Introduction
Angiogenesis is a vital and complicated process
involving endothelial cells, pericytes and the environment, which
is executed to ensure normal physiological responses, including
wound healing, embryonic development and bone remodeling (1,2). On
the contrary, it also plays a critical role in the pathogenesis of
multiple diseases and cancer progression (3). Tumor-induced angiogenesis not only
provides the essential blood supply but also allows cancer cell
metastasis by allowing cells to enter the circulation (4,5).
Without a blood circulation supplement, a tumor is unable to grow
beyond a critical size or to metastasize to another organ. Thus,
tumor neovascularization has become a potential therapeutic target.
Anti-angiogenetic strategies attempt to destroy existing vessels
and inhibit new vessel formation to starve and prison tumor cells
(6,7). Angiogenesis inhibitors have now been
approved for clinical application, and vascular endothelial growth
factor (VEGF) was initially identified as a critical angiogenesis
promoter (5,8). Bevacizumab (Avastin; Roche/Genentech)
is a specific VEGF antibody used as an angiogenesis inhibitor
(9), and this agent has been used
with chemotherapy or cytokine therapy for various advanced
metastatic cancers (10–12). Furthermore, multiple-targeted
pan-VEGF receptor tyrosine kinase inhibitors were subsequently
approved for the treatment of advanced cancer and age-related
macular degeneration. With treatment, the survival of cancer
patients is generally prolonged by several months (13,14).
However, targeting of the VEGF pathway has not
proven as efficacious as hoped. Tumor cells are able to evade
anti-VEGF therapy after a period of treatment paricularly certain
late-stage cancers (1,8). Multiple plausible mechanisms of escape
and resistance have been reported, including upregulation of
alternative pathways in selected tumor clones, providing vascular
progenitors and modulators with resilient systems to support a
neovascular response (15) and
tumor-associated endothelial cell genetic instability and
resistance to anti-VEGF therapy (16). Moreover, induction of endothelial
cell apoptosis leads to dysfunction of blood vessel to support
non-nutrients during angiogenesis (6,7). As a
result, engineering new inhibitors with which to target
angiogenesis through pathways other than VEGF signaling is
increasingly important.
Quinazoline derivatives have been found to possess
various pharmacological effects, including anti-inflammatory and
anticancer activities (17–20). In our laboratory, we synthesized a
series of quinazoline compounds with fluorine as human anticancer
candidates (21). One of these
compounds, 6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino) quinazoline
(HMJ-30), has been reported to induce apoptotic death through
induction of oxidative stress and upregulation of ataxia
telangiectasia mutated (ATM)/p53 signaling in U-2 OS human
osteosarcoma cells (20).
Additionally, xenograft tumor growth of osteosarcoma in nude mice
was inhibited by HMJ-30 (unpublished data). Since angiogenesis
contributes to a poor prognosis in human osteosarcoma (23), the study of the anti-angiogenic
mechanism of osteosarcoma cells may lead to the development of
novel and successful strategies for the treatment of osteosarcoma.
The inhibitory effects on the angiogenic response by HMJ-30 and the
molecular mechanisms of its cytotoxic effects on HUVECs remain
unclear; thus these issues were investigated in the present study.
We focused on the vascular targeting effects and resulted in
apoptosis of endothelial cells triggered by HMJ-30.
Materials and methods
Chemicals and reagents
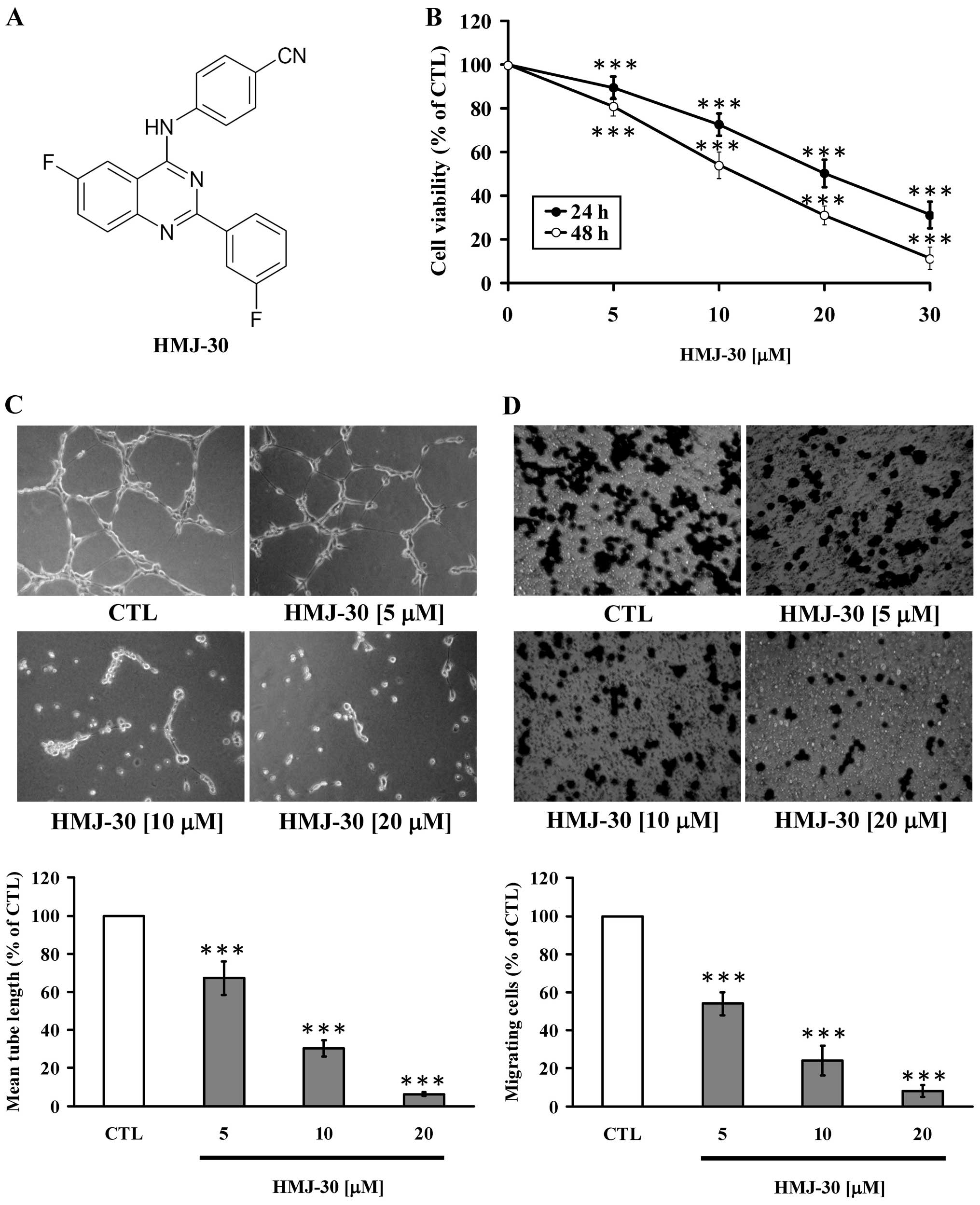
HMJ-30 was synthesized by Dr Mann-Jen Hour, and its
chemical structure is shown in Fig.
