Introduction
Osteosarcoma is the most common primary malignant
bone malignancy and usually occurs in children and adolescents, and
is characterized by an aggressive clinical course (1). Although the combination treatment of
chemotherapy and surgery has improved the prognosis of patients
with primary bone osteosarcoma, the most effective drugs are still
those developed decades ago, and there is the risk of local relapse
after chemotherapy for a number of patients with osteosarcoma
(1–3). For this reason, understanding the
mechanisms of multiple genetic abnormalities synergistically
contributing to osteosarcoma progression and investigating novel
strategies for osteosarcoma treatment are necessary.
Recently, growing evidence suggests that miRNAs, an
abundant class of small noncoding RNAs, play critical roles in the
regulation of diverse biological processes (4). miRNAs post transcriptionally regulate
gene expression through binding to the 3′ untranslated regions
(3′UTR) of target mRNAs and thus can function either as oncogenes
or tumor-suppressor genes in tumorigenesis (5). Although dysregulation of miRNAs,
including miR-34a (6), miR-376c
(7) and miR-133a (8), has been associated with the initiation
and progression of osteosarcoma, a clear understanding of the
detailed roles and molecular mechanisms of miRNAs in osteosarcoma
is lacking.
microRNA-1 (miR-1) is a highly conserved miRNA and
plays critical role in skeletal and cardiac muscle development
(9). Recent studies have shown that
downregulation of miR-1 is a frequent event in various types of
cancer including osteosarcoma and functions as a tumor-suppressor
gene (10,11). In osteosarcoma, miR-1 has been shown
to affect cell proliferation and the cell cycle (12). Several oncogenic genes have been
validated to be the targets of miR-1, such as the met
proto-oncogene (MET) (13,14), cyclin D2 (CCND2) (15) and prothymosin α (PTMA) (16). This implies that miR-1 is involved
in tumorigenesis via a complex regulatory network, which, however,
has not been totally elucidated in osteosarcoma.
In the present study, we confirmed two target genes
of miR-1, Med1 and Med31, in osteosarcoma. Both Med1 and Med31 were
overexpressed in osteosarcoma and involved in cell proliferation.
Our data indicate that MET may mediate the downstream signaling and
suggest the potential therapeutic application of miR-1 which
functions by targeting multiple oncogenes in osteosarcoma.
Materials and methods
Patients and osteosarcoma tissues
Surgically resected paired osteosarcoma tumor
tissues and adjacent normal bone and myeloid tissues were collected
from 30 primary osteosarcoma patients who underwent surgical
resection following informed consent between 2006 and 2009 at
Shanghai 6th People’s Hospital (Shanghai, China). Surgically
removed tissues were snap-frozen in liquid nitrogen for further
use. Both tumor and non-tumor samples were confirmed by
pathological examinations. The experiments were approved by the
Ethics Committee of Shanghai Jiaotong University (Shanghai,
China).
Cell culture and transfection
Human osteoblasts hFOB and three human osteosarcoma
cell lines, MG-63, U2OS and Saos-2, were purchased from the
American Type Culture Collection. The cells were maintained in DMEM
(MG-63 and U2OS) and RPMI-1640 medium (Saos-2) (Gibco, Carlsbad,
CA, USA) with 10% fetal bovine serum (Gibco) and antibiotics (100
U/ml penicillin and 100 μg/ml streptomycin) at 37°C.
For transfection, cells were plated in 6-well
clusters or 96-well plates and incubated overnight, and then
transfected with miRNA mimics or negative control (Ambion, Austin,
TX, USA) at a final concentration of 100 nM using Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s
instructions.
The expression of Med1 and Med31 was inhibited in
MG-63 and U2OS cell lines using a lentiviral shRNA system (Lv-con,
Lv-Med1 shRNA, Lv-Med31 shRNA) from Santa Cruz Biotechnology
(Dallas, Texas, USA) with supporting siRNA transfection reagent
(Santa Cruz). MG-63 and U2OS cells stably transfected with Med
shRNA were selected in medium containing puromycin.
For overexpression of Med1 and Med31, the cDNA
clones were purchased from OriGene Technologies (Rockville, MD,
USA), and recombinant adenoviruses (Ad-Med1 and Ad-Med31) were
constructed by Hanbio Co. Ltd. (Shanghai, China).
For the detection of MET signaling, hepatocyte
growth factor (HGF; Sigma-Aldrich) was added to the medium as a MET
ligand.
RNA extraction and real-time PCR
Total RNA, including miRNA, was extracted using the
miRNeasy kit (Qiagen, Valencia, CA, USA). MicroRNA levels were
quantified using the TaqMan MicroRNA assay kit (Applied Biosystems,
Foster City, CA, USA) according to the manufacturer’s instructions.
After reverse transcription, mRNA levels were analyzed using SYBR
Green PCR master mix (Applied Biosystems) in the ABI PRISM 7900
Sequence Detection System (Applied Biosystems).
All samples were normalized to U6 small nuclear RNA
or β-actin mRNA, and fold changes were calculated using the
2−ΔΔCt method.
The primers used are listed as follows:
5′-CTAATGCTGG TCCCTTGGATAA-3′ and 5′-AGGTCAGAAGGAGAGACA TAGT-3′ for
Med1; 5′-GTCGCTATGGAGACAGATGAT-3′ and 5′-AGTAACCTCTTTGGGCAAGAA-3′
for Med31; and 5′-CACTCTTCCAGCCTTCCTTC-3 and 5′-GTACAGGTCT
TTGCGGATGT-3 for β-actin.
Cell proliferation assays
MG-63 and U2OS cells were seeded into 96-well plates
(5×103 cells/well), transfected with the miRNA mimics or
negative control and further incubated at 37°C. At the indicated
time periods, medium was replaced with fresh medium containing 0.5
mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT; Sigma-Aldrich, St. Louis, MO, USA). Cells were then incubated
at 37°C for 4 h and resolved by dimethyl sulfoxide (DMSO;
Sigma-Aldrich). The absorbance was measured at 490 nm using an
ELISA reader (Bio-Rad Laboratories, Hercules, CA, USA). Each
experiment was performed in triplicate and repeated three
times.
