1. Clinical landscape
Malignant melanoma is responsible for ~75% of all
skin cancer-related deaths with 250,000 new cases diagnosed yearly
worldwide. The 5-year survival rate is ~95% for patients diagnosed
with stage I disease and markedly falls to <10% for patients
with metastatic stage IV disease (1). Until recently, available treatments
were limited. The most commonly used chemotherapeutic agent to
treat patients with advanced melanoma has for many years been
dacarbazine, an alkylating agent licenced in 1975 to treat
unresectable stage IV disease. Dacarbazine works by inducing cell
cycle arrest and tumour cell apoptosis and treatment results in a
response rate of ~15–20% and median response duration of ~4 months
(2). Immunotherapeutic strategies
in the form of the recombinant cytokines IL-2 and IFNα, although
not universally applied in standard practice due to their
side-effects, support the notion that activating immunity in cancer
may have a positive clinical impact (3). The recent regulatory approval of two
classes of agents has transformed clinical care: the pathway
inhibitor small drugs vemurafenib, dabrafenib and trametinib; and a
monoclonal antibody, known as ipilimumab, that targets the T cell
checkpoint molecule CTLA4 to promote T cell activation (Table I) (4–7).
 | Table IFood and drug administration-approved
agents for the treatment of malignant melanoma. |
Table I
Food and drug administration-approved
agents for the treatment of malignant melanoma.
| Agent (brand
name) | Year of
approval | Specificity | Class | Mechanisms of
action |
|---|
| Dacarbazine
(DTIC-Dome®) | 1975 | Non-specific | Chemotherapy | Alkylating agent
leading to DNA damage, inducing cell cycle arrest and tumour cell
apoptosis |
| IFNα2b
(INTRON®A) | 1995 | IFNα receptor 1 and
2 | Immunotherapy
(cytokine) | Multifunctioning
immunoactivatory cytokine enhances antitumoural response,
anti-angiogenic, anti-proliferative and pro-apoptotic
properties |
| High dose IL-2
(Aldesleukin, Proleukin®) | 1998 | IL-2 receptor
expressed on lymphocytes | Immunotherapy
(cytokine) | Immune activating,
increases activation and proliferation of immune cells (e.g. T, NK,
B cells) |
| Pegylated IFNα2b
(PEG INTRON®A) | 2011 | IFNα receptor 1 and
2 | Immunotherapy
(cytokine) | Modified
(pegylated) form of IFNα2b with increased half-life and enhanced
therapeutic efficacy |
| Ipilimumab
(Yervoy®) | 2011 | CTLA4 expressed on
T cells | Immunotherapy
(mAb) | Humanised mAb
targeting the inhibitory receptor CTLA4 activates immune system
enhancing T cell activation and targeting CTLA4-expressing
Tregs |
| Verumafenib
(Zelboraf®) | 2011 | BRAF V600E, mutated
form of BRAF | Small molecule
inhibitor | Blocks
mitogen-activated protein kinase pathway reducing protein
proliferation of melanoma cells carrying mutation |
| Dabrafenib
(Tafinlar®) | 2013 | BRAF V600E mutated
form of BRAF protein | Small molecule
inhibitor | Blocks
mitogen-activated protein kinase pathway reducing proliferation of
melanoma cells carrying mutation |
| Trametinib
(Mekinist®) | 2013 | BRAF V600E or V600K
mutated forms of BRAF protein | Small molecule
inhibitor | Blocks
mitogen-activated protein kinase pathway reducing proliferation of
melanoma cells carrying mutation |
Insights from the interplay with immune
responses
Melanoma has for decades been considered an
immunogenic tumour. The presence of specific immune cell subsets
such as T lymphocyte infiltrates within melanoma lesions is thought
to correlate with favourable patient outcomes (8). Clinical observations of partial or
whole regressions of melanoma lesions, clinical evidence of
spontaneous complete remissions, the increased prevalence of
melanoma in immunocompromised patients and the mixed success of
immune-based therapies dating back over many years, further support
an important role for the tumour-immunity interface in
melanoma.
Melanoma cells are thought to potentiate an
inflammatory-based response constituting various cell subsets such
as cytotoxic T cells (CTLs), dendritic cells (DCs), macrophages,
neutrophils, mast cells, T and B lymphocytes and also an array of
cytokines and antibodies within the tumour and systemically
(9–11). However, local and systemic immunity
often fails to stem cancer progression. It has been postulated that
the immune system struggles to balance mounting a sufficient immune
response against altered self-proteins expressed on tumours on the
one hand, while aiming to avoid unwanted responses against self, on
the other (12). Tumour-associated
immunosuppressive components such as regulatory T cells (Tregs),
myeloid-derived suppressor cells (MDSCs) and alternatively
activated (M2d) macrophages, along with mediators such as IL-10,
transforming growth factor (TGF-β) and vascular endothelial growth
factor (VEGF) promote inflammation and immune suppression,
diverting T and B cell responses and antibody expression in favour
of inactive or suppressive subclasses such as IgG4 and preventing
potentially cytotoxic T cells, DC and macrophages from launching
potent antitumoural functions (13–16).