1A. Caspase-3, caspase-8 and caspase-9 colorimetric assay kits,
caspase-3 inhibitor Z-DEVD-FMK, caspase-8 inhibitor Z-IETD-FMK and
recombinant human VEGF were purchased from R&D Systems Inc.
(Minneapolis, MN, USA). Materials and chemicals for electrophoresis
were obtained from Bio-Rad Laboratories, Inc. (Hercules, CA, USA).
Primary antibodies (Fas/CD95, DR4, DR5, TNFR and actin) and
horseradish peroxidase (HRP)-conjugated secondary antibodies
against rabbit or mouse immunoglobulin were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). The other antibodies for
immunoblotting were purchased from Cell Signaling Technology
(Beverly, MA, USA). All other chemicals were of analytical grade
and were obtained from Sigma-Aldrich Corp. (St. Louis, MO, USA)
unless otherwise stated.
Cell culture
Human umbilical vein endothelial cells (HUVECs,
CD31+ >99%) were purchased from the Bioresource
Collection and Research Center (BCRC, Hsinchu, Taiwan) and
maintained in Medium 200 (Gibco Life Technologies, Carlsbad, CA,
USA) supplemented with low serum growth supplement (LSGS; Gibco
Life Technologies) at 37°C in a humidified atmosphere with 5%
CO2. HUVECs were used between the second and the fourth
generation.
Cell viability
HUVECs in 96-well plates at a density of
5×103 cells/well were exposed to HMJ-30 at various
concentrations (5, 10, 20 and 30 μM) for 24 and 48 h. The effects
of HMJ-30-induced cytotoxicity were measured using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay following previously reported methods (24,25).
Endothelial tube formation assay
HUVECs (1×105 cells/well) were seeded on
a 48-well culture plate precoated with Matrigel (BD Biosciences,
Bedford, MA, USA) containing 20 ng/ml VEGF as previously described
(26) and were subsequently treated
with or without different concentrations of HMJ-30 (5, 10 and 20
μM). At 16 h post-seeding, cells were fixed with 4%
paraformaldehyde prior to capturing images under an inverted
phase-contrast microscope.
Endothelial cell migration assay
HUVECs (5×104 cells/ml) were initially
incubated in a Transwell (Millicell Cell Culture Insert,
polycarbonate, 8.0 μm; EMD Millipore Corporation, Billerica, MA,
USA) for 4 h. Subsequently, different concentrations of HMJ-30 (5,
10 and 20 μM) were added into individual wells and the procedure
was followed as described in the protocols by Chiang et al
(27). The migrating cells were
quantified by counting the cell number after being stained in three
random fields/well with a microscope.
Chick embryo assay
In vivo angiogenesis assays have made
important progress in elucidating the mechanism of action of
angiogenesis-influencing factors (28). After 7–9 days, the chick
chorioallantoic membrane (CAM) in embryos was exposed to final
concentrations of 10 and 20 μM of HMJ-30 by making a window in the
egg shell. The window was sealed and eggs were re-incubated for an
appropriate incubation period.
Ex vitro rat aortic ring assay
The aortic ring explant cultures were carried out as
previously described by Pyun et al with various modification
(29). Six-week old male
Sprague-Dawley (SD) rats were obtained from BioLASCO Taiwan Co.,
Ltd. (Taipei, Taiwan) and subsequently the aortic rings were
collected to test vessel sprouting as previously described
(27,30).
Matrigel plug assay
Matrigel basement membrane matrix (BD Biosciences)
containing 200 ng/ml VEGF was injected subcutaneously into the
dorsal region of 6- to 8-week old BALB/c nude mice obtained from
the National Laboratory Animal Center (Taipei, Taiwan). After
Matrigel plug solidification according to a previous method
(27,31), HMJ-30 at 10 and 20 mg/kg was
administered intraperitoneally daily before mice were sacrificed.
On day 7, Matrigel pellets were harvested and processed for
staining and then photographed. Neovascularization was quantified
by measuring the hemoglobin of the plugs using the Drabkin method
(Drabkin’s reagent; Sigma-Aldrich Corp.).
Detection of HUVEC morphology and DNA
content analysis by flow cytometry
Following HMJ-30 treatment (5, 10 or 20 μM), cells
in 24-well plates at a density of 1×105 cells/well for
24 h with VEGF stimulation were harvested before morphological
changes were examined by a phase-contrast microscope. The collected
cells were fixed in 75% ethanol overnight at −20°C before being
stained with 0.1 M phosphate/citric acid buffer (0.2 M
NaHPO4, 0.1 M citric acid, pH 7.8) and 40 μg/ml
propidium iodide for 30 min at room temperature in darkness. The
stained cells were determined with FACSCalibur (BD Biosciences,
Franklin Lakes, NJ, USA).
Apoptotic assay
Following treatment with 5, 10 and 20 μM of HMJ-30
for 24 h or without treatment, apoptosis in the HUVECs was
determined by terminal deoxynucleotidyl transferase dUTP nick end
labeling (TUNEL) assay, using an In Situ Cell Death
Detection kit, fluorescein (Roche Diagnostics GmbH, Mannheim,
Germany) and flow cytometric analysis. Following HMJ-30 exposure,
cells were prepared for detecttion of DNA fragmentation following a
previously reported method (32,33).
Measurements of ROS production and cell
viability following pretreatment with N-acetylcysteine (NAC) or
catalase
HUVECs were cultured with 20 μM HMJ-30 for 0, 3, 6
and 12 h. Cells were then harvested and labeled with 20 μM
2,7-dichlorodihydrofluorescein diacetate (H2DCF-DA) (a
specific ROS fluorescence probe) at 37°C for 30 min. Consequently,
ROS production was analyzed for fluorescence intensity by flow
cytometry. Cells were pre-incubated with or without 10 mM NAC (an
antioxidant) or 5 μg/ml catalase for 1 h before exposure to 20 μM
HMJ-30 for 24 h. Cell viability was determined by MTT assay as
described above.
Determination of caspase-3, caspase-8 and
caspase-9 activities and the effect of their specific
inhibitors
HUVECs (5×106 cells) were incubated in
75-T flasks and treated with 20 μM of HMJ-30 for 0, 12 and 24 h.
Cells were then harvested to assess the relative caspase activity
using caspase-3, caspase-8 and caspase-9 colorimetric assay kits
(R&D Systems Inc.) following the manufacturer’s instructions.
Cells were pretreated with 10 μM Z-DEVD-FMK (a specific caspase-3
inhibitor) or 10 μM Z-IETD-FMK (a specific caspase-8 inhibitor) for
1 h and MTT assay was performed as detailed above.