Cell cycle analysis
After being serum-starved in 1% FBS-containing
medium for 12 h, cells were transfected with or without the miRNA
and incubated in complete medium for 48 h. For flow cytometric
analysis, cells were washed twice with 1X phosphate-buffered saline
(PBS), fixed with ice-cold 70% ethanol, resuspended in 1 ml
solution containing 0.4 mM sodium citrate, 25 μg/ml propidium
iodide (PI) and 50 μg/ml RNase. After being stained for 1 h, cells
were analyzed in a FACScan flow cytometer (BD Biosciences) using
ModFIT program (Verity Software House, Topsham, ME, USA).
3′UTR luciferase reporter assay
A fragment of 3′UTR of Med1 or Med31 containing the
putative miR-1 binding site (420–427 or 1059–1069, respectively)
was amplified by PCR using the following primers: wt-Med1
(forward), 5′-GAA AGGCATATCCAGACCCTATT-3′ and wt-Med1 (reverse),
5′-GGAAGGCTGTCCTACACTAAAC-3′; wt-Med31 (forward),
5′-TCTCCTGGAACCTTACTGTCT-3′ and wt-Med31 (reverse),
5′-GCAACTGATGATATTCCTGA AACC-3′. The PCR product was subcloned into
a pMIR-REPORT vector (Ambion) to generate the
pMIR-Report-Med1/Med31 wt plasmid. Site-directed mutagenesis of
miR-1 binding sites was carried out using a QuikChange
site-directed mutagenesis kit (Stratagene, Santa Clara, CA, USA).
All constructs were verified by DNA sequencing.
For reporter assays, U2OS cells were plated in
96-well clusters, then cotransfected with 0.3 μg wt or mutant
reporter plasmids and 60 nM miR-1 precursors. At 48 h after
transfection, luciferase activity was measured and normalized by
the control vector containing Renilla luciferase.
Western blotting
Cultured or transfected cells were lysed using M-PER
protein extraction reagent (Thermo Fisher Scientific Inc., Waltham,
MA, USA) supplemented with complete proteinase inhibitor mixture.
Equal amounts of the extracts were loaded onto an SDS-PAGE,
transferred to a PVDF membrane and then incubated with the
following primary antibodies: mouse monoclonal Med1 and Med31
antibodies (both from Santa Cruz), rabbit monoclonal MET antibody
(Abcam, Cambridge, MA, USA), mouse monoclonal β-actin antibody and
rabbit monoclonal p-Erk1/2 (Thr202/Tyr204) and Erk1/2 antibodies
(all from Cell Signaling Technology, Inc., Danvers, MA, USA). After
incubation with HRP-conjugated secondary antibodies (Cell
Signaling), protein bands were visualized using ECL substrates
(Millipore, Billerica, MA, USA).
Statistical analysis
All data from three independent experiments are
presented as the mean ± SD. Statistical comparisons between two
groups were analyzed using the two-tailed Student’s t-test.
Variance analysis between multiple groups was performed using
one-way ANOVA followed by a Student-Newman-Keuls test. The
correlation between miR-1 and Med1 or Med31 expression was assessed
by Pearson’s correlation test. P-values of <0.05 were considered
to indicate statistically significant results.
Results
miR-1 is downregulated in
osteosarcoma
To determine the clinicopathologic significance of
miR-1 in osteosarcoma development, we evaluated the expression
level of miR-1 in 30 paired clinical human osteosarcoma tissues and
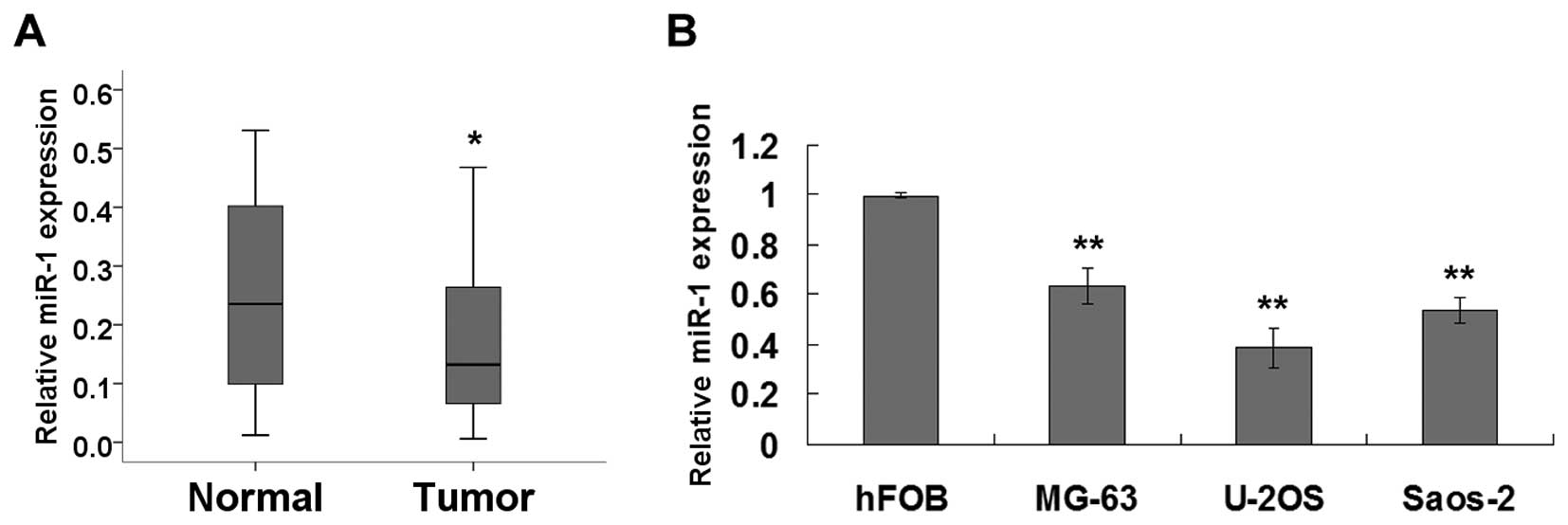
adjacent normal tissue. As shown in Fig. 1A, miR-1 expression was significantly
reduced in the tumor tissues as compared with that in the adjacent
normal tissues. Furthermore, we extended our test to human
osteosarcoma cell lines. Real-time PCR revealed that miR-1
expression was significantly decreased in the MG63, U2OS and Saos-2
osteosarcoma cells compared with the hFOB osteoblast cells
(Fig. 1B).