Therefore, interactions between tumours and immunity are often
tipped in favour of tumour growth.
The limited positive impact of standard
chemotherapeutics on advanced disease survival and evidence that
host immunity is capable of detecting the presence of tumours have
long provided the motivation to design new therapy approaches,
including those with the capacity to activate or redirect immune
responses against melanoma.
Cytokine therapies
Early efforts to harness immunity focused on
recombinant cytokines IFNα2b and IL-2 (3,17).
IL-2 was used on the basis that it could have antitumour effects by
inducing expansion of tumour-specific T cell populations (18). However, treatment with high dose
IL-2 for advanced disease resulted in an objective response rate of
5–27% and complete responses of up to 4% of patients in randomised
controlled trials (19). High dose
treatments have serious side-effects, such as hepatic and renal
toxicities and high mortality rates (Table I) (20).
IFNα, a member of the type I-IFN family, is an
antiviral and powerful pro-inflammatory cytokine produced by a wide
variety of cells (including T cells, NK cells, macrophages). Shown
to inhibit proliferation of B16 murine melanoma cells in
vivo and in vitro, it is thought to have antitumoural
effects via anti-angiogenic and pro-apoptotic functions and by
enhancing tumour antigen presentation and potentiating
CD4+ T cell-mediated tumour cell targeting (21,22).
High doses of IFNα have been shown to prolong disease-free survival
and, when used in the adjuvant setting, the agent could increase
overall survival in high-risk patients (23), although treatment was not
demonstrated to result in significant survival improvements when
compared with chemotherapy in randomised trials. Although approved
for clinical use and used in the adjuvant setting in Europe, INFα
is not standard of care in the United Kingdom (24,25).
Recent investigations have shown that a modified form of IFNα
(pegylated IFNα2β) with a longer half-life in vivo has
enhanced therapeutic efficacy and improved tolerability, leading to
FDA approval as an adjuvant therapy in 2011 (Table I) (26).
Vaccine approaches
Vaccination strategies have been investigated using
peptides, proteins, cells, DNA and viral vaccines or various forms
of modified cell therapies such as adoptive DC and T cell
therapies; a DC therapy is available for the treatment of prostate
cancer, offering hope for similar treatments in melanoma (27). Tumour cells used as immunogens,
melanoma peptide and protein recombinant antigens or DNA viral
vector vaccines have been designed to stimulate different
components of the immune response and these efforts continue to
date (28). A recent study showed
induction of immunity against metastatic melanoma through
vaccination with mature DCs loaded with melanoma antigens (MART-1,
MAGE-3, gp100 and tyrosinase) processed through melanoma
constitutive proteasomes for presentation by MHC class I to cognate
T cells. Treatment enhanced Ag-specific T cell responses and
reduced levels of circulating tumour cells in all patients
(29). A phase I study of a murine
gp100 DNA vaccine in malignant melanoma patients showed that the
delivery of xenogeneic melanoma antigens (Tyr, gp100) can activate
a specific CTL response to these proteins, with low associated
toxicity and gp100-reactive T cell responses were reported in some
patients, but without improving median survival (30). Although limited by their individual
patient-specific nature, reliance on high expertise and high
associated costs, adoptive cell strategies are now accelerated by
the emergence of new technologies. To date, however, vaccines
demonstrating clinical benefits have not reached clinical utility
in melanoma.