Western blot analysis
HUVECs (5×106 cells) were incubated in 20
μM HMJ-30 for 0, 3, 6, 12 or 24 h. Cells were harvested, and an
equal amount of protein extract from the cell lysate was separated
on 10% SDS-polyacrylamide electrophoresis gels (SDS-PAGE) as
previously described (27,34). The appropriate primary antibodies
were hybridized and the specific protein signals were then observed
using the Immobilon Western HRP substrate kit (Merck Millipore)
after using the HRP-conjugated secondary antibodies. Actin served
as an internal control to ensure equal loading, and NIH ImageJ 1.47
software was used to perform the densitometric quantification of
each band.
Statistical analysis
Data are represented as mean ± standard error of the
mean (SEM) from at least three separate experiments. Statistical
calculations of the data were carried out using the Student’s
t-test. p<0.001 was considered to indicate a statistically
significant result.
Results
HMJ-30 inhibits the angiogenesis of
HUVECs in vitro
To evaluate the angiogenesis targeting potential of
HMJ-30 in vitro, we carried out a series of angiogenic
cellular functional assays in HUVECs. The viability of the HUVECs
was significantly decreased in the HMJ-30-treated groups in a
concentration- and time-dependent manner (Fig. 1B). Subsequently, we conducted a tube
formation assay to determine the effect of HMJ-30 on tube-like
network formation of HUVECs. Cells were placed on a Matrigel-coated
plate with endothelial cell growth media with VEGF. HUVECs formed
robust and elongated tube-like structures in the control group. In
contrast, treatment with HMJ-30 concentration-dependently inhibited
the formation of tube-like networks (Fig. 1C). Transwell migration assay was
performed to evaluate the migratory behavior of HUVECs. The
migration of endothelial cells is a pivotal step in the formation
of new vessels. HMJ-30 significantly suppressed cell migration in a
concentration-dependent manner (Fig.
1D). Overall, these results provide evidence that HMJ-30
inhibited the angiogenic activity of HUVECs in vitro.
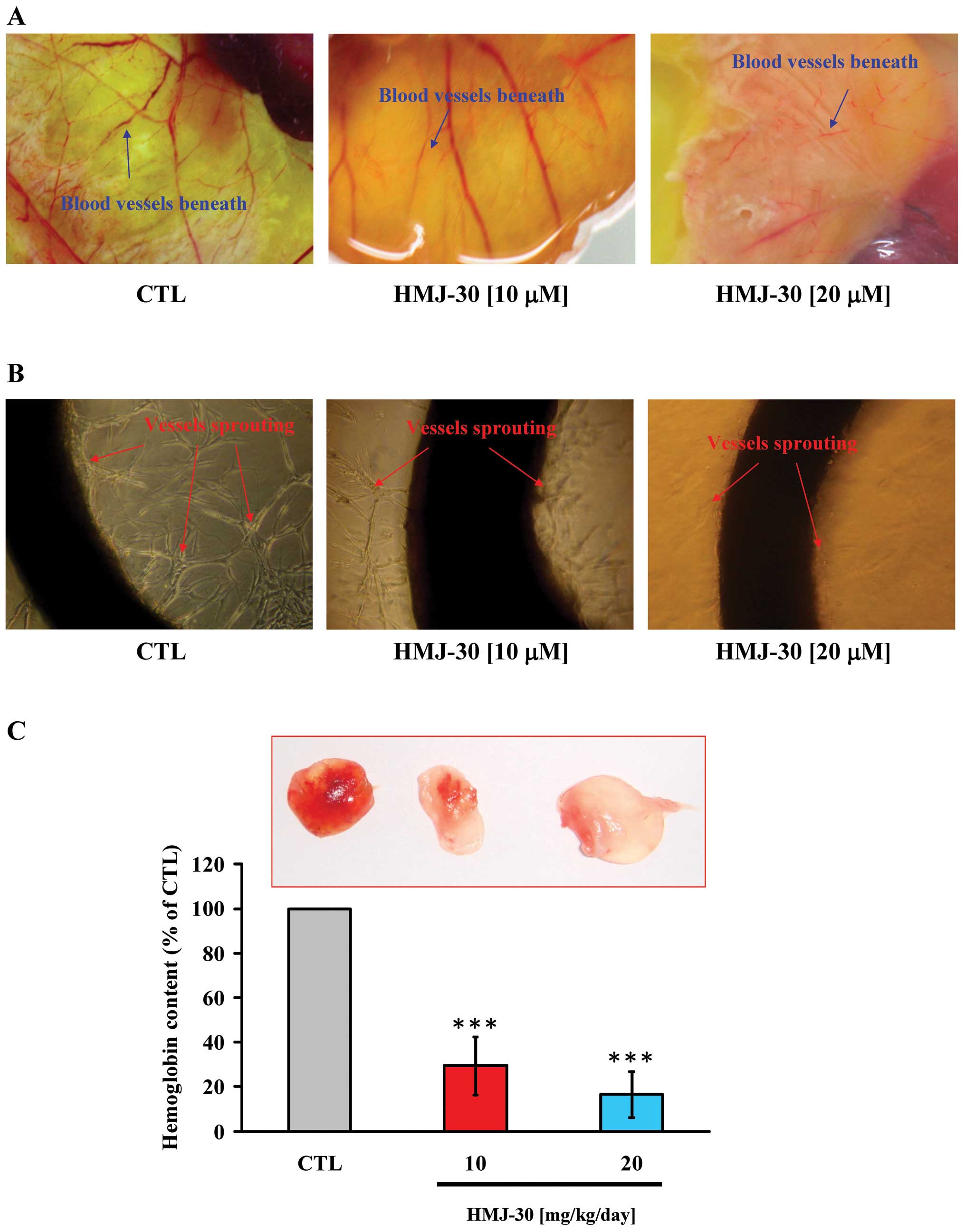
HMJ-30 suppresses angiogenesis in vivo
and ex vivo
Among various animal model systems established to
investigate the mechanisms underlying angiogenesis, the chick
embryo model and the mouse Matrigel plug have been well developed
to analyze the anti-angiogenic or pro-angiogenic potential of
compounds (27,31). Both assays were conducted to verify
the anti-angiogenic activity of HMJ-30 in vivo. The
chorioallantoic membrane is the specialized and highly vascularized
tissue of the avian embryo. The capillary plexus was dense and
appeared as a honeycomb network in the control group. However, in
the HMJ-30 treatment groups, vessel branching and sprouting were
significantly reduced (Fig. 2A).
Aortic ring vessel sprouting around the aortic rings stimulated by
VEGF was observed in the untreated control, but this was markedly
disrupted by HMJ-30 at 10 and 20 μM to form a network of vessels
(Fig. 2B). Angiogenesis in Matrigel
plugs can be induced by VEGF in the mouse Matrigel plug assay. The
plug in the control group contained abundant erythrocyte-filled
vessels, indicating the formation of neovascularization, whereas
only few vessels were observed in the HMJ-30-treated plugs
(Fig. 2C). Additionally, we also
measured the amount of hemoglobin contained in the plugs for
quantification. The hemoglobin quantity was markedly reduced after
treatment with HMJ-30 as compared to the control group (Fig. 2C). Taken together, these findings
provide strong evidence of HMJ-30 to exhibit an anti-angiogenic
response in vivo.