Restoration of miR-1 inhibits cell
proliferation
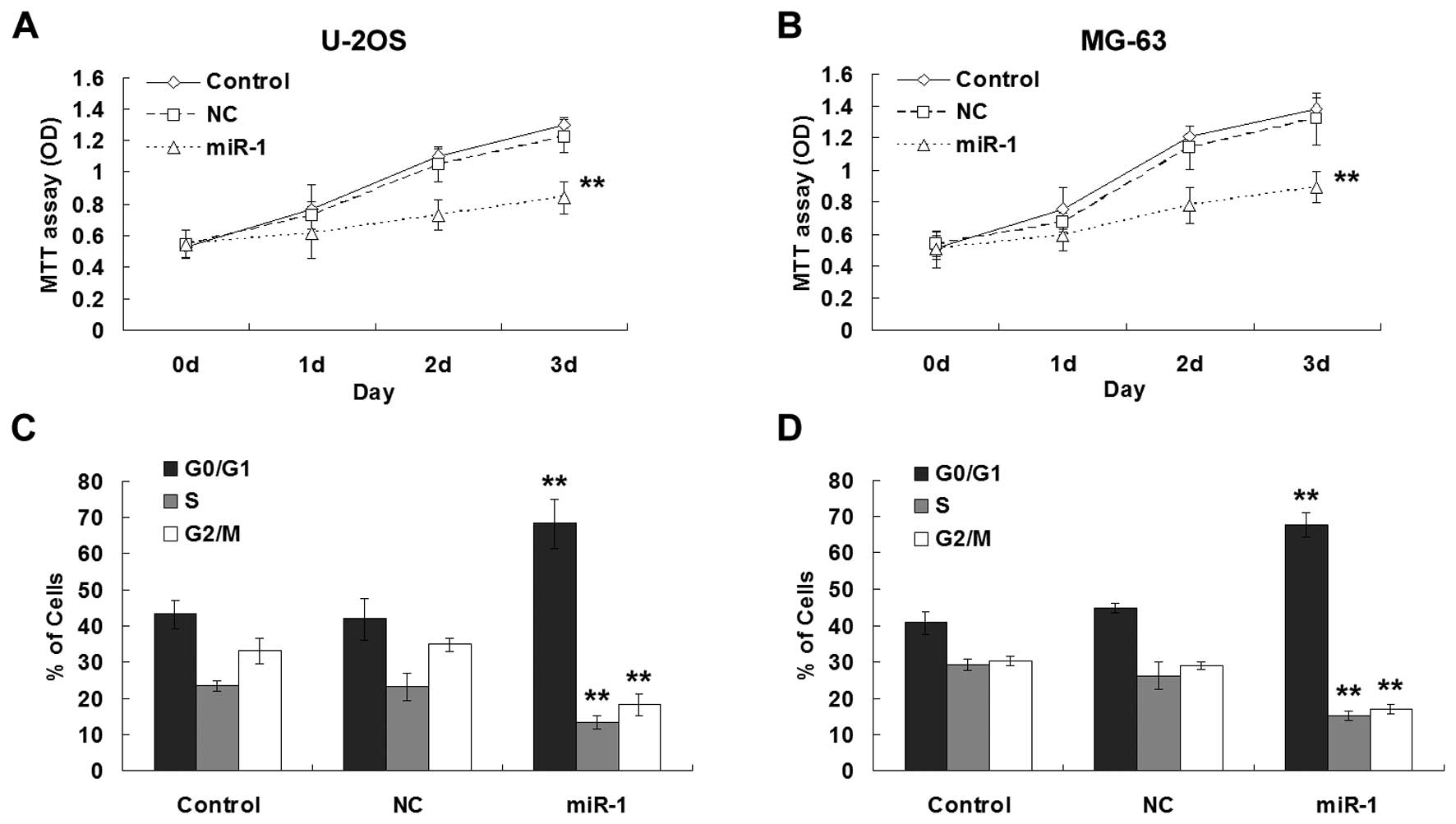
To investigate whether miR-1 functions as a tumor
suppressor in osteosarcoma, we assessed the effect of miR-1 on
osteosarcoma cell growth. In the U2OS and MG63 cells, transfection
of miR-1 mimics significantly inhibited cell proliferation
(Fig. 2A and B). Furthermore,
analysis of DNA uptake by flow cytometry showed that miR-1
decreased the S and G2/M phase populations, with a concomitant
increase in the proportion of cells in the G0/G1 phase in both
osteosarcoma cell lines (Fig. 2C and
D).
miR-1 directly targets Med1 and
Med31
To further investigate the molecular mechanism, we
aimed to identify the molecular targets of miR-1. Among the
predicted targets of miR-1 from three programs, including miRDB,
TargetScan and microrna.org, we were interested in Med1 and Med31.
Both of them associate with the mediator complex (MED), which
functions as a component of the RNA polymerase II-mediated
transcription machinery and plays an important role in
transcription activation of eukaryotes (17).