2. Monoclonal antibodies
Antibody-based agents have been increasingly used as
therapies for a wide range of human malignancies, including some
solid tumour indications such as breast, colorectal and lung
cancers (31). Antibodies can exert
their antitumoural functions directly by specific recognition of
cell surface antigen-expressing target cells, such as signalling
proliferation arrest, inducing apoptosis, blocking cytokine
receptor interactions to starve tumour cells of vital growth
signals, or preventing tumour cell-extracellular matrix
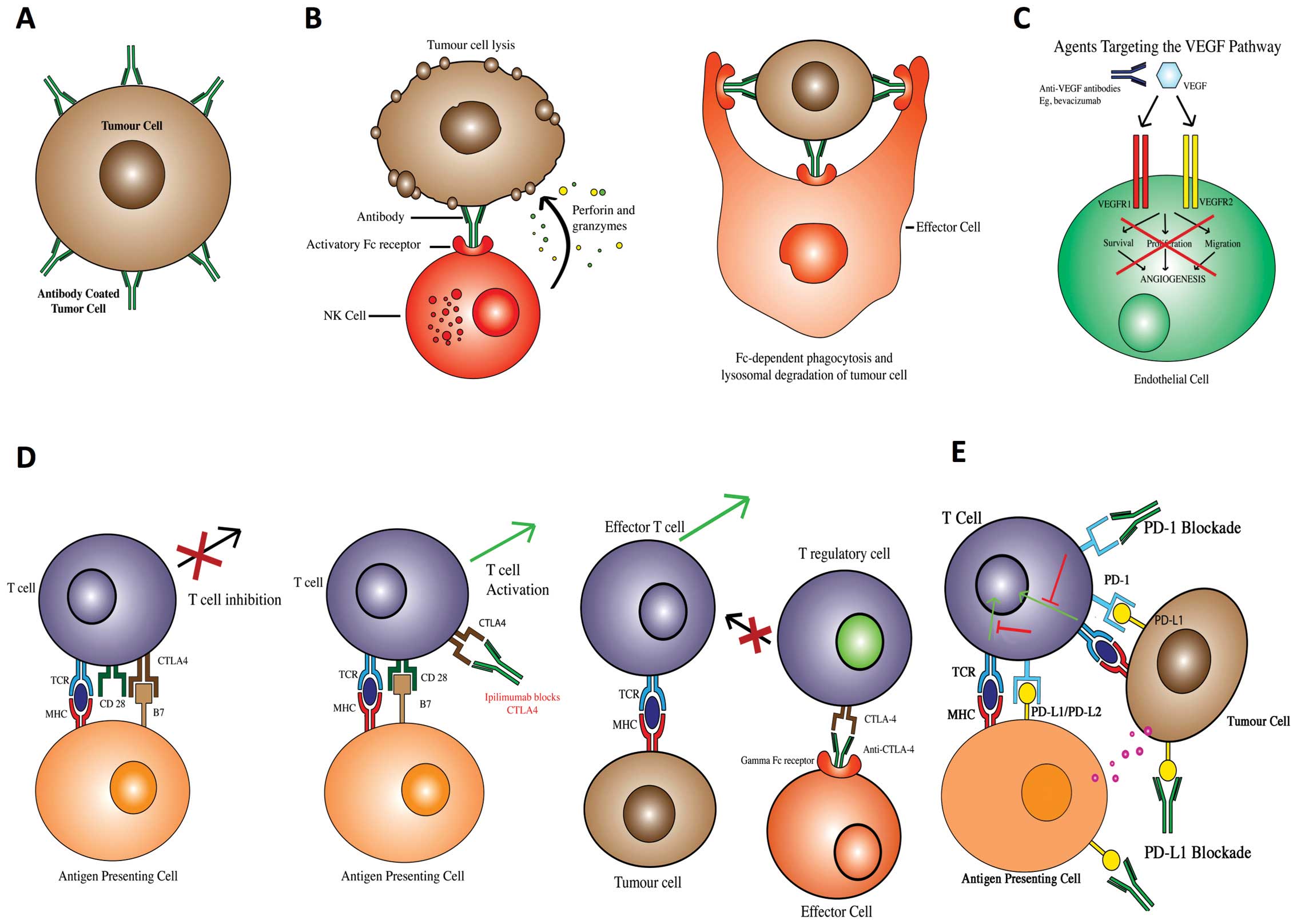
interactions to restrict migration and metastasis (Fig. 1A). Antibodies can also link target
antigen expressing cells (such as tumour cells) with immune
effector cells bearing Fc receptors, potentiating effector cell
activation and target-neutralising functions (Fig. 1B) by engendering antibody-dependent
effector cell-mediated cytotoxicity (ADCC), phagocytosis (ADCP) or
complement activation (CDC). Antibodies can also be used as
immunogens, to promote antigen presentation and initiate adaptive
immune responses against cancer cells, or by targeting key elements
of immune modulatory pathways to overcome effector cell anergy.
Another function may entail targeting critical events in the tumour
microenvironment, such as VEGFs to inhibit angiogenesis,
restricting tumours of vital nutrient supply and/or escape of
metastatic cells into the circulation (Fig. 1C).
Studies on the merits of antibody therapies for the
treatment of melanoma date back a few decades, but the clinical
utility of this modality has only recently been demonstrated with
the approval of the first antibody in 2011. Renewed attention is
now focused towards melanoma immunotherapies including monoclonal
antibodies that can target key cancer pathways and activate
immunity (32). Here, we discuss
examples of different antibody therapeutic strategies studied for
melanoma.
Targeting cell surface antigens on tumour
cells: CSPG4
A widely used approach for antibody therapies
entails selective recognition of specific molecules such as
proteins, sugar or lipid moieties, which are overexpressed or
mutated on the surface of cancer cells. In the context of melanoma,
a notable example of this strategy includes antibodies designed to
recognise the tumour-associated antigen chondroitin sulphate
proteoglycan 4 (CSPG4). Otherwise known as melanoma-associated
chondroitin sulphate proteoglycan (MCSP) or human high molecular
weight-melanoma associated antigen (HMW-MAA), CSPG4 is a
2322-residue membrane-bound protein overexpressed on 80–85% of
melanoma lesions and also on a proportion of basal cell and breast
carcinomas and several leukaemia types (33,34).