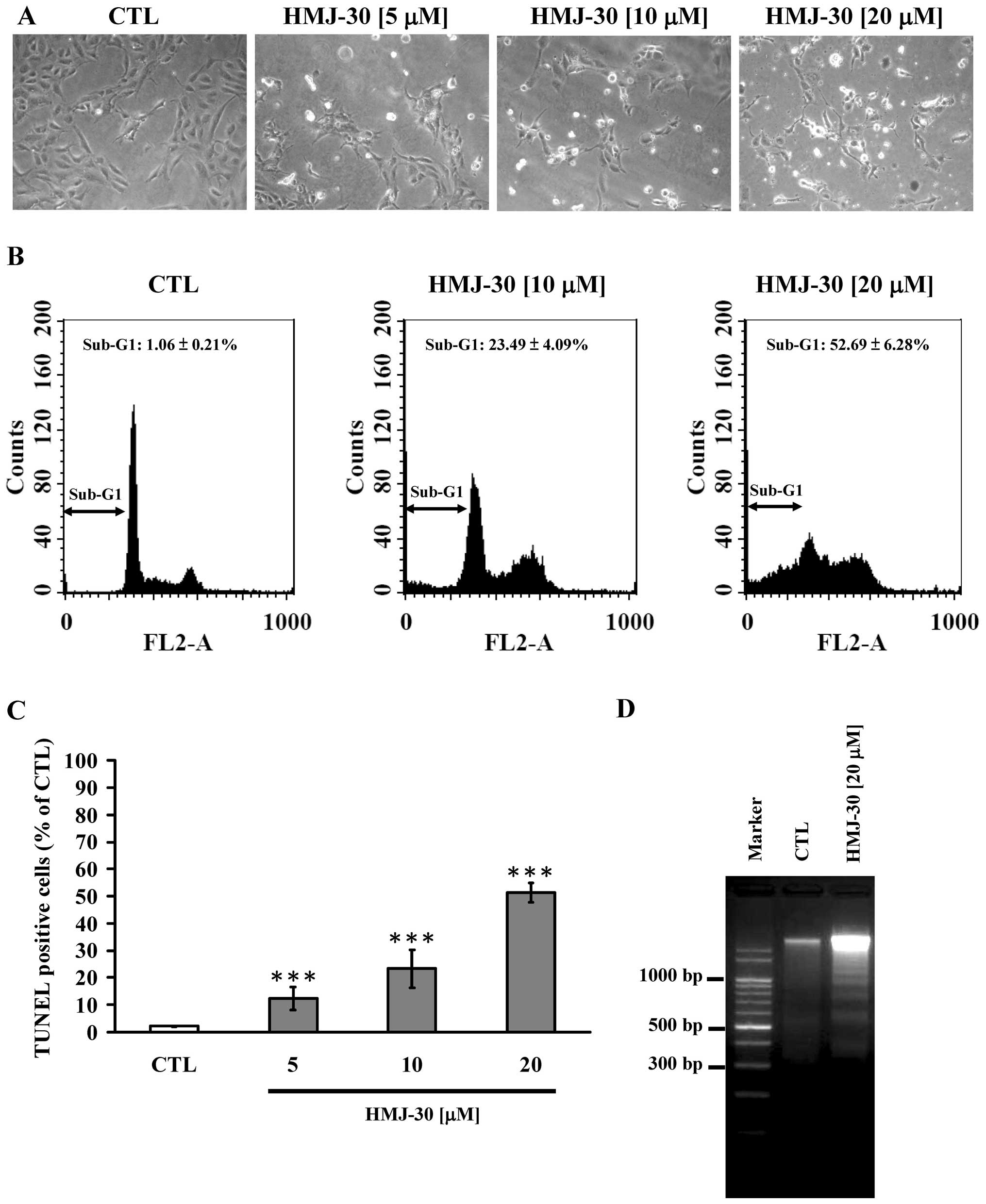
HMJ-30 induces apoptosis in HUVECs
We earlier confirmed that HMJ-30 inhibited cell
proliferation of HUVECs in vitro. HUVECs detached from the
surface of the plate and morphologic shrinkage was noted in the
HMJ-30-treated cells, whereas cells of the control group were well
spread with normal morphology (Fig.
3A). On the other hand, to determine the mechanism triggered by
HMJ-30, we investigated the DNA content and apoptotic population by
flow cytometry. HMJ-30 induced an increase in hypodiploid (sub-G1
phase) cells (Fig. 3B). In
addition, we conducted TUNEL assay and DNA agarose gel
electrophoresis to evaluate DNA fragmentation. An increase in
TUNEL-positive cells (Fig. 3C) and
oligonucleosomal fragments (Fig.
3D) was observed in the HMJ-30-treated cells. Overall, these
results demonstrated that HMJ-30 induced apoptotic cell death in
the HUVECs.