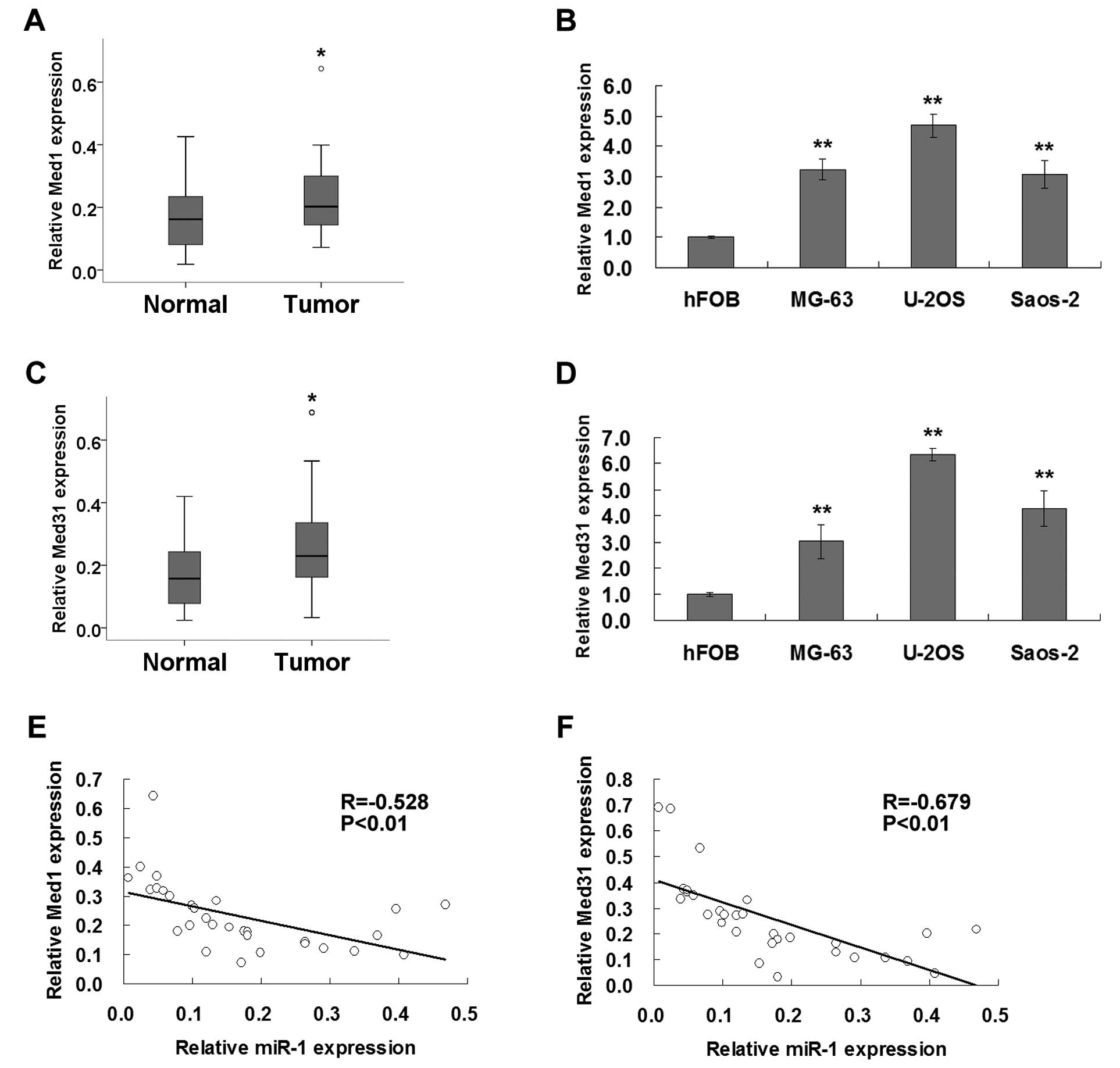
We next tested the expression of Med1 and Med31 in
clinical tissues and cell lines using real-time PCR. As shown in
Fig. 3A–D, the expression levels of
both Med1 and Med31 in the osteosarcoma tissues or osteosarcoma
cell lines were significantly higher than the levels in normal
tissue or osteoblast cells. Correlations between expression levels
of miR-1 and Med1 or Med31 were further examined in primary human
osteosarcoma tissues. Pearson’s correlation analysis suggested that
the expression levels of Med1 and Med31 were both significantly
inversely correlated with miR-1 expression in the osteosarcoma
tissues (Fig. 3E and F).
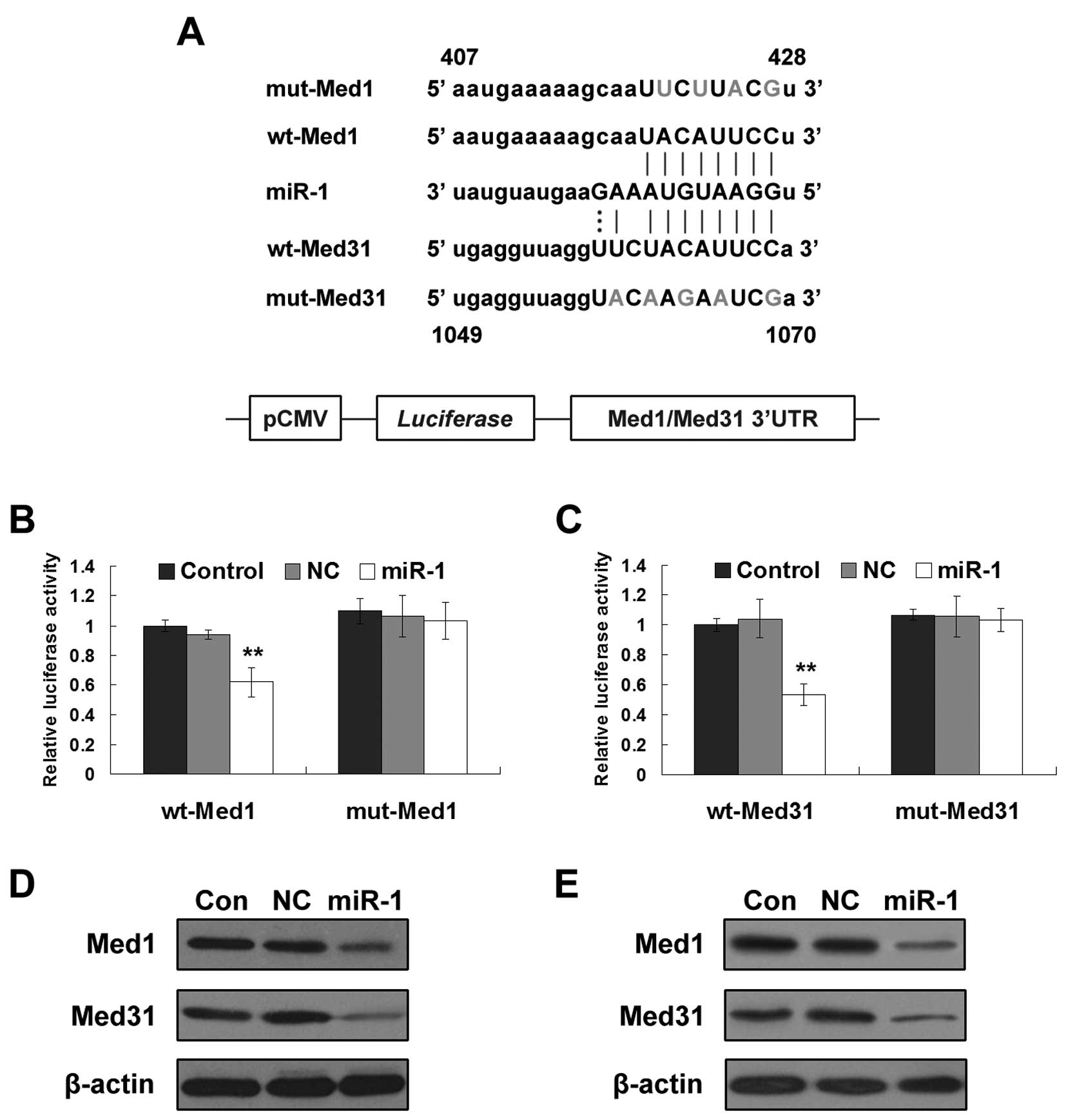
To verify whether miR-1 directly targets the 3′UTR
of Med1 and Med31, a dual-luciferase reporter system was employed.
miR-1 and luciferase reporter plasmids containing wild-type or
mutated miR-1 binding sites in 3′UTR of Med1 or Med31 (Fig. 4A) were co-transfected into U2OS
cells. Overexpression of miR-1 significantly suppressed the
activity of the luciferase reporter containing wild-type Med1 or
Med31 3′UTR, but not the activity of a reporter containing mutant
Med1 or Med31 3′UTR (Fig. 4B and
C). These data suggest that both Med1 and Med31 are directly
targeted by miR-1. Moreover, enhanced miR-1 suppressed endogenous
protein expression of Med1 and Med31 in the U2OS and MG63 cells
(Fig. 4D and E).
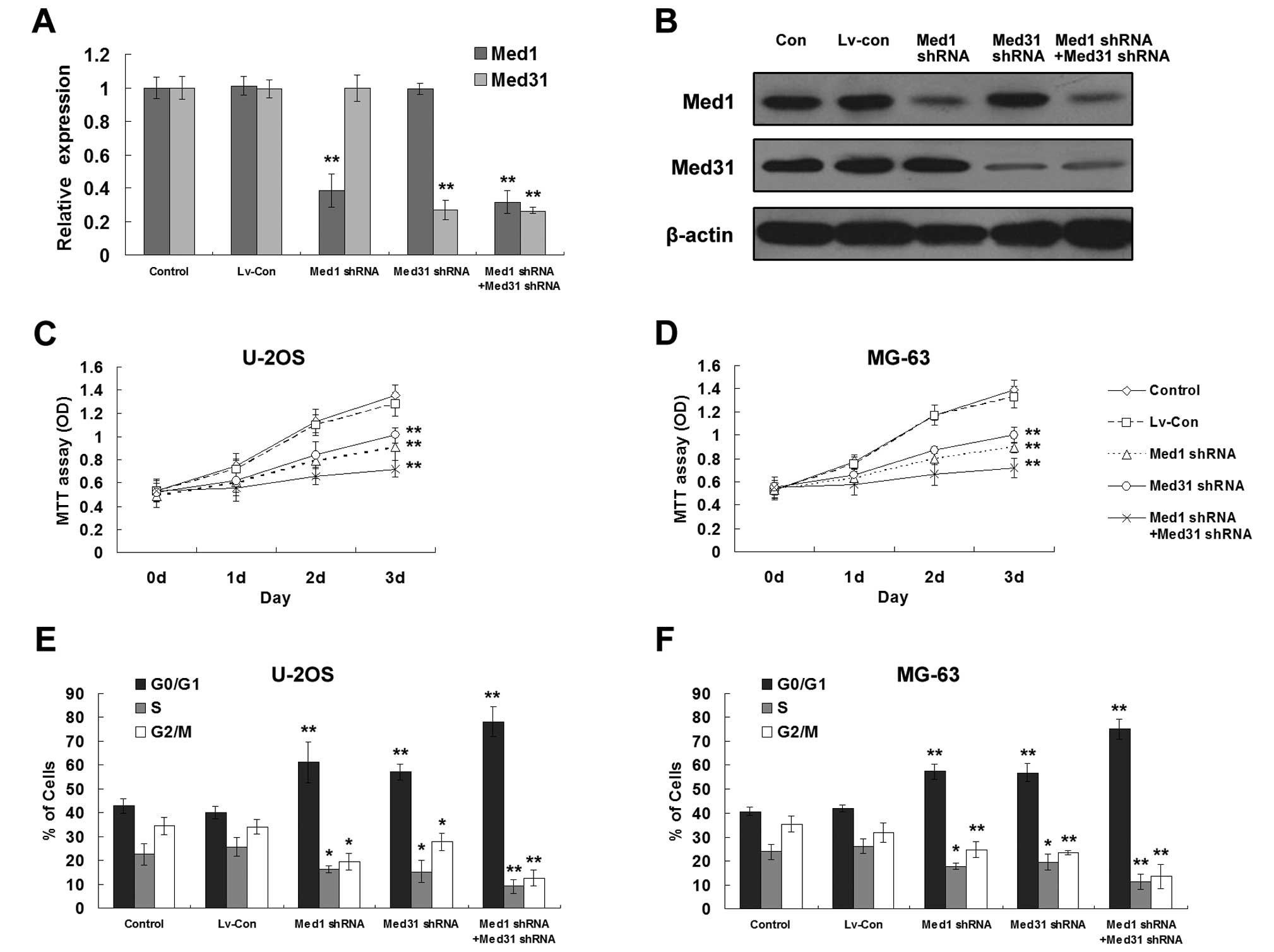
Downregulation of Med1 and/or Med31
suppresses the proliferation of osteosarcoma cells
To elucidate the potential role of Med1 and Med31 in
osteosarcoma development, we established stable Med1 and/or
Med31-silenced osteosarcoma cell lines (Fig. 5A and B). MTT assay showed that
separate or simultaneous silencing of Med1 and Med31 significantly
inhibited proliferation of both U2OS and MG63 cells (Fig. 5C and D). Similar to induction of
miR-1, reduced expression of Med1 and/or Med31 blocked the cell
cycle progression at the G0/G1 phase (Fig. 5E and F).