CSPG4 is known to play roles in the adhesion,
spreading and migration of melanoma cells through activation of
intracellular signalling cascades such as the focal adhesion kinase
(FAK), PI3K/AKT, NFκB and MAPK/ERK 1/2 pathways, promoting
sustained high levels of activatory signals required for malignant
progression (35–37). In addition to melanoma cells, CSPG4
is expressed at high levels in tumour angiogenic vasculature,
providing the potential for a targeted therapy to also restrict
tumour blood supply (38).
Overexpression in melanomas at different stages, restricted tissue
distribution in normal tissues and a central role in cancer cell
motility, metastasis and tissue invasion, render CSPG4 an
attractive therapeutic target for applications, including
monoclonal antibodies that may be used in adjuvant and in advanced
disease settings.
Monoclonal antibodies and antibody fragments have
been examined with the view to develop targeted therapies against
CSPG4-expressing tumours. Antibodies conjugated to radioisotopes or
toxins (purothionin, pseudomonas exotoxin A, methotrexate) could
induce tumour cell clearance by antibody-targeted delivery of these
moieties to melanoma cells. Another early approach aimed at
overcoming immunological unresponsiveness to CSPG4 entailed
designing anti-idiotypic antibodies to act as immunogens by
mimicking tumour antigen epitopes known to be recognised by patient
humoral responses. Early clinical trials reported the development
of anti-CSPG4 antibodies associated with prolonged survival of
melanoma patients who responded to antibody therapy (39–43).
Host anti-CSPG4 antibodies were detected in ~60% of patients after
treatment with an anti-idiotypic antibody in conjunction with an
adjuvant (BCG) and those individuals who developed antibodies also
showed longer median survival (42–44).
A humanised bi-specific BiTE antibody (bi-specific
T-cell engaging), able to bind both CSPG4 and human CD3 to engage
the T cell receptor (TCR) complex, was designed to redirect
CD3+ T cells (especially CTL) against melanoma cells
(45). This strategy aimed at
triggering T cell activation in the absence of specific T cell
clones or antigen presentation by APC. When peripheral blood
mononuclear cells (PMBCs) from healthy donors co-cultured with
melanoma cells were treated with MCSP-BiTE antibodies at different
doses, PBMCs lysed CSPG4-expressing melanoma cells in an
antigen-specific manner. Notably, similar cytotoxic activity was
observed with T cells from melanoma patients, although the
proportions of CD3+ T cells in patient blood were lower
compared to those from healthy volunteers.
An anti-CSPG4 scFv antibody fragment fused with
human TRAIL (TNF-related apoptosis-inducing ligand) was designed to
deliver pro-apoptotic TRAIL-signalling activity when bound to
CSPG4+ melanoma cells, while inhibiting antitumourigenic
signalling via CSPG4 downstream signal blocking. Previous studies
showed the inhibition of CSPG4+ melanoma cells in
vitro and in vivo with this agent and reported that
antitumoural activity was enhanced in the presence of the sigma
receptor (σR) rimcazole, also known to have selective antitumoural
activity (46,47). A more recent approach entails
engineered T cells with a CSPG4-specific chimaeric antigen receptor
also encoding the CD28 co-stimulatory endodomain.
CAR-CSPG4-expressing T cells demonstrated antitumoural activities
in vitro and in mouse models of melanoma, mesothelioma,
breast and head and neck squamous cell carcinomas (48). Several years of promising findings
with a variety of approaches based on CSPG4 as a tumour target,
together with recent technical, translational and clinical
developments in the field of antibody therapeutics for solid
tumours, now provide a strong case for revisiting the concept of an
antibody modality centred on this promising tumour antigen.
Targeting tumour vasculature: VEGF
Malignant melanoma progression from the radial to
the vertical growth phases has been associated with increased
microvessel density and poorer outcomes for patients with high
tumour-associated vasculature. Melanoma tumours feature increased
production of pro-angiogenic factors such as the VEGF family
molecules, VEGF-A involved in angiogenesis, VEGF-B associated with
embryonic angiogenesis, VEGF-C with known roles in
lymphangiogenesis, VEGF-D participating in lung bronchiole
lymphatic vasculature, and VEGF-E encoded from a gene of viral
origin with endothelial cell proliferative properties. Production
of VEGFs, especially VEGF-A, VEGF-C and VEGF-D, and their receptors
VEGFR-1, VEGFR-2 and VEGFR-3 trigger changes in endothelial cell
proliferation, support formation of de novo blood vessels
and vascular permeability at different tumour sites, to allow a
higher degree of tumour cell survival and distal metastases. During
tumour growth, VEGF-A, overexpressed in many solid tumours
including melanoma, can stimulate a hypoxia-driven cascade of
endothelial cell proliferation and migration leading to
angiogenesis via recognition of the class III tyrosine kinase
receptors (VEGFR-1 and 2) (49,50).
Melanoma lymph node metastases involve higher expression and
engagement of the VEGF-C/D and VEGFR-3 system.
The hypothesis that blocking VEGF-VEGFR interactions
may be beneficial for patients led to the development of
bevacizumab, a humanised monoclonal antibody recognising a high
affinity epitope expressed on all VEGF-A isoforms. The antibody
blocks VEGF binding to both receptors (Fig. 1C) (51,52)
and has been approved by FDA for the first-line treatment of
metastatic colorectal and breast cancers. A phase II study of this
antibody as a first-line treatment for metastatic melanoma, in
combination with the cytotoxic nitrosourea alkylating agent
fotemustine, significantly reduced systemic levels of VEGF-A and
VEGF-C, and also of the receptors VEGFR-1 and VEGFR-2. These
findings suggested that this therapy contributes to the inhibition
of angiogenesis and lymphangiogenesis, both of which are highly
relevant in the context of melanoma progression (53). The mean time to progression was
reported to be 8 months and overall survival was 20.5 months in
previously untreated patients with advanced disease. Other trials,
such as a phase II trial testing bevacizumab in combination with
low dose IFN-α2b or standard chemotherapy, showed low activity and
general minimal toxicity in addition to a stable disease state
prolonged in some patients (54,55).
The adjuvant avastin trial in high-risk melanoma (AVAST-M)
sponsored by Cancer Research UK, a large phase III study for
patients with stage IIB, IIC and III cutaneous melanoma following
resection aimed to evaluate the capacity of bevacizumab to prevent
disease recurrence in the adjuvant setting, when early angiogenesis
may be prevented from assisting formation of early metastases.
Results from the AVAST-M trial demonstrated good tolerability but
noted no significant differences in the overall survival between
treatment and observation groups (56). Other approaches may include
antibodies directly targeting VEGF receptors (e.g. combinations of
antibodies such as bevacizumab with chemotherapies or
immunotherapies (57–59).
Focus on T cell checkpoint cascades:
CTLA4
The cytotoxic T-lymphocyte antigen-4, CTLA4,
expressed on the surface of activated T lymphocytes, acts as an
inhibitory molecule of T cell activation by competing with CD28 for
binding to the co-stimulatory B7 family members on
antigen-presenting cells (60,61).
This leads to inhibition of TCR activity, reducing IL-2 gene
transcription and T cell proliferation. CTLA4 acts as a negative
regulator of T cell activation, contributing to antigen tolerance
and limiting T cell mediated autoimmunity and homeostasis
maintenance. Since tumour antigens are largely self-antigens, it
was hypothesised that blocking CTLA4-B7 interactions to enhance T
cell activation could help overcome tumour antigen tolerance and
consequently potentiate enhanced antitumoural immune responses.
Ipilimumab, a human IgG1 antibody recognising CTLA4,
interferes with CTLA4-B7 interactions on the surface of antigen
presenting cells, permitting CD28-B7 complex formation. The
antibody has been shown to operate by reducing CTLA4-induced T cell
inhibitory functions, but also by targeting activatory Fc
receptor-expressing effector cells against CTLA4-expressing Tregs,
thereby blocking their immunosuppressive functions in tumours
(Fig. 1D) (62–64).
In early clinical studies, this agent showed adequate safety and an
indication of efficacy, leading to subsequent trials at the
National Cancer Institute (USA) that demonstrated sustained
responses (>2 years) in a proportion of patients. Grade 3 or 4
adverse events such as colitis, rash and liver function
abnormalities were observed in 19% of patients. A randomised,
multi-institution, double blind, dose-ranging clinical study showed
encouraging results with response rates as high as 11.1% and
survival data as high as 30% (65–67).
Combination studies of administration of ipilimumab with
dacarbazine indicated increased response rates with combination
treatment when compared with single treatment controls (68,69). A
phase III trial was the first randomised study to show a survival
benefit in patients with metastatic melanoma: ipilimumab (3 mg/kg
dose) demonstrated median overall survival of 10 months and 10.1
months when combined with an HLA-A*0201-restricted gp100
vaccine peptide, while gp100 peptide treatment alone showed a
median overall survival of 6.4 months (70).
Ipilimumab (at 3 mg/kg once every three weeks for
four times) was the first antibody to be approved in 2011 by the
FDA in the USA and soon afterwards in Europe for the treatment of
patients with unresectable or metastatic melanoma (stage III and IV
disease) (Table I) (71). This was a significant milestone as
this was the first antibody to demonstrate significant survival
benefits in patients with advanced melanoma (72). In clinical application, ~40% of
patients experience immune-related adverse events (irAEs) through
the universal activation of T cells, leading to tissue-specific
inflammation and autoimmune-related side-effects, such as
dermatitis, colitis and hepatitis. These side-effects can be
managed with systemic steroid treatments without significant
effects on benefit from the antibody therapy, but highlight the
need for careful patient selection in addition to frequent
monitoring. Comprehensive toxicity management algorithms have been
developed to help manage patients, especially as most of the
toxicities are reversible with early intervention. It is noteworthy
that patients who experience autoimmune-related adverse events are
more likely to benefit from treatment with ipilimumab, pointing to
a fine balance between autoimmunity and the clearance of tumours
(73).
Tremelimumab, a human IgG2 anti-CTLA4 monoclonal
antibody, was also examined. Although it demonstrated durable
objective tumour regressions in early clinical trials, it was less
effective than ipilimumab, with lower response rates and median
survival and a phase III study indicated no superior benefits
relative to those observed with dacarbazine (74).
Although ipilimumab is the only antibody therapy to
date that has shown significant impact on overall survival in
advanced melanoma, ongoing late phase clinical trials are presently
exploring anti-CTLA4 antibodies alone or in combination with other
antibodies, immunotherapies and chemotherapeutic agents (Tables I and II).
| Table IIUSA-registered phase III clinical
trials of antibody therapies for melanoma.a |
Table II
USA-registered phase III clinical
trials of antibody therapies for melanoma.a
|
Drug/intervention | Drug type | Sequence of drug
administration | Stage/cancer
type | Identifier |
|---|
| Ipilimumab (i) vs.
MD-1379 (ii) vs. Ipilimumab (i) + MD-1379 (ii) | Anti-CTLA4
monoclonal antibody (i) melanoma peptide vaccine (ii) | IV ipilimumab every
3 weeks for 4 doses
MDX-1379 2 ml (2 subcutaneous injections of 2 ml each, 1 to each
thigh), every 3 weeks for 4 doses | Unresectable or
metastatic melanoma | NCT00094653 |
| Ipilimumab (i) vs.
recombinant IFNα2b (ii) | Anti-CTLA4
monoclonal antibody (i) IFNα2b (ii) | High dose
ipililmumab IV over 90 min every 21 days for 4 courses. Then
maintenance high-dose ipilimumab IV over 90 min every 90 days for a
maximum of 4 courses
High dose recombinant IFNα2b IV on days 1–5, 8–12, 15–19 and 22–26
then maintenance high dose on days 1,3 and 5 | Resected high-risk
melanoma | NCT01274338 |
| CP-675, 206 (i) vs.
Dacarbazine (ii) or Tremozolomide (iii) | Anti-CTLA4 human
monoclonal antibody (i) alkylating chemotherapy agent (ii &
iii) | CP-675, 206 15
mg/kg IV Q 90 days × 4
Dacarbazine 1,000 mg/kg IV Q 90 days × 4
Temozolomide 200 mg/m2 orally on days 1–5 every 28 days
× 12 | Advanced
melanoma | NCT00257205 |
| Ipilimumab (i) vs.
placebo | Anti-CTLA4
monoclonal antibody (ii) placebo (ii) | IV ipilimumab every
21 days for 4 doses, then starting from week 24 every 12 weeks
until week 156 or progression
IV placebo 4 × every 21 days then starting from week 24 every 12
weeks until week 156 or progression | High-risk
melanoma | NCT00636168 |
| Nivolumab (i) vs.
Nivolumab (i) + Ipilimumab (ii) vs. Ipilimumab (ii) | Anti-PD-1
monoclonal antibody (i) anti-CTLA4 monoclonal antibody (ii) | IV nivolumab every
2 weeks IV nivolumab with IV ipilimumab every 3 weeks for 4 doses
then nivolumab IV every 2 weeks. IV ipilimumab every 3 weeks for a
total of 4 doses | Untreated advanced
melanoma | NCT01844505 |
The PD-1/PD-L1 axis
Programmed death-1 (PD-1, CD279, or B7-H1), a member
of the B7:CD28 group of cell surface molecules with homology to
CTLA4, is an inhibitory cell surface protein expressed on the
surface of mature, antigen-experienced T and B lymphocytes and
myeloid cells following activation. Its ligand, PD-L1, is
upregulated on the surface of antigen presenting cells in addition
to epithelial cells and vascular endothelial cells upon antigenic
stimulation with inflammatory signals such as IFNγ. PD-L1 has also
been shown to be expressed on melanoma cells and other immune cells
in tumours. Engagement of PD-1 to PD-L1 can inhibit T cell growth
and cytokine secretion. PD-1 may play a critical role in tumour
immune escape by engaging with PD-L1 and negatively regulating both
cellular and humoral immune responses, permitting tumours to evade
immune surveillance. PD-1 and PD-L1 expression is also linked to
poor clinical prognosis. Therefore, blocking their interactions has
been investigated as a possible immunotherapy strategy (Fig. 1E) (75–77).
A phase I clinical trial of the human IgG4 antibody
nivolumab (MDX-1106) recognising PD-1 in melanoma (among other
refractory cancers) has shown encouraging clinical activity and
safety profiles (78). Nivolumab
was also recently tested in further clinical studies, with reported
median overall survival of 16.8 months, 62% 1-year and 43% 2-year
survival rates. Grade 3 and 4 toxicities were found in only 5% of
patients and clinical responses persisted even after cessation of
therapy (79,80). A recent study showed that treatment
with another anti-PD-1 humanised IgG4 antibody lambrolizumab in
patients with advanced melanoma led to a tumour regression response
rate of 38% (according to the response evaluation criteria in solid
tumours, RECIST), with a durable effect and a median
progression-free survival >7 months. Common, mostly low grade
adverse effects observed during the treatment were fatigue,
pruritus and rash (81).
Antibodies targeting PD-L1 found to be upregulated
on tumour cells and on tumour-associated APCs triggered a high
level of infiltrating T cells within tumour environments. A
fully-human IgG4 antibody to PD-L1 that blocks association with
PD-1 as well as with CD80 was tested in early clinical studies in
different solid tumours including melanoma. Objective responses
were observed in a proportion of patients with melanoma and durable
responses were reported in a small proportion of patients (82). Any therapeutic effects of the
PD-1/PD-L1 blockade continue to be examined in ongoing experimental
and clinical studies.
Depleting Tregs: anti-CD25
Tregs are considered a key suppressive cell subset
which has high relevance in modulated antitumour responses. It has
been suggested that Tregs may be partly responsible for the limited
efficacy of adjuvant immunotherapies and vaccines (83). Tregs have been identified in primary
melanomas, different metastatic sites and lymph nodes with melanoma
cell metastases, and higher densities of Tregs correlate with
thicker primary melanomas in the vertical growth phase (84,85).
Increased numbers of skin resident Tregs are also observed in the
skin of older people, indicating that increased age may entail less
effective immune activation (86).
Studies on tumour models suggest a link between Treg depletion and
enhanced antitumour immunity. Targeting this cell subset has
therefore been considered as a potential strategy to enhance the
potency of immunotherapies. Daclizumab, a human anti-CD25 antibody
was found to be successful in deleting circulating Tregs in
patients with melanoma; however, depletion did not enhance the
efficacy of a DC vaccine (87). An
important limitation is that CD25 is not exclusively expressed by
Tregs, but is also expressed on other activated T cell subsets,
with potential harmful consequences associated with an anti-CD25
targeted therapy in weakening other crucial components of
immunity.
3. Examples of combination strategies with
antibodies
Combination of anti-CTLA4 and
anti-PD-1
Another approach is based on combining the blockade
of both CTLA4 and PD-1 with antibodies. This strategy resulted in
reduced Treg density in addition to enhanced effector T cell
infiltration in tumour lesions in a mouse melanoma model (88). The concept was recently examined in
a phase I clinical trial, administering nivolumab (IgG4 antibody to
PD-1) and ipilimumab (IgG1 antibody to CTLA4) in patients with
stage III or IV melanoma. Different regimens were used: concurrent
(both mAbs, followed bynivolumab alone and subsequent combined
treatments), while patients pre-treated with ipilimumab were
administered nivolumab (sequenced). A 2.5 year follow-up showed 53%
objective responses in the concurrent treatment group with marked
tumour reductions (the majority showing a tumour regression of
≥80%) compared to 20% objective response (OR) rate for sequenced
treatments. Over half of patients administered concurrent
treatments showed grade 3 and 4 toxicities (89). Thus, important advantages of this
combination therapy were response durability and the increased
proportion of patients experiencing responses, compared to
monotherapies; further late phase clinical trials are now examining
this concept (Tables II and
III).
| Table IIIEuropean- and UK-registered phase III
clinical trials of antibody therapies for melanoma.a |
Table III
European- and UK-registered phase III
clinical trials of antibody therapies for melanoma.a
|
Drug/intervention | Drug type | Stage/cancer
type | Identifier |
|---|
| MK-3475 (i) vs.
Ipilimumab (ii) | Anti-PD-1
monoclonal antibody (i) anti-CTLA4 monoclonal antibody (ii) | Advanced
melanoma | 2012-004907-10
(EU) |
| Response and/or
toxicity vs. Ipilimumab (i) | Anti-CTLA4
monoclonal antibody (i) | Unresectable stage
III or IV malignant melanoma | 2005-002126-64
(EU) |
| Nivolumab (i) vs.
investigator’s choice | Anti-PD-1
monoclonal antibody (i) | Unresectable or
metastatic melanoma progressing post anti-CTLA4 therapy | 13396 (UKCRN ID)
(UK) |
| Nivolumab (i) vs.
Ipilimumab (ii) vs. Nivolumab (i) + Ipilimumab (ii) | Anti-PD-1
monoclonal antibody (i) anti-CTLA4 monoclonal antibody (ii) | Unresectable or
metastatic melanoma | 14725 (UKCRN ID)
(UK) |
Combination of mAb and vaccines
Combining mAb and vaccines comprising long-peptides
of known melanoma- or melanocyte-associated antigens, such as gp100
and the tumour-differentiation antigens tyrosinase-related proteins
(TRP)-1 and TRP-2 has been studied. In a melanoma mouse model, the
altered long-peptide gp10025–33 vaccine together with the TLR7
ligand imiquimod (to activate gp100-specific CTL responses), and
the mAb TA99 (specific for the melanocyte protein TRP-1) were used
in combination (90). This led to
activation of tissue-resident FcγR+ immune cells (e.g.
macrophages), creating a critical window of time that allowed
development of peptide vaccine-induced T cell responses. Delayed
tumour growth and long-term survival responses were observed in
>50% of the treated animals.
Combination of anti-GD2, agonist CD40
mAbs and CpG
A combination of two monoclonal antibodies and a
TLR9 ligand was examined. One antibody targeted the tumour antigen
disialoganglioside-GD2, a glycosphingolipid overexpressed in
neuroblastoma and melanoma. This antibody was engineered with
reduced complement-dependent toxicity and improved ADCC functions.
The second antibody was agonistic to CD40, a member of the
TNF-receptor superfamily expressed on several cells such as DCs and
macrophages. By targeting CD40, the antibody promoted tumour cell
apoptosis, activated DC and macrophage activation and maturation.
The antibodies were administered with the TLR9 ligand class B CpG
ODN 1826 recognised by B cells, DCs and NK cells to enhance innate
cell responses. This combination immunotherapy was more efficacious
than single therapies in a model of GD2-expressing B16 melanoma at
minimum tumour burden (91).
Specifically, peritoneal macrophages from mice treated with
anti-CD40 antibody and CpG were able to inhibit tumour cell
proliferation in vitro and suggested a potential role in
vivo in an antigen-dependent manner. Moreover, the antitumour
efficacy of anti-GD2 in vivo was increased by anti-CD40 plus
CpG therapy combinations.
4. Signalling pathway inhibitors: novel
non-immunological treatments with immunological relevance
A proportion (~60%) of melanomas harbour mutations
in the serine-threonine kinase BRAF, resulting in constitutive
activation of the RAS/RAF/MEK/ERK pathway and irregular cell
proliferation. The most common mutations (90%) involve a valine
instead of a glutamic acid in the amino acid position 600, termed
V600E mutation (92). Vemurafenib
is licenced for patients whose tumours express the mutant form of
the BRAF gene. This treatment has markedly greater response rates,
overall survival rates and progression-free survival than
dacarbazine, but most patients develop resistance and experience
disease relapse (93). The BRAF
inhibitors vemurafenib and dabrafenib, both selectively recognising
mutant forms of BRAF, are now approved for clinical use for
advanced stage disease (94).
Individually, ipilimumab and BRAF inhibitors improve
overall survival and each has different clinical profiles; BRAF
inhibition can result in quick but mostly temporary clinical
responses, with median progression usually <7 months, whereas
ipilimumab has a slow onset and a low rate of objective, but
patients who respond to treatment experience long-lasting
responses. It has also emerged that BRAF-inhibitors may improve
antigen presentation and have been associated with reduced
immunomodulatory responses and activated tumour-reactive T cells
while patients respond to therapy (95,96).
Thus, suggestions to combine an oncogene directed therapy with
immunotherapy triggered substantial interest (97). Ribas et al conducted a Phase
I study aiming to evaluate the safety and the best administration
schedule for patients with metastatic BRAF V600-mutant melanoma.
After treatment with both agents at the full approved doses
(starting with vemurafenib for one month, then ipilimumab and
concurrent vemurafenib), a few patients developed toxic effects
such as increased aminotransferase levels and hepatic adverse
events, both asymptomatic and reversible through administration of
glucocorticoids or temporary termination of the therapy. These
toxicities resulted in study closure (98). However, ongoing clinical trials
continue to examine different strategies based on this concept.
5. Conclusions
Melanoma has historically been considered an
immunogenic malignancy and has for long been resistant to standard
treatments. Antibodies have always constituted a promising
modality; however, with the development of checkpoint blockade
antibodies and the approval of the first antibody for clinical use,
their capacity to transform the treatment of patients is now better
appreciated