell
lines were randomly infected with one of the vector types. To
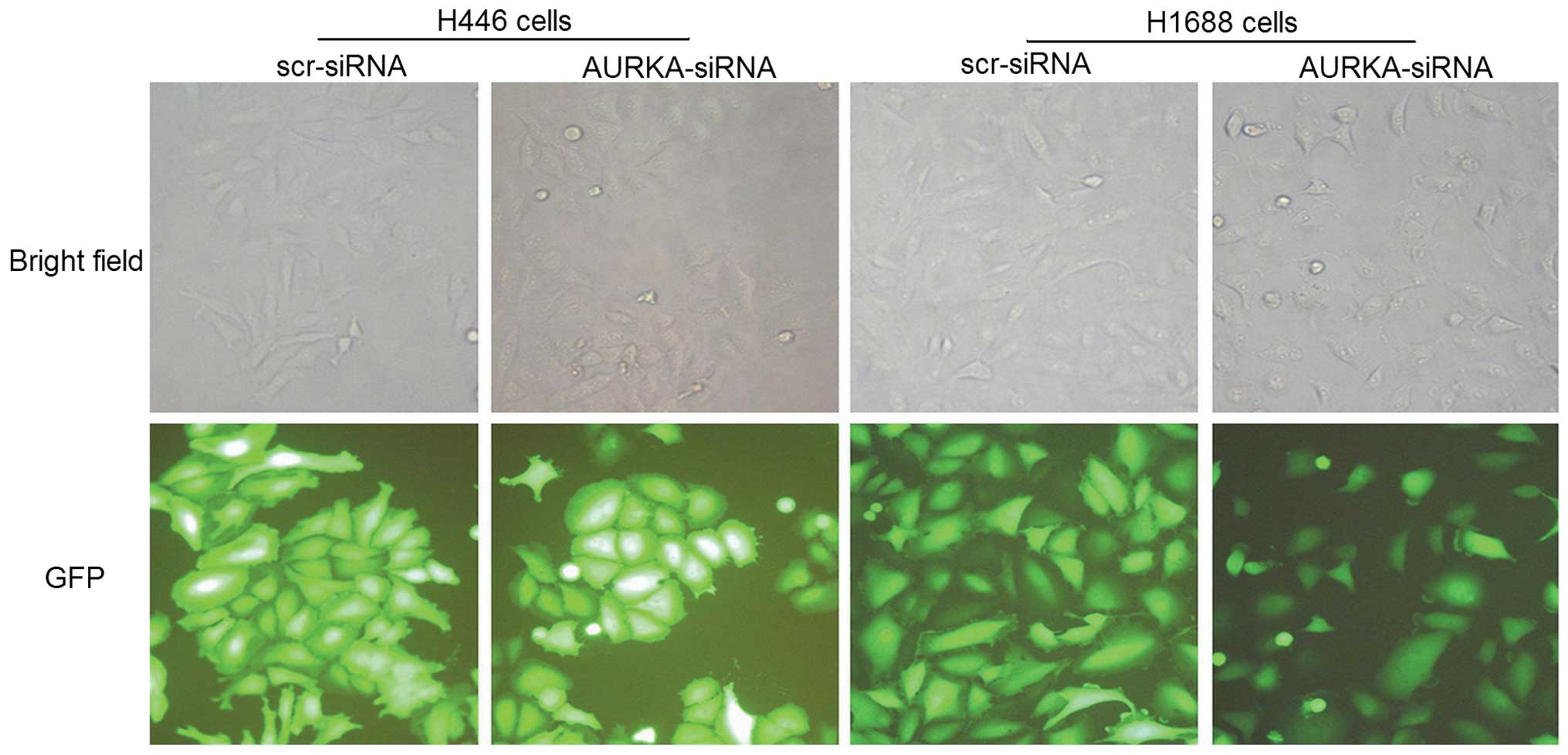
investigate the lentiviral infection efficiency, an
immunofluorescence assay was conducted, which showed that >90%
of the cells exhibited the green fluorescence indicative of
infection (Fig. 1). To confirm
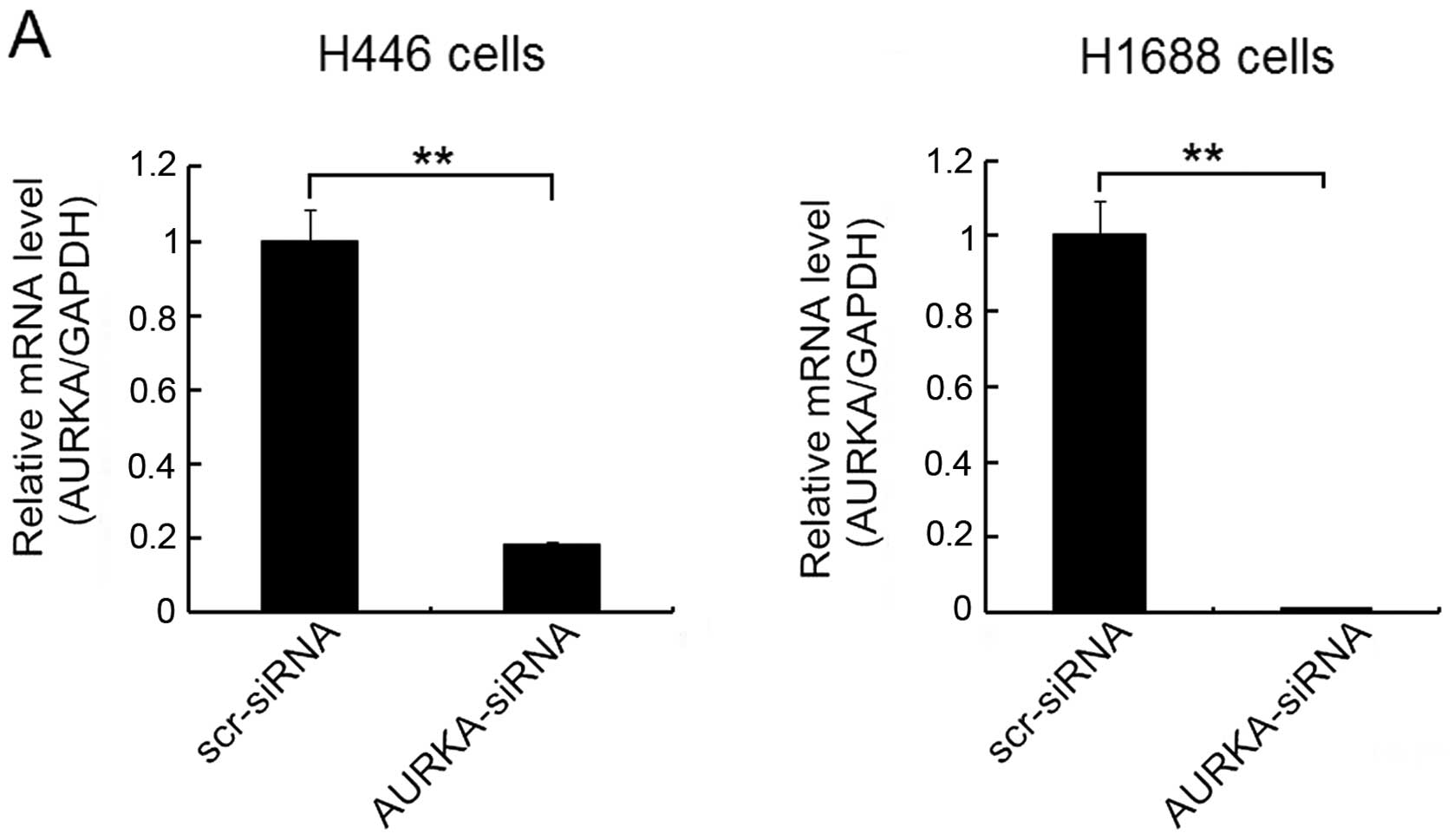
whether the AURKA-siRNA had silenced the expression of AURKA and
dampened AURKA protein levels, real-time qPCR and western blot
analyses were performed on the lentivirus-infected cells. The
results indicated that the quantity of AURKA mRNA (Fig. 2A) and protein (Fig. 2B) were significantly reduced by the