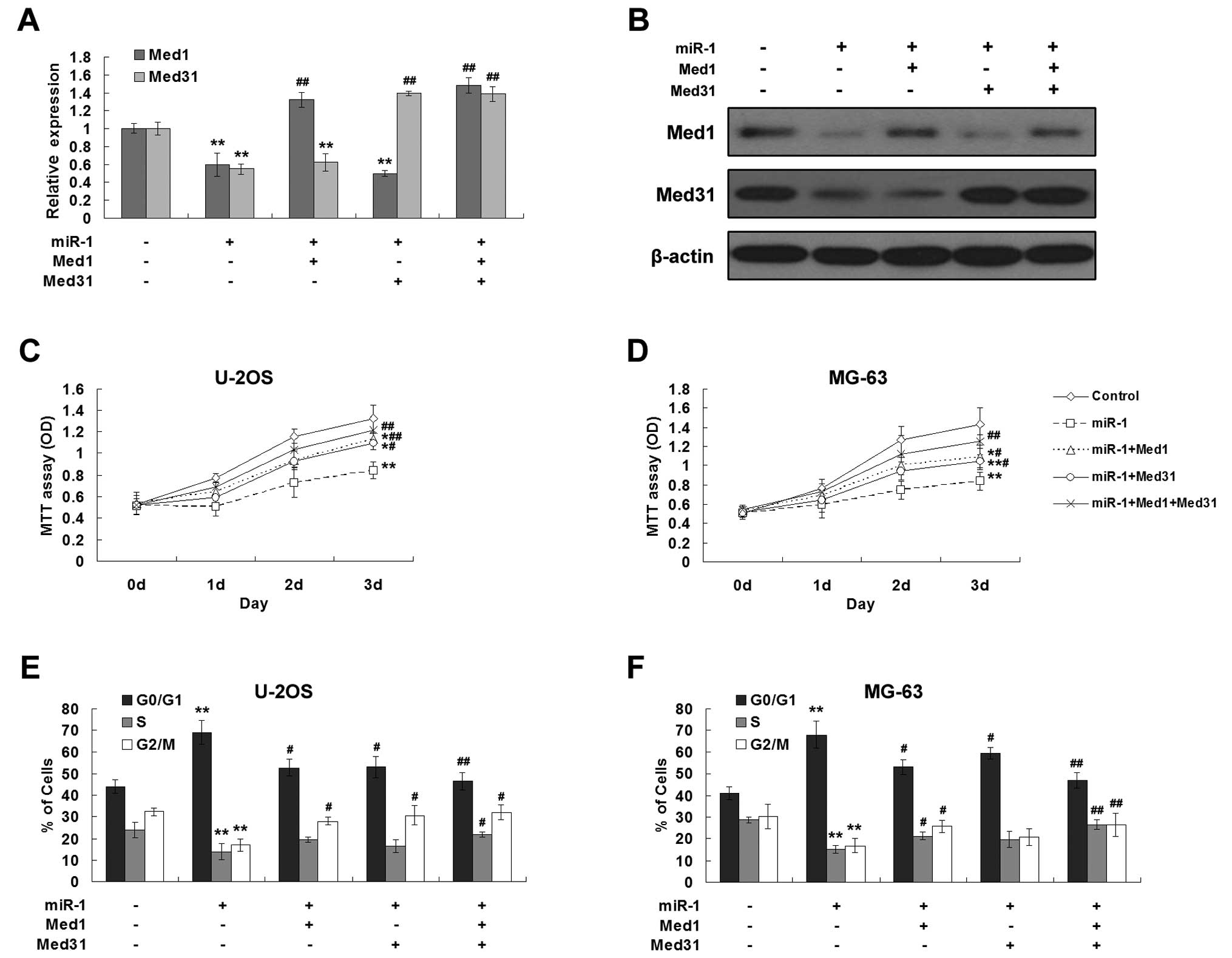
Overexpression of Med1 and/or Med31
abrogates the effects of miR-1
To further confirm that the tumor suppressive effect
of miR-1 is mediated by supression of Med1 and Med31 in
osteosarcoma cells, recombinant Ad-Med1 and Ad-Med31 were used to
infect the U2OS and MG63 cells before miR-1 transfection. The
decreased levels of Med1 and/or Med31 by miR-1 were significantly
rescued via the infection of recombinant adenoviruses (Fig. 6A and B). Similarly, ectopic
expression of Med1 and/or Med31 abrogated the growth suppressive
effect induced by miR-1 in both U2OS and MG63 cells (Fig. 6C and D). Meanwhile, the restoration
of expression of Med1 and Med31 significantly increased the
proportion of cells in the S and G2/M phases (Fig. 6E and F). These data suggest that the
growth suppressive effects of miR-1 were chiefly through inhibition
of Med1 and Med31.
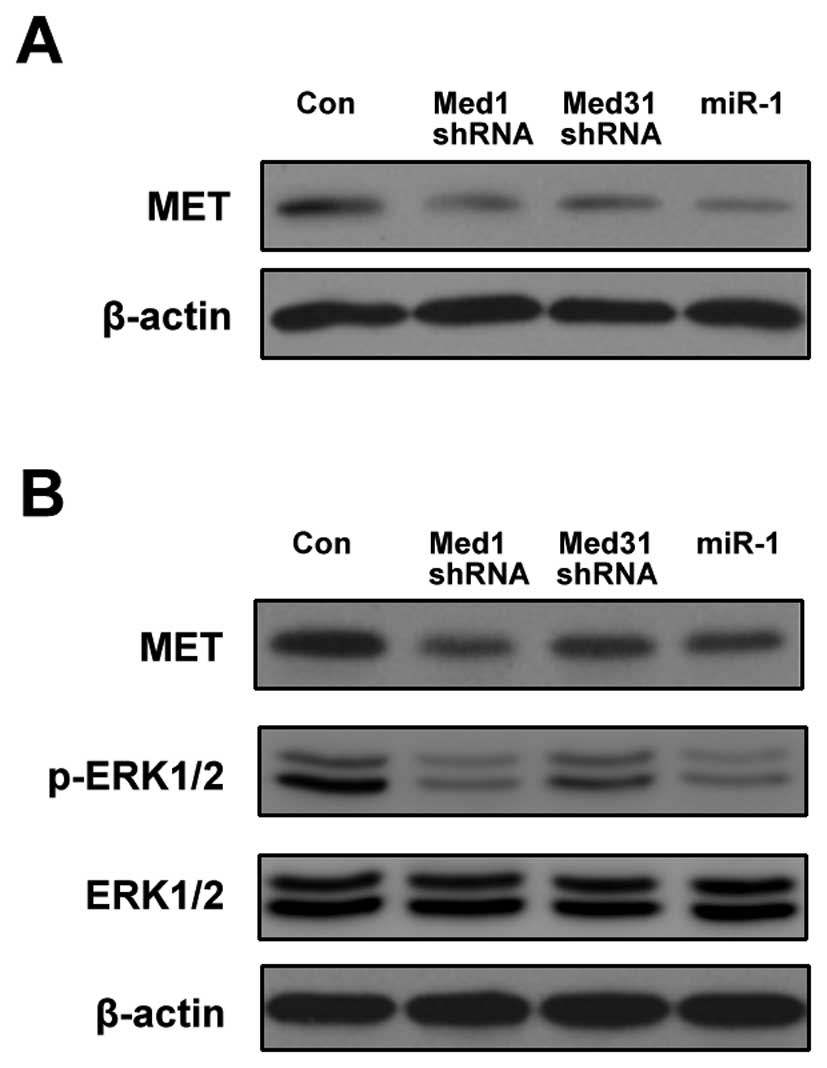
Med1 and Med31 modulate MET
signaling
To further investigate the mechanism involved in the
effect on cell proliferation by Med1 and Med31, we focused on MET
signaling. Novello et al (12) showed that miR-1 could reduce the
expression of MET in U2OS cells, implying that miR-1 modulates MET
signaling in osteosarcoma. In contrast, Med1 deficiency resulted in
the reduction of MET mRNA level and the block of the response to
hepatocyte growth factor (HGF)/scatter factor (SF) in hepatocytes
(18).
As shown in Fig. 7A,
the downregulation of either Med1 or Med31 suppressed the
expression of MET. Even in the presence of HGF, the expression of
MET was still decreased by Med1/Med31 shRNA compared with the
control (Fig. 7B). The
phosphorylation of ERK1/2, a downstream target of MET, was also
inhibited by Med1/Med31 shRNA (Fig.
7B). miR-1 exhibited effects similar to Med1/Med31 shRNA
(Fig. 7A and B).
Discussion
In recent years, studies on the molecular mechanisms
contributing to osteosarcoma carcinogenesis have revealed the
critical role of miRNAs in the process. Several miRNAs have been
found deregulated and related to osteosarcoma development (19). However, the detailed roles of these
miRNAs in osteosarcoma progression are largely unknown. Here, we
found that miR-1 was downregulated in osteosarcoma and targets Med1
and Med31. Both Med1 and Med31 were involved in the proliferation
of osteosarcoma cells through influencing cell cycle progression.
Furthermore, MET signaling may be an important downstream target of
Med1 and Med31.
Previously studies have found that the expression
levels of miR-1 are significantly reduced in lung cancer (13), colorectal cancer (20) and rhabdomyosarcoma (21). DNA methylation may partly explained
miR-1 silencing (14). Our study
showed that miR-1 was downregulated in osteosarcoma and restoration
of miR-1 reduced cell proliferation, which was consistent with the
findings of other researchers (11,12).
Although several target genes of miR-1 have been validated in other
cancers (13–16), no explicit target has been confirmed
in osteosarcoma. In the present study, we found two new targets of
miR-1, Med1 and Med31. Notably, both are subunits of MED.
MED is a multiprotein complex which plays a critical
role in the regulation of eukaryotic mRNA synthesis through direct
interactions with RNA Pol II and other transcriptional regulators,
such as activators and transcription factors (17,22).
To date, 30 distinct MED subunits (MEDs) have been found and
different MED subunits may interact with the activation domain of
different activators (17,23). Recent studies have suggested that a
number of subunits, other than the complex itself, have a role in
tumorigenesis. The deregulation of MEDs has been found in a variety
of tumors including osteosarcoma (24–26).
Med1 has equivocal functions in different tumors. It functions as a
tumor-suppressor gene and inhibits invasion and metastasis in lung
carcinomas (27) and melanoma cells
(28). Conversely, deficiency of
Med1 protects hepatocytes from chemical carcinogen-induced
hepatocarcinogenesis (29), and
loss of Med1 significantly decreased proliferation of prostate
cancer cells (30). Med31 has not
been found to be associated with tumorigenesis, but a recent study
suggests that it is required for cell proliferation during
mammalian development (31).
We found that both Med1 and Med31 were upregulated
in osteosarcoma tissues or cell lines when compared with normal
tissue or cells. Although Med1 and Med31 are located in different
modules of MED (17), they showed
similar effects in osteosarcoma progression. Downregulation of both
Med1 and Med31 or either of them suppressed the proliferation of
osteosarcoma cells, suggesting that they function as oncogenes in
osteosarcoma. Both Med1 and Med31 were identified to be new direct
targets of miR-1 in osteosarcoma, and overexpression abrogated the
effects of miR-1, suggesting that miR-1 exerts its antitumor
function via inhibition of Med1 and Med31 expression.
We further investigated the potential signaling
pathway linking these Meds and cell proliferation and found that
MET was a possible pathway. The Met gene encodes the tyrosine
kinase receptor for HGF/SF. Met activation may induce
proliferation, angiogenesis or stimulate motility to form
micrometastases in tumor (32).
Loss of Med1 resulted in the reduction of MET mRNA level in
hepatocytes, suggesting Med1 could induce the expression of MET.
Our data showed that the downregulation of either Med1 or Med31
suppressed the expression of MET and blocked the downstream
signaling of MET responding to HGF. In combination with previous
studies in which MET was identified as a target of miR-1 in other
types of cancer (13,14), MET signaling may play a crucial role
in osteosarcoma cell proliferation and may be regulated by miR-1
directly or through Med1 and Med31 indirectly.
In conclusion, taken together, our results
demonstrate that miR-1 has great biological effect on the
proliferation of osteosarcoma cells. This effect of miR-1 was
mediated by the direct inhibition of Med1 and Med31. Both Med1 and
Med31 were overexpressed in osteosarcoma and downregulation of Med1
and Med31 suppressed the proliferation of osteosarcoma cells.
Furthermore, MET signaling may be involved in osteosarcoma cell
proliferation regulated by miR-1. The present findings suggest that
MEDs may serve as potential gene therapeutic targets in
osteosarcoma and miR-1 may prove to be a promising agent.
References
|
1
|
Longhi A, Errani C, De Paolis M, Mercuri M
and Bacci G: Primary bone osteosarcoma in the pediatric age: state
of the art. Cancer Treat Rev. 32:423–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chou AJ, Geller DS and Gorlick R: Therapy
for osteosarcoma: where do we go from here? Paediatr Drugs.
10:315–327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferguson WS and Goorin AM: Current
treatment of osteosarcoma. Cancer Invest. 19:292–315. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yan K, Gao J, Yang T, et al: MicroRNA-34a
inhibits the proliferation and metastasis of osteosarcoma cells
both in vitro and in vivo. PLoS One. 7:e337782012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jin Y, Peng D, Shen Y, et al:
MicroRNA-376c inhibits cell proliferation and invasion in
osteosarcoma by targeting to transforming growth factor-alpha. DNA
Cell Biol. 32:302–309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ji F, Zhang H, Wang Y, et al:
MicroRNA-133a, downregulated in osteosarcoma, suppresses
proliferation and promotes apoptosis by targeting Bcl-xL and Mcl-1.
Bone. 56:220–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Townley-Tilson WH, Callis TE and Wang D:
MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac
muscle development, function, and disease. Int J Biochem Cell Biol.
42:1252–1255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nohata N, Hanazawa T, Enokida H and Seki
N: microRNA-1/133a and microRNA-206/133b clusters: dysregulation
and functional roles in human cancers. Oncotarget. 3:9–21.
2012.PubMed/NCBI
|
|
11
|
Namlos HM, Meza-Zepeda LA, Baroy T, et al:
Modulation of the osteosarcoma expression phenotype by microRNAs.
PLoS One. 7:e480862012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Novello C, Pazzaglia L, Cingolani C, et
al: miRNA expression profile in human osteosarcoma: Role of miR-1
and miR-133b in proliferation and cell cycle control. Int J Oncol.
42:667–675. 2013.PubMed/NCBI
|
|
13
|
Nasser MW, Datta J, Nuovo G, et al:
Down-regulation of micro-RNA-1 (miR-1) in lung cancer: suppression
of tumorigenic property of lung cancer cells and their
sensitization to doxorubicin-induced apoptosis by miR-1. J Biol
Chem. 283:33394–33405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Datta J, Kutay H, Nasser MW, et al:
Methylation mediated silencing of microRNA-1 gene and its role in
hepatocellular carcinogenesis. Cancer Res. 68:5049–5058. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leone V, D’Angelo D, Rubio I, et al: MiR-1
is a tumor suppressor in thyroid carcinogenesis targeting CCND2,
CXCR4, and SDF-1alpha. J Clin Endocrinol Metab. 96:E1388–E1398.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu CD, Kuo YS, Wu HC and Lin CT:
MicroRNA-1 induces apoptosis by targeting prothymosin alpha in
nasopharyngeal carcinoma cells. J Biomed Sci. 18:802011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Napoli C, Sessa M, Infante T and
Casamassimi A: Unraveling framework of the ancestral mediator
complex in human diseases. Biochimie. 94:579–587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsumoto K, Yu S, Jia Y, et al: Critical
role for transcription coactivator peroxisome
proliferator-activated receptor (PPAR)-binding protein/TRAP220 in
liver regeneration and PPARalpha ligand-induced liver tumor
development. J Biol Chem. 282:17053–17060. 2007. View Article : Google Scholar
|
|
19
|
Miao J, Wu S, Peng Z, Tania M and Zhang C:
MicroRNAs in osteosarcoma: diagnostic and therapeutic aspects.
Tumour Biol. 34:2093–2098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sarver A, French A, Borralho P, et al:
Human colon cancer profiles show differential microRNA expression
depending on mismatch repair status and are characteristic of
undifferentiated proliferative states. BMC Cancer. 9:4012009.
View Article : Google Scholar
|
|
21
|
Rao PK, Missiaglia E, Shields L, et al:
Distinct roles for miR-1 and miR-133a in the proliferation and
differentiation of rhabdomyosarcoma cells. FASEB J. 24:3427–3437.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ansari SA and Morse RH: Mechanisms of
mediator complex action in transcriptional activation. Cell Mol
Life Sci. 70:2743–2756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Collins SR, Miller KM, Maas NL, et al:
Functional dissection of protein complexes involved in yeast
chromosome biology using a genetic interaction map. Nature.
446:806–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang H, Jiang H, Wang W, et al:
Expression of Med19 in bladder cancer tissues and its role on
bladder cancer cell growth. Urol Oncol. 30:920–927. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoon NK, Maresh EL, Elshimali Y, et al:
Elevated MED28 expression predicts poor outcome in women with
breast cancer. BMC Cancer. 10:3352010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schiano C, Rienzo M, Casamassimi A and
Napoli C: Gene expression profile of the whole mediator complex in
human osteosarcoma and normal osteoblasts. Med Oncol. 30:7392013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gade P, Singh AK, Roy SK, Reddy SP and
Kalvakolanu DV: Down-regulation of the transcriptional mediator
subunit Med1 contributes to the loss of expression of
metastasis-associated dapk1 in human cancers and cancer cells. Int
J Cancer. 125:1566–1574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
de Ndong JL, Jean D, Rousselet N and Frade
R: Down-regulation of the expression of RB18A/MED1, a cofactor of
transcription, triggers strong tumorigenic phenotype of human
melanoma cells. Int J Cancer. 124:2597–2606. 2009.PubMed/NCBI
|
|
29
|
Matsumoto K, Huang J, Viswakarma N, et al:
Transcription coactivator PBP/MED1-deficient hepatocytes are not
susceptible to diethylnitrosamine-induced hepatocarcinogenesis in
the mouse. Carcinogenesis. 31:318–325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vijayvargia R, May MS and Fondell JD: A
coregulatory role for the mediator complex in prostate cancer cell
proliferation and gene expression. Cancer Res. 67:4034–4041. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Risley MD, Clowes C, Yu M, Mitchell K and
Hentges KE: The mediator complex protein Med31 is required for
embryonic growth and cell proliferation during mammalian
development. Dev Biol. 342:146–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao CF and Woude GF: HGF/SF-Met signaling
in tumor progression. Cell Res. 15:49–51. 2005. View Article : Google Scholar : PubMed/NCBI
|