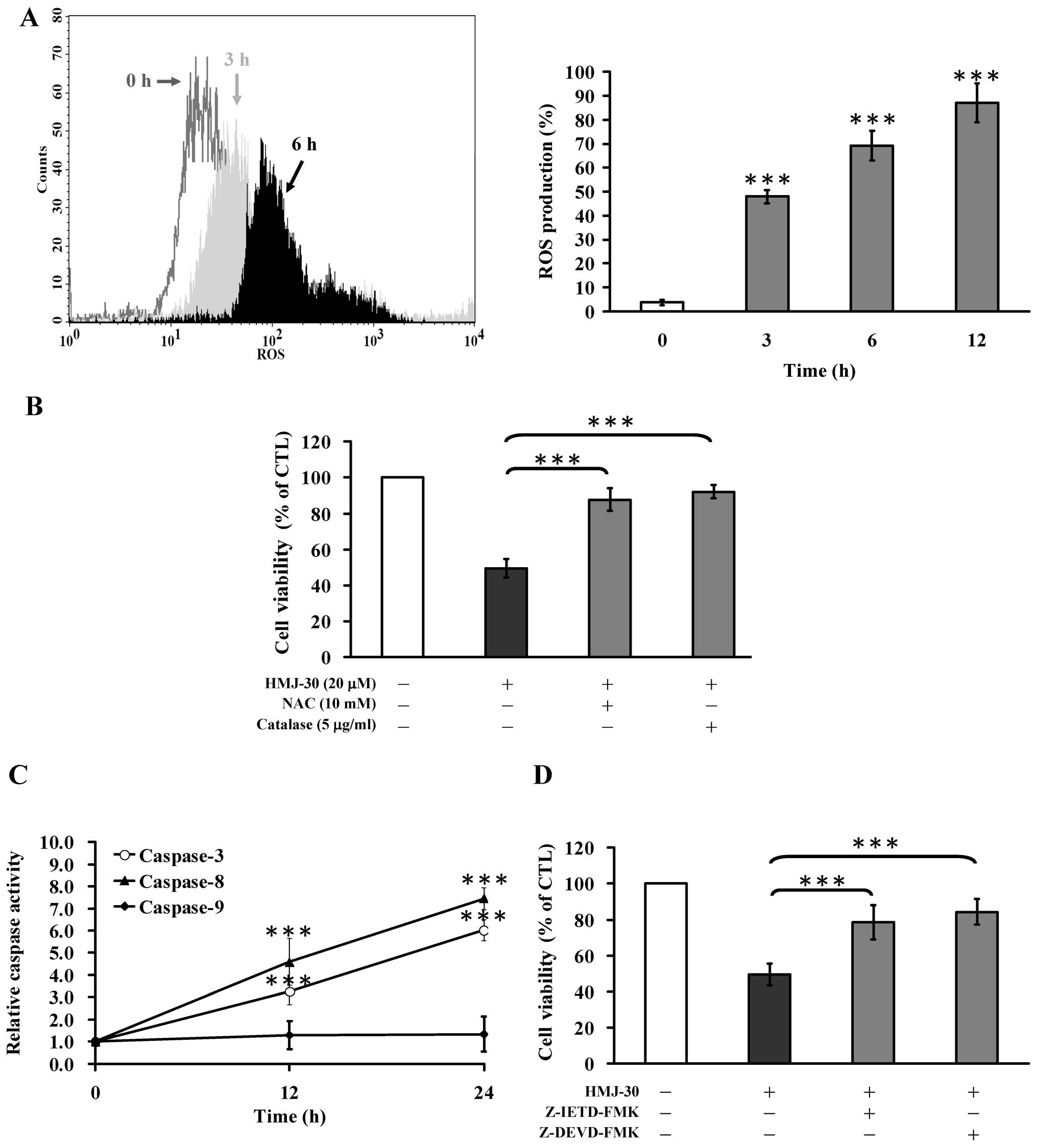
HMJ-30 increases ROS generation in
HUVECs
An increase in intracellular ROS has been
demonstrated to play a critical role in eliciting an early response
of apoptosis (35,36). HMJ-30 time-dependently increased
intracellular ROS levels (Fig. 4A).
Furthermore, pretreatment with NAC or catalase significantly
reduced HMJ-30-induced cell death (Fig.
4B). Based on these results, the induction of oxidative stress
was required for HMJ-30-induced apoptosis in HUVECs.
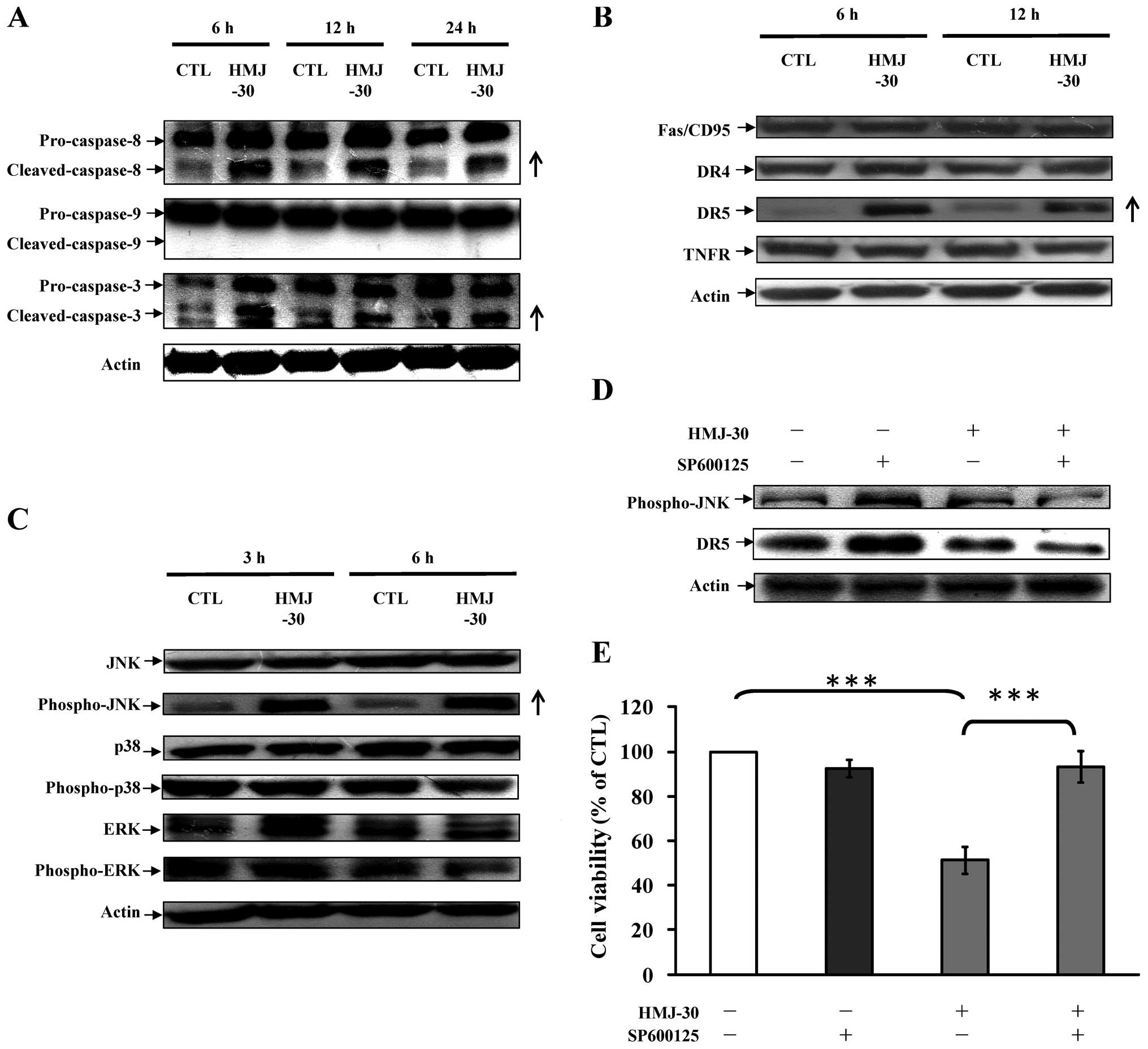
HMJ-30 activates extrinsic apoptosis in
HUVECs
To determine the molecular mechanism of apoptotic
death induced by HMJ-30, we explored the activities of caspase-9,
caspase-8, and caspase-3, respectively. The activities of caspase-3
and caspase-8 were significantly increased after treatment with
HMJ-30 in a time-dependent manner (Fig.
4C). Moreover, pre-incubation with specific inhibitors of
Z-IETD-FMK or Z-DEVD-FMK strongly increased cell viability compared
with HMJ-30 treatment alone (Fig.
4D). However, caspase-9 activity was not significantly affected
by HMJ-30 treatment (Fig. 4C).
Overall, these data demonstrated that caspase-3/-8-dependent
signaling plays a crucial role in HMJ-30-triggered apoptosis of
HUVECs.
c-Jun N-terminal kinase (JNK)-mediated
death receptor pathway in HUVECs is involved in HMJ-30-induced
apoptosis
To elucidate the possible signaling pathway involved
in HMJ-30-mediated apoptosis, the levels of associated proteins
were evaluated. Treatment of HMJ-30 increased the cleaved forms of
caspase-3 and caspase-8 rather than caspase-9 (Fig. 5A), indicating activation of the
extrinsic apoptotic pathway. DR5 (a death receptor-associated
protein) was upregulated (Fig. 5B)
and subsequently stimulated caspase-8 expression to promote
downstream effectors caspase-3 to induce cell apoptosis (37). Additionally, mitogen-activated
protein kinase (MAPK) pathway-associated proteins, including JNK,
p38, extracellular signal-regulated kinase (ERK) and their
phosphorylated molecules, were evaluated (38,39).
HMJ-30 increased the protein level of phosphorylated JNK (Fig. 5C), followed by an increase in DR5
dependent on the exposure time. Pretreatment with SP600125, a
selective JNK inhibitor, effectively attenuated the phosphorylation
of JNK and DR5 protein expression as well as markedly reversed the
inhibition of cell viability in the HMJ-30-treated group. The
results from our experimental approaches conclude that
HMJ-30-induced apoptosis of HUVECs is mediated through DR5 and the
JNK pathway.
Discussion
Blood vessels remove waste, deliver oxygen and
nutrients to every part of the body; they also nourish cancer and
provide pathways for malignant tumor cells to spread to other
organs (1,3). Bevacizumab, a recombinant human
monoclonal antibody against VEGF, was approved for clinical
treatment to target the angiogenesis of cancer (9). However, approval of bevacizumab war
questioned due to concerns about its toxicity and efficacy
(15). Appearance of drug
resistance ultimately results in the failure of VEGF-targeted
therapies. HMJ-30 is able to prevent revascularization and disrupt
existing vessels through induction of endothelial cell apoptosis.
The different mechanism of this vascular targeting agent was
therefore reported in this study.
Endothelial cells are able to disrupt the
surrounding basement membrane to migrate toward angiogenic stimuli
(6,7,27).
These cells subsequently form the necessary three-dimensional
vessel structures to create new vessels through cell proliferation
and reorganization (6,27). The results of the tube-formation
assay (Fig. 1C), migration assay
(Fig. 1D), chick embryo assay
(Fig. 2A), ex vivo rat
aortic ring assay (Fig. 2B), and
Matrigel plug assay (Fig. 2C)
clearly indicated that HMJ-30 possessed strong anti-angiogenic
activity in these in vitro, ex vivo and in
vivo experiments.
Apoptosis is the process of programmed cell death,
which plays a crucial role in diverse biological phenomenon and
diseases (7,44). Accumulating evidence indicates that
endothelial cell apoptosis causes the disruption of blood vessel
formation, resulting in the suppression of tumor progression
(7,43). Activation of the extrinsic initiator
caspase-8 with death effector domain would activate downstream of
caspase-3, causing cell apoptosis (44). Our results showed that ROS
generation played an important role in HMJ-30-induced apoptosis in
HUVECs (Fig. 4A and B). Similar
results were found by Chiu et al (22). HMJ-30 not only induced apoptotic
death in HUVECs (Fig. 3), but also
increased the activities and protein levels of caspase-8 and
caspase-3 in HMJ-30-treated HUVECs. Furthermore, pre-incubation
with their specific inhibitors restored the cell viability
following HMJ-30 treatment. Thus, activation of the death
receptor-associated caspase cascade was required for HMJ-30-induced
apoptosis. Li et al (45)
demonstrated that DR4 and DR5 proteins can be strongly expressed in
HUVECs and human dermal microvessels. Incubation with tumor
necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)
leads to caspase-8-dependent apoptosis death in HUVECs (45). The protein levels of TNF receptor
superfamily members, including Fas, DR4, DR5 and TNFR, were
therefore investigated in our study (Fig. 5B). Our data indicated that only DR5
was elevated following HMJ-30 treatment, suggesting that
HMJ-30-induced apoptosis was mediated by the death-receptor
pathway.
JNK signaling has been implicated in diverse cell
physiological processes, including differentiation, proliferation
and cellular stress-induced apoptosis. JNK signaling induces
apoptosis by leading to secretion of death ligands to promote
cytochrome c release from mitochondria, or by
phosphorylation of downstream pro-apoptotic proteins (46). Zhou et al (47) demonstrated that activation of JNK
upregulates DR5 expression, which in turn leads to the activation
of caspase-8-dependent apoptosis cascade in cancer cells. Our
results showed that HMJ-30 increased the protein level of
phospho-JNK, followed by an increase in DR5. Pretreatment with
SP600125 effectively reversed the inhibition of cell viability
(Fig. 5). These results indicated
that HMJ-30-induced apoptosis and DR5 expression was mediated
via a JNK-mediated mechanism. This is the first report to
show the correlation between JNK signaling and DR5 in HUVECs.
The diverse pharmacological properties and
anticancer activity of quinazoline have been previously reported
(18,19,21).
The inhibition of tumor progression by quinazoline-like compounds
is due to i) suppression of microtubule polymerization, ii)
downregulation of the tyrosine kinase signaling pathway and iii)
activation of apoptotic signaling cascades caused by intracellular
stress (24,26,33,36–38).
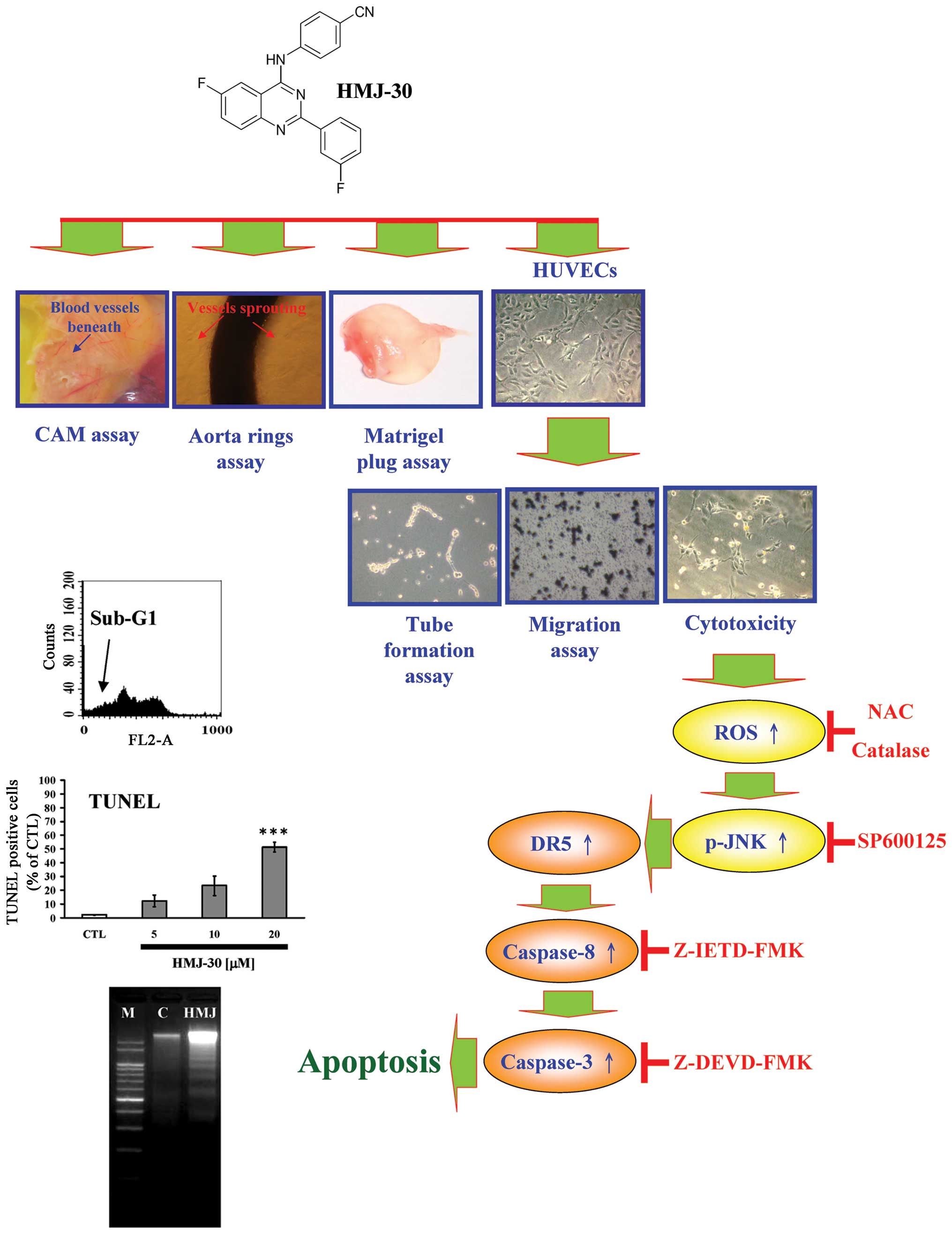
In conclusion, we first demonstrated that HMJ-30 exhibits a potent
anti-angiogenesis effect. The possible working mechanism of this
agent might include i) prevention of new blood vessel formation,
ii) elimination of existing vessels in tumors and iii) induction of
apoptosis in HUVECs through upreregulation of DR5 and JNK-mediated
extrinsic apoptosis signaling pathway (Fig. 6). These collective results provide
pharmacological basis for the therapeutic application of HMJ-30 in
pathological angiogenesis-related diseases such as cancer.
Acknowledgements
The present study was supported in part by a
research grant from the National Science Council of Taiwan, R.O.C.
(NSC 102-2320-B-039-028-MY3 to T-S.W.).
References
|
1
|
Kerbel R and Folkman J: Clinical
translation of angiogenesis inhibitors. Nat Rev Cancer. 2:727–739.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tian T, Nan KJ, Wang SH, et al: PTEN
regulates angiogenesis and VEGF expression through
phosphatase-dependent and -independent mechanisms in HepG2 cells.
Carcinogenesis. 31:1211–1219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hasan J, Shnyder SD, Bibby M, Double JA,
Bicknel R and Jayson GC: Quantitative angiogenesis assays in vivo -
a review. Angiogenesis. 7:1–16. 2004. View Article : Google Scholar
|
|
4
|
Stoletov K, Kato H, Zardouzian E, et al:
Visualizing extravasation dynamics of metastatic tumor cells. J
Cell Sci. 123:2332–2341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferrara N: VEGF and the quest for tumour
angiogenesis factors. Nat Rev Cancer. 2:795–803. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xuan H, Zhao J, Miao J, Li Y, Chu Y and Hu
F: Effect of Brazilian propolis on human umbilical vein endothelial
cell apoptosis. Food Chem Toxicol. 49:78–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Folkman J: Angiogenesis and apoptosis.
Semin Cancer Biol. 13:159–167. 2003. View Article : Google Scholar
|
|
8
|
Plate KH, Breier G, Weich HA, Mennel HD
and Risau W: Vascular endothelial growth factor and glioma
angiogenesis: coordinate induction of VEGF receptors, distribution
of VEGF protein and possible in vivo regulatory mechanisms. Int J
Cancer. 59:520–529. 1994. View Article : Google Scholar
|
|
9
|
Strickler JH and Hurwitz HI:
Bevacizumab-based therapies in the first-line treatment of
metastatic colorectal cancer. Oncologist. 17:513–524. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sandler AB, Johnson DH and Herbst RS:
Anti-vascular endothelial growth factor monoclonals in non-small
cell lung cancer. Clin Cancer Res. 10:4258s–4262s. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang JC, Haworth L, Sherry RM, et al: A
randomized trial of bevacizumab, an anti-vascular endothelial
growth factor antibody, for metastatic renal cancer. N Engl J Med.
349:427–434. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferrara N, Hillan KJ, Gerber HP and
Novotny W: Discovery and development of bevacizumab, an anti-VEGF
antibody for treating cancer. Nat Rev Drug Discov. 3:391–400. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scott BJ, Quant EC, McNamara MB, Ryg PA,
Batchelor TT and Wen PY: Bevacizumab salvage therapy following
progression in high-grade glioma patients treated with VEGF
receptor tyrosine kinase inhibitors. Neuro Oncol. 12:603–607. 2010.
View Article : Google Scholar
|
|
14
|
Gild ML, Bullock M, Robinson BG and
Clifton-Bligh R: Multikinase inhibitors: a new option for the
treatment of thyroid cancer. Nat Rev Endocrinol. 7:617–624. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bottsford-Miller JN, Coleman RL and Sood
AK: Resistance and escape from antiangiogenesis therapy: clinical
implications and future strategies. J Clin Oncol. 30:4026–4034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trowe T, Boukouvala S, Calkins K, et al:
EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib
resistance and neoplastic transformation. Clin Cancer Res.
14:2465–2475. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raghav N and Singh M: Design, synthesis
and docking studies of bischalcones based quinazoline-2(1H)-ones
and quinazoline-2(1H)-thiones derivatives as novel inhibitors of
cathepsin B and cathepsin H. Eur J Pharm Sci. 54:28–39. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bilbro J, Mart M and Kyprianou N:
Therapeutic value of quinazoline-based compounds in prostate
cancer. Anticancer Res. 33:4695–4700. 2013.PubMed/NCBI
|
|
19
|
Xu L and Russu WA: Molecular docking and
synthesis of novel quinazoline analogues as inhibitors of
transcription factors NF-κB activation and their anti-cancer
activities. Bioorg Med Chem. 21:540–546. 2013.PubMed/NCBI
|
|
20
|
Chatterjee N, Das S, Bose D, et al:
Exploring the anti-inflammatory activity of a novel
2-phenylquinazoline analog with protection against inflammatory
injury. Toxicol Appl Pharmacol. 264:182–191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hour MJ, Huang LJ, Kuo SC, et al:
6-Alkylamino- and 2,3-dihydro-3′-methoxy-2-phenyl-4-quinazolinones
and related compounds: their synthesis, cytotoxicity, and
inhibition of tubulin polymerization. J Med Chem. 43:4479–4487.
2000.
|
|
22
|
Chiu YJ, Hour MJ, Lu CC, et al: Novel
quinazoline HMJ-30 induces U-2 OS human osteogenic sarcoma cell
apoptosis through induction of oxidative stress and up-regulation
of ATM/p53 signaling pathway. J Orthop Res. 29:1448–1456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren T, Qing Y, Dai N, et al:
Apurinic/apyrimidinic endonuclease 1 induced upregulation of
fibroblast growth factor 2 and its receptor 3 induces angiogenesis
in human osteosarcoma cells. Cancer Sci. 105:186–194. 2014.
View Article : Google Scholar
|
|
24
|
Wu PP, Liu KC, Huang WW, et al: Triptolide
induces apoptosis in human adrenal cancer NCI-H295 cells through a
mitochondrial-dependent pathway. Oncol Rep. 25:551–557.
2011.PubMed/NCBI
|
|
25
|
Yang JS, Hour MJ, Huang WW, Lin KL, Kuo SC
and Chung JG: MJ-29 inhibits tubulin polymerization, induces
mitotic arrest, and triggers apoptosis via cyclin-dependent kinase
1-mediated Bcl-2 phosphorylation in human leukemia U937 cells. J
Pharmacol Exp Ther. 334:477–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pan SL, Guh JH, Peng CY, et al: YC-1
[3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole] inhibits
endothelial cell functions induced by angiogenic factors in vitro
and angiogenesis in vivo models. J Pharmacol Exp Ther. 314:35–42.
2005.
|
|
27
|
Chiang JH, Yang JS, Lu CC, et al: Newly
synthesized quinazolinone HMJ-38 suppresses angiogenetic responses
and triggers human umbilical vein endothelial cell apoptosis
through p53-modulated Fas/death receptor signaling. Toxicol Appl
Pharmacol. 269:150–162. 2013. View Article : Google Scholar
|
|
28
|
Kiriakidis S, Hogemeier O, Starcke S, et
al: Novel tempeh (fermented soyabean) isoflavones inhibit in vivo
angiogenesis in the chicken chorioallantoic membrane assay. Br J
Nutr. 93:317–323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pyun BJ, Choi S, Lee Y, et al: Capsiate, a
nonpungent capsaicin-like compound, inhibits angiogenesis and
vascular permeability via a direct inhibition of Src kinase
activity. Cancer Res. 68:227–235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pang X, Yi Z, Zhang X, et al:
Acetyl-11-keto-beta-boswellic acid inhibits prostate tumor growth
by suppressing vascular endothelial growth factor receptor
2-mediated angiogenesis. Cancer Res. 69:5893–5900. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pang X, Yi Z, Zhang J, et al: Celastrol
suppresses angiogenesis-mediated tumor growth through inhibition of
AKT/mammalian target of rapamycin pathway. Cancer Res.
70:1951–1959. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chung JG, Yang JS, Huang LJ, et al:
Proteomic approach to studying the cytotoxicity of YC-1 on U937
leukemia cells and antileukemia activity in orthotopic model of
leukemia mice. Proteomics. 7:3305–3317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang WW, Chiu YJ, Fan MJ, et al:
Kaempferol induced apoptosis via endoplasmic reticulum stress and
mitochondria-dependent pathway in human osteosarcoma U-2 OS cells.
Mol Nutr Food Res. 54:1585–1595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu CC, Yang JS, Chiang JH, et al: Novel
quinazolinone MJ-29 triggers endoplasmic reticulum stress and
intrinsic apoptosis in murine leukemia WEHI-3 cells and inhibits
leukemic mice. PLoS One. 7:e368312012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yodkeeree S, Sung B, Limtrakul P and
Aggarwal BB: Zerumbone enhances TRAIL-induced apoptosis through the
induction of death receptors in human colon cancer cells: Evidence
for an essential role of reactive oxygen species. Cancer Res.
69:6581–6589. 2009. View Article : Google Scholar
|
|
36
|
Prasad S, Yadav VR, Ravindran J and
Aggarwal BB: ROS and CHOP are critical for dibenzylideneacetone to
sensitize tumor cells to TRAIL through induction of death receptors
and downregulation of cell survival proteins. Cancer Res.
71:538–549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aggarwal BB, Bhardwaj U and Takada Y:
Regulation of TRAIL-induced apoptosis by ectopic expression of
antiapoptotic factors. Vitam Horm. 67:453–483. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Iwai A, Shiozaki T and Miyazaki T:
Relevance of signaling molecules for apoptosis induction on
influenza A virus replication. Biochem Biophys Res Commun.
441:531–537. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kannaiyan R, Shanmugam MK and Sethi G:
Molecular targets of celastrol derived from Thunder of God Vine:
potential role in the treatment of inflammatory disorders and
cancer. Cancer Lett. 303:9–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chiang JH, Yang JS, Lu CC, et al: Effect
of DNA damage response by quinazolinone analogue HMJ-38 on human
umbilical vein endothelial cells: Evidence for γH2A.X and
DNA-PK-dependent pathway. Hum Exp Toxicol. 33:590–601.
2014.PubMed/NCBI
|
|
41
|
Yang JS, Hour MJ, Kuo SC, Huang LJ and Lee
MR: Selective induction of G2/M arrest and apoptosis in HL-60 by a
potent anticancer agent, HMJ-38. Anticancer Res. 24:1769–1778.
2004.PubMed/NCBI
|
|
42
|
Lu CC, Yang JS, Chiang JH, et al:
Inhibition of invasion and migration by newly synthesized
quinazolinone MJ-29 in human oral cancer CAL 27 cells through
suppression of MMP-2/9 expression and combined down-regulation of
MAPK and AKT signaling. Anticancer Res. 32:2895–2903.
2012.PubMed/NCBI
|
|
43
|
Tozer GM, Kanthou C and Baguley BC:
Disrupting tumour blood vessels. Nat Rev Cancer. 5:423–435. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Portt L, Norman G, Clapp C, Greenwood M
and Greenwood MT: Anti-apoptosis and cell survival: a review.
Biochim Biophys Acta. 1813:238–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li JH, Kirkiles-Smith NC, McNiff JM and
Pober JS: TRAIL induces apoptosis and inflammatory gene expression
in human endothelial cells. J Immunol. 171:1526–1533. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Herr I and Debatin KM: Cellular stress
response and apoptosis in cancer therapy. Blood. 98:2603–2614.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zou W, Liu X, Yue P, et al: c-Jun
NH2-terminal kinase- mediated up-regulation of death receptor 5
contributes to induction of apoptosis by the novel synthetic
triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in
human lung cancer cells. Cancer Res. 64:7570–7578. 2004. View Article : Google Scholar
|