infection of AURKA-siRNA in both H446 and H1688 cells compared to
scr-siRNA treatment groups. These results suggest that the
AURKA-siRNA effectively suppressed the AURKA expression in SCLC
cells.
Effects of AURKA-siRNA on cell
proliferation, colony formation
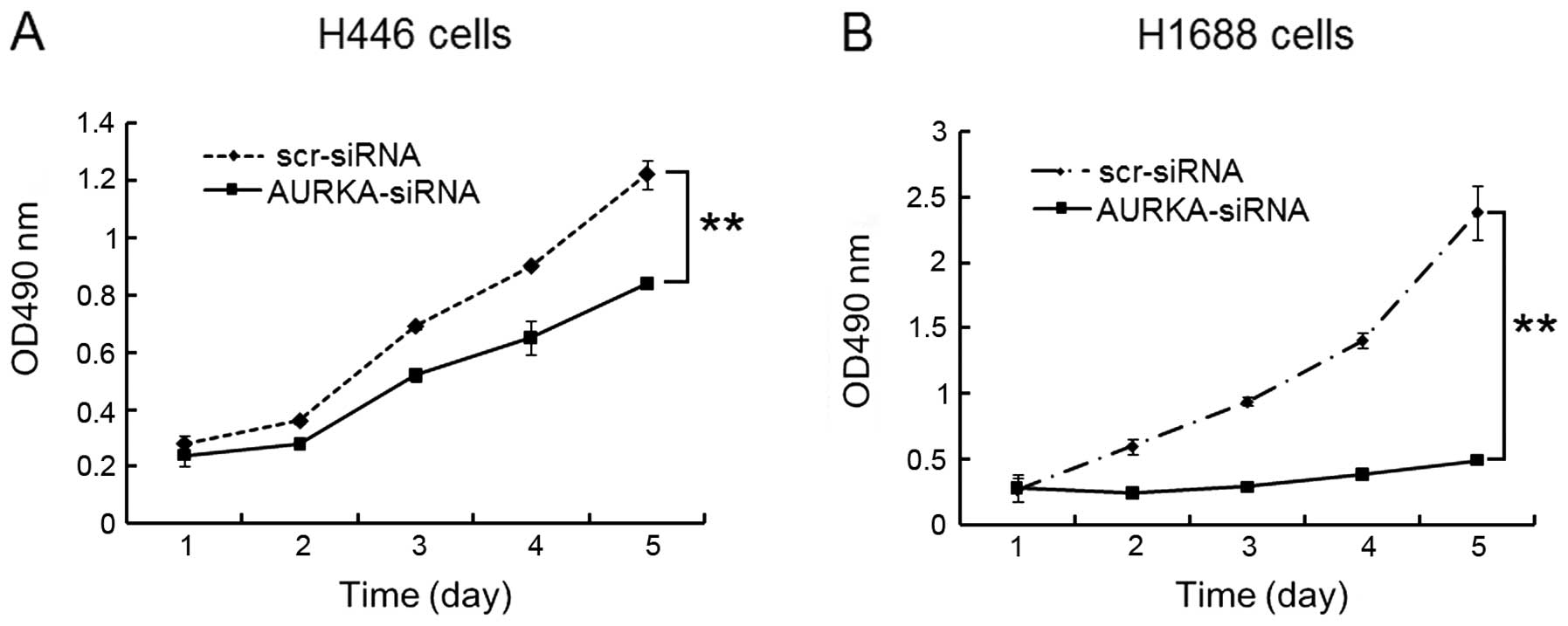
An MTT assay was performed to study the effect of
AURKA-siRNA on H446 and H1688 cell growth. As shown in Fig. 3, both H446 (Fig. 3A) and H1688 cells (Fig. 3B) showed significant (p<0.01)
reduction in cell viability. These results indicate that the
proliferation rate of cells is significantly dampened when the
AURKA gene was silenced. AURKA is a facilitator of cell division,
and the frequency of cell division mirrors the magnitude of AURKA
protein presence in the cell in a parallel fashion.
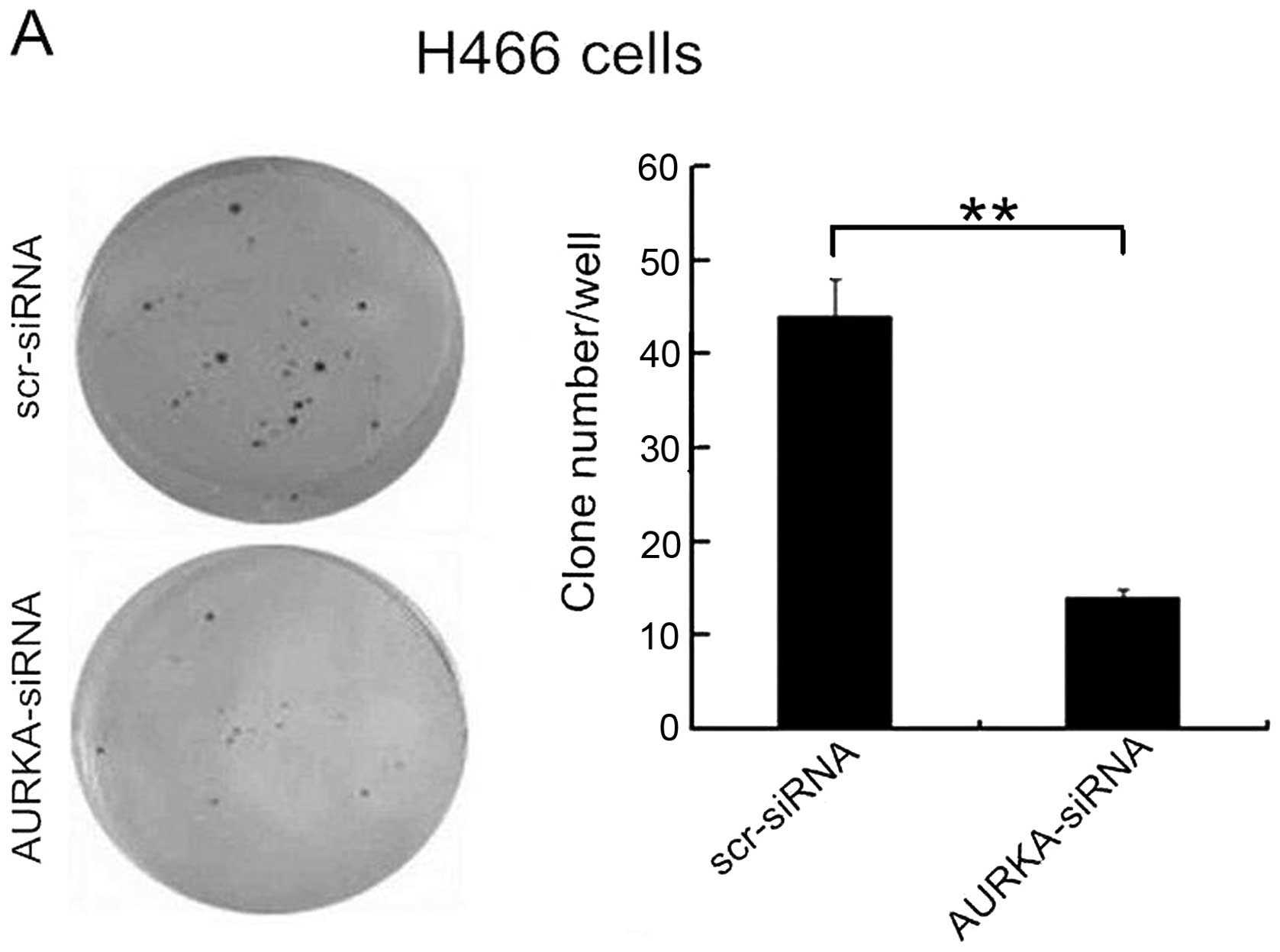
Heightened colony formation is an essential
morphologic feature of the SCLC cells. The result of a colony
formation assay showed that in H446 cells the number of colonies of
the AURKA-siRNA group (14±1) was significantly less than that of
the scr-siRNA group (44±4) (p<0.01) (Fig. 4A). In H1688 cells, the number of
colonies of the AURKA-siRNA group was 1±1, markedly lower than that
in the scr-siRNA group (41±2) (p<0.01) (Fig. 4B). These results demonstrated that a
reduction in AURKA expression resulted in a decreased ability of
the H446 and H1688 cell types to form colonies.
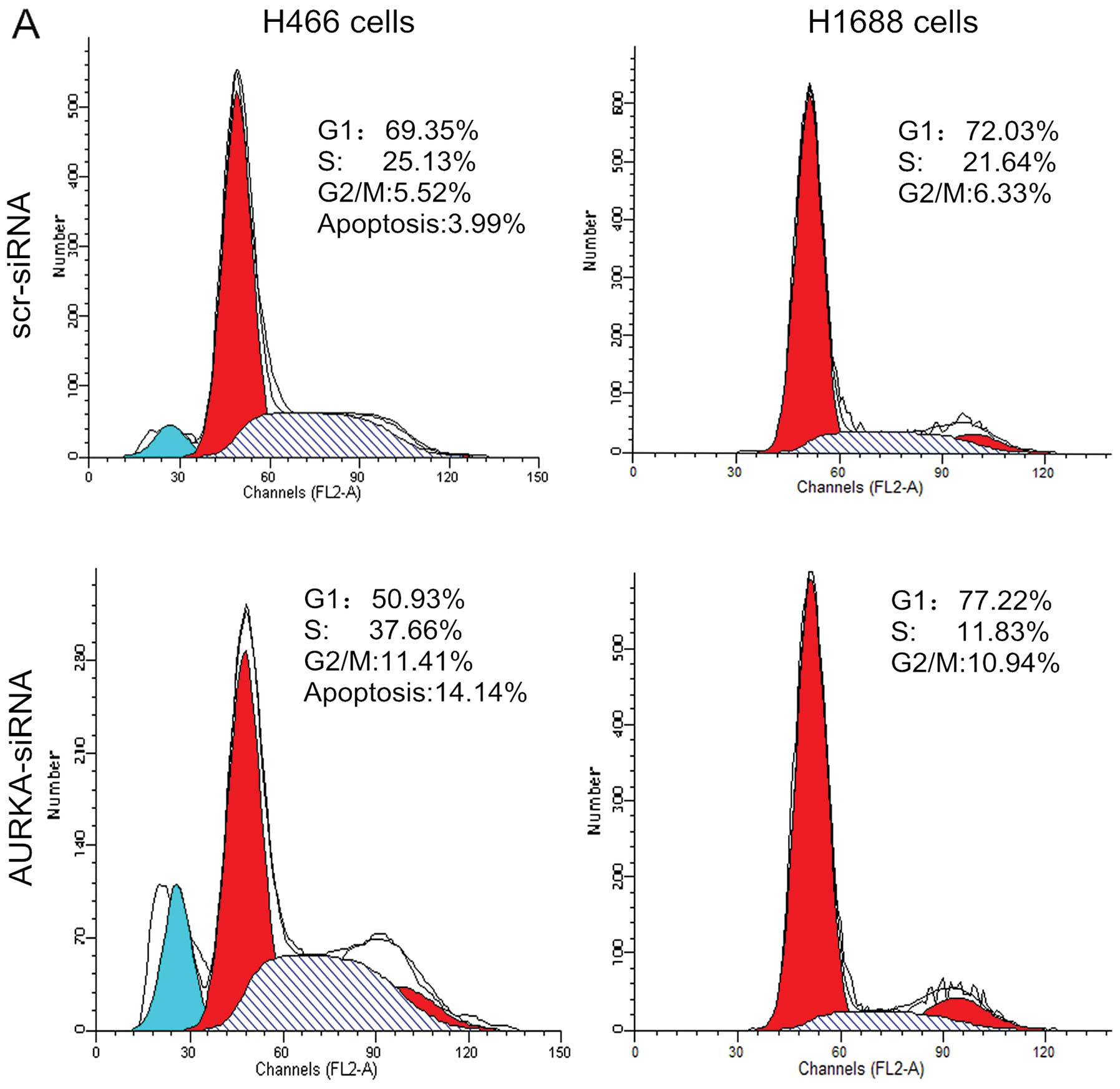
Cell cycle profile analysis after
knockdown of AURKA in SCLC cells
To determine if cell growth inhibition was the
upshot of a shift in the cell cycle, we examined cell cycle phase
distribution of SCLC cells after the silencing of AURKA. As shown
in Fig. 5, treatment with
AURKA-siRNA resulted in an increase in the percentage of H446 cells
in the G2/M phase from 3.13±0.29% to 11.62±0.19% (p<0.01).
Concomitant with this increase in the percentage of cells in the
G2/M phase was a significant decrease in the percentage of cells in
G1 phase, from 64.10±0.30% to 51.44±0.72% (p<0.01). Treatment
with AURKA-siRNA resulted in an increase in the percentage of H1688
cells in the G2/M phase, from 6.77±0.70% to 10.35±0.56%
(p<0.01). This was accompanied by a significant reduction of the
fraction of S phase cells from 20.65±0.93% to 12.93±1.09%
(p<0.01). These results together suggest that the depletion of
AURKA inhibited the proliferation of SCLC cells by prompting arrest
in G2/M phase of the cell cycle.
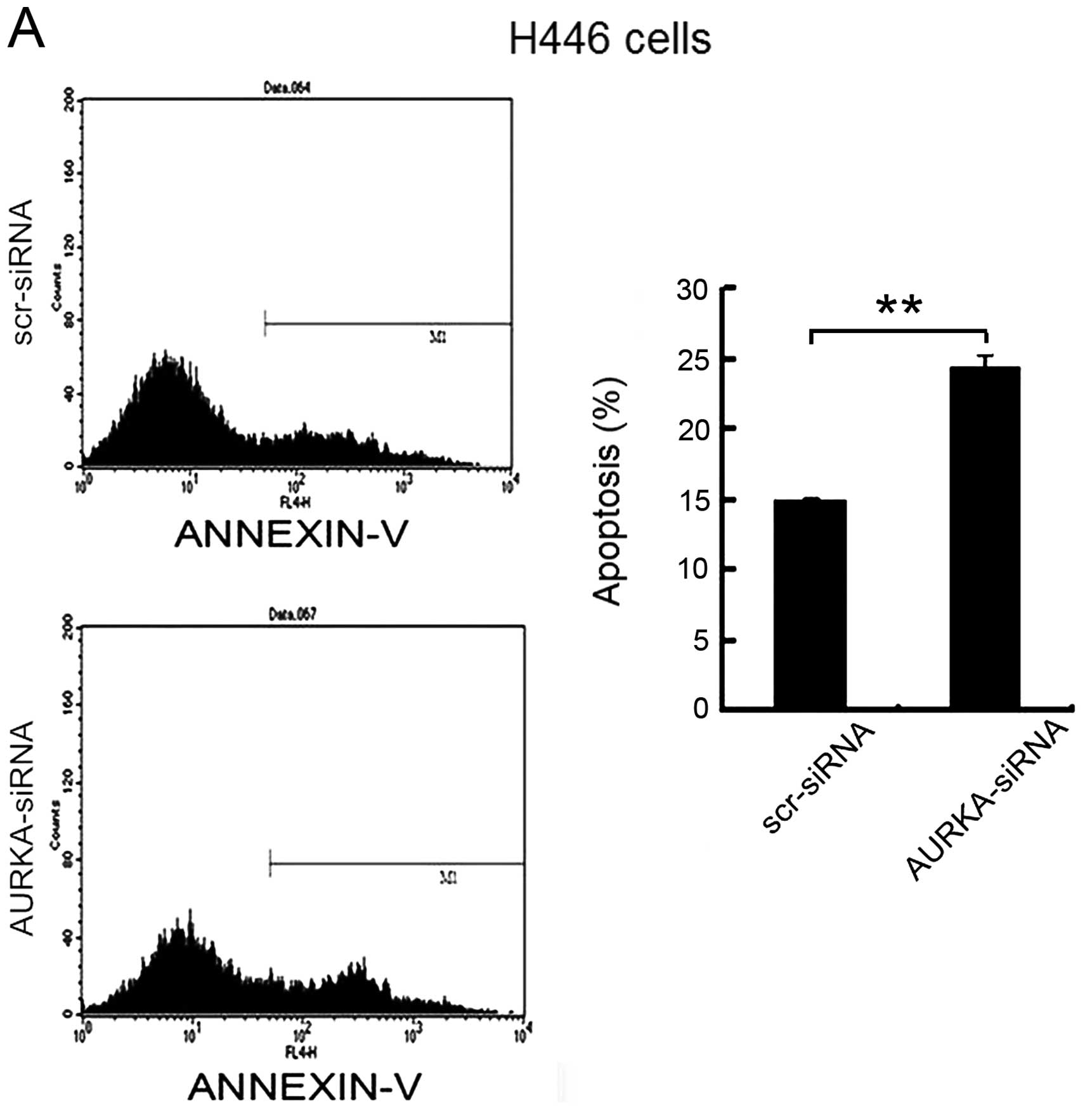
To determine whether the depletion of AURKA induces
cell apoptosis, flow cytometry was used to analyze the apoptosis of
H446 and H1688 cells after infection with AURKA-siRNA for 72 h. As
shown in Fig. 6, flow cytometry
analysis showed that the percentage of apoptotic H446 cells was
14.97±0.56% in scr-siRNA group and the percentage of apoptotic
cells increased to 24.29±1.07% in the AURKA-siRNA group (p<0.01)
(Fig. 6A). The percentage of
apoptotic H1688 cells was 4.41±0.39% in scr-siRNA group cells and
increased to 9.56±0.38% in AURKA-siRNA group (p<0.01) (Fig. 6B). These data suggest that the
depletion of AURKA specifically induced apoptosis of the SCLC
cells.
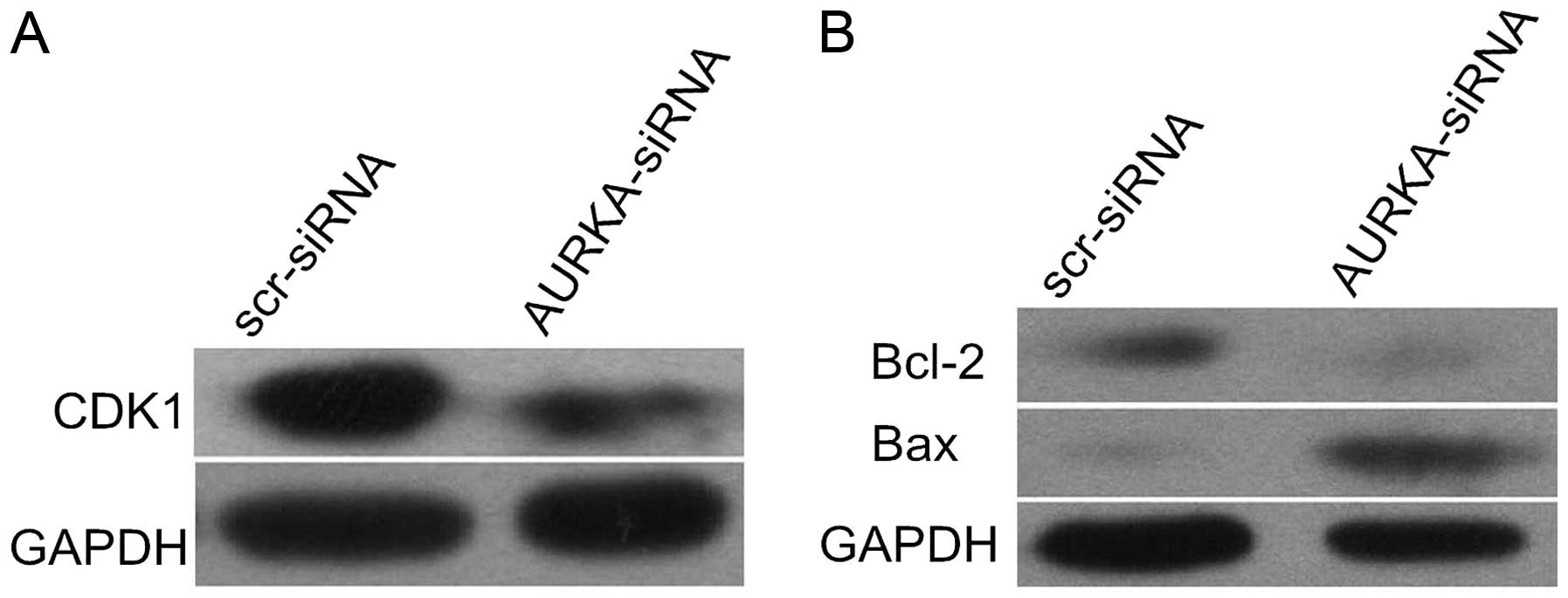
Apoptosis of H446 cells following AURKA
inhibition are associated with downregulated Bcl-2 and upregulated
Bax
Western blot assay showed that AURKA inhibition
resulted in decreased expression of CDK1 protein, reinforcing the
likelihood of G2/M cell cycle arrest in H446 cells (Fig. 7A). In order to examine the molecular
mechanism of AURKA in cell apoptosis, we analyzed two key apoptotic
mediators of the Bcl-2 family, Bcl-2 and Bax. Our results revealed
that the knockdown of AURKA downregulated Bcl-2 and upregulated Bax
expression in H446 cells. This result indicates that the heightened
apoptosis associated with AURKA downregulation may be partly
mediated by these Bcl-2 family proteins in H446 cells (Fig. 7B).
Discussion
SCLC is a type of highly malignant lung tumor that
metastasizes markedly quickly after its initial genesis (16). In a previous study, ectopic
expression of AURKA in NIH 3T3 cells and Rat1 fibroblasts led to
centrosome amplification and cell transformation in vitro
and manifested in tumorigenesis in vivo (17). The expression of AURKA mRNA and
protein is associated with the tumor stage and metastasis in head
and neck squamous cell carcinoma (18). Aneuploidy and overexpression of
AURKA have been shown to predict poor outcome in serous ovarian
carcinoma (19). These findings
suggest that AURKA is a potential target for diagnosis and
treatment for various types of cancer. However, little is known
about the mechanisms of action of AURKA in SCLC.
RNA interference (RNAi) is a widely used technique
to reduce the expression level of target proteins (20). The lentivirus vector is an effective
vehicle for introducing RNAi into cells under controlled conditions
(21). The vector is capable of
integrating a vast amount of genetic information into the host’s
genome. The host cell permanently expresses the viral vector gene
as if it was its own. We used this methodology in our study to
downregulate the expression of AURKA gene, resulting in partial to
complete loss of function gene (22).
In the present study, we observed that suppression
of AURKA expression by AURKA-siRNA led to prompted mitotic arrest
of the H446 and H1688 cells in the G2/M phase, and induced
apoptosis. Other studies have found that the knockdown of AURKA
inhibits tumor cell proliferation and invasion, and enhances
apoptosis in other types of cancer, such as human esophageal
squamous cell carcinoma (ESCC) (23). Wang et al found that the
knockdown of AURKA led to increased genomic stabilization and
slowed the progression of the cell cycle in addition to the
aforementioned processes in ESCC cells (24). Another study, by Yang et al,
demonstrated that the knockdown of AURKA in ovarian cancer cell
lines disabled the cancer’s ability to thrive, which is also
through the same molecular processes (25). Our findings confirm AURKA’s role in
SCLC cells.
CDK1 is necessary for cells to exit the G2 phase and
enter mitosis (26). CDK1 activity
is determined by the relative levels of CDK1 activators and
repressors in the cellular environment. The major activator of CDK1
is cyclin B1 (27). Activated AURKA
is required for the recruitment of CDK1-cyclin B1 to the centrosome
and thus the commitment of a cell to mitosis. Moreover, AURKA
phosphorylates CDC25B at the centrosome and contributes to G2/M
transition in cancer cells (28).
Our findings demonstrated that AURKA inhibition resulted in
decreased CDK1 protein expression. This may be associated with the
noted G2/M arrest. Moreover, apoptosis is partially modulated by
the Bcl-2 family, including both apoptotic enabling as well as
inhibiting factors (29,30). Bcl-2 and Bax are among the most
widely recognized pro-survival pro-apoptotic proteins,
respectively. Bcl-2 and Bax can form homodimers or heterodimers
with one another (31). Reduced
Bcl-2 expression in the presence of increased Bax expression likely
generates a dominant signal in favor of cell death (32). In the present study, we found that
the knockdown of AURKA decreased Bcl-2 expression and increased Bax
expression. This indicates that the Bcl-2 family may be implicated
in the apoptosis of H446 cells following knockdown of AURKA.
In conclusion, our results demonstrated that the
significant downregulation of AURKA expression by AURKA siRNA in
SCLC cells inhibited cell proliferation and induced cell apoptosis.
A potential mechanism of the mitotic suppression was widespread
arrest in the G2/M phase of the cell cycle. In addition, the
increased cell apoptosis rate after knockdown of the AURKA gene may
be partially through downregulation of Bcl-2 and upregulation of
Bax. Our data therefore elucidate the potentially therapeutic roles
of AURKA in SCLC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 81172347) and the Science
and Technology Foundation of Kunshan city (KSZ1309). The authors
thank Dr Wenxiang Wei (Soochow University, Suzhou 215123, China)
for his help and technical support.
References
|
1
|
van Meerbeeck JP, Fennell DA and De
Ruysscher DK: Small-cell lung cancer. Lancet. 378:1741–1755.
2011.
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
3
|
Lally BE, Urbanic JJ, Blackstock AW,
Miller AA and Perry MC: Small cell lung cancer: have we made any
progress over the last 25 years? Oncologist. 12:1096–1104.
2007.PubMed/NCBI
|
|
4
|
Oze I, Hotta K, Kiura K, et al:
Twenty-seven years of phase III trials for patients with extensive
disease small-cell lung cancer: disappointing results. PLoS One.
4:e78352009.PubMed/NCBI
|
|
5
|
Vader G and Lens SM: The Aurora kinase
family in cell division and cancer. Biochim Biophys Acta.
1786:60–72. 2008.PubMed/NCBI
|
|
6
|
Nikonova AS, Astsaturov I, Serebriiskii
IG, Dunbrack RL Jr and Golemis EA: Aurora A kinase (AURKA) in
normal and pathological cell division. Cell Mol Life Sci.
70:661–687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Katayama H, Brinkley WR and Sen S: The
Aurora kinases: role in cell transformation and tumorigenesis.
Cancer Metastasis Rev. 22:451–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang Y, Zhang Y, Lees E and Seghezzi W:
AuroraA overexpression overrides the mitotic spindle checkpoint
triggered by nocodazole, a microtubule destabilizer. Oncogene.
22:8293–8301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Anand S, Penrhyn-Lowe S and Venkitaraman
AR: AURORA-A amplification overrides the mitotic spindle
assembly checkpoint, inducing resistance to Taxol. Cancer Cell.
3:51–62. 2003. View Article : Google Scholar
|
|
10
|
Zhou H, Kuang J, Zhong L, et al: Tumour
amplified kinase STK15/BTAK induces centrosome amplification,
aneuploidy and transformation. Nat Genet. 20:189–193. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang X, Zhou YX, Qiao W, et al:
Overexpression of aurora kinase A in mouse mammary epithelium
induces genetic instability preceding mammary tumor formation.
Oncogene. 25:7148–7158. 2006. View Article : Google Scholar
|
|
12
|
Burnett JC and Rossi JJ: RNA-based
therapeutics: current progress and future prospects. Chem Biol.
19:60–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Watts JK and Corey DR: Silencing disease
genes in the laboratory and the clinic. J Pathol. 226:365–379.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Castanotto D and Rossi JJ: The promises
and pitfalls of RNA-interference-based therapeutics. Nature.
457:426–433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ketting RF: The many faces of RNAi. Dev
Cell. 20:148–161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stovold R, Blackhall F, Meredith S, Hou J,
Dive C and White A: Biomarkers for small cell lung cancer:
neuroendocrine, epithelial and circulating tumour cells. Lung
Cancer. 76:263–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bischoff JR, Anderson L, Zhu Y, et al: A
homologue of Drosophila aurora kinase is oncogenic and
amplified in human cancers. EMBO J. 17:3052–3065. 1998.PubMed/NCBI
|
|
18
|
Reiter R, Gais P, Jütting U, et al: Aurora
kinase A messenger RNA overexpression is correlated with tumor
progression and shortened survival in head and neck squamous cell
carcinoma. Clin Cancer Res. 12:5136–5141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lassus H, Staff S, Leminen A, Isola J and
Butzow R: Aurora-A overexpression and aneuploidy predict poor
outcome in serous ovarian carcinoma. Gynecol Oncol. 120:11–17.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stein EV, Price DK and Figg WD: shRNA
technology: investigating Ras-dependent cancer. Cancer Biol Ther.
8:1798–1799. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo J, Emanuele MJ, Li D, et al: A
genome-wide RNAi screen identifies multiple synthetic lethal
interactions with the Ras oncogene. Cell. 137:835–848. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brummelkamp TR, Bernards R and Agami R: A
system for stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Dong L, Xie J, Tong T and Zhan Q:
Stable knockdown of Aurora-A by vector-based RNA interference in
human esophageal squamous cell carcinoma cell line inhibits tumor
cell proliferation, invasion and enhances apoptosis. Cancer Biol
Ther. 8:1852–1859. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Lu N, Niu B, Chen X, Xie J and
Cheng N: Overexpression of Aurora-A enhances invasion and matrix
metalloproteinase-2 expression in esophageal squamous cell
carcinoma cells. Mol Cancer Res. 10:588–596. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang G, Chang B, Yang F, et al: Aurora
kinase A promotes ovarian tumorigenesis through dysregulation of
the cell cycle and suppression of BRCA2. Clin Cancer Res.
16:3171–3181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Riabowol K, Draetta G, Brizuela L, Vandre
D and Beach D: The cdc2 kinase is a nuclear protein that is
essential for mitosis in mammalian cells. Cell. 57:393–401. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dutertre S, Cazales M, Quaranta M, et al:
Phosphorylation of CDC25B by Aurora-A at the centrosome contributes
to the G2-M transition. J Cell Sci. 117:2523–2531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qi F, Inagaki Y, Gao B, et al: Bufalin and
cinobufagin induce apoptosis of human hepatocellular carcinoma
cells via Fas- and mitochondria-mediated pathways. Cancer Sci.
102:951–958. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oltersdorf T, Elmore SW, Shoemaker AR, et
al: An inhibitor of Bcl-2 family proteins induces regression of
solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gu X, Yao Y, Cheng R, et al: Plasminogen
K5 activates mitochondrial apoptosis pathway in endothelial cells
by regulating Bak and Bcl-xL subcellular distribution.
Apoptosis. 16:846–855. 2011. View Article : Google Scholar : PubMed/NCBI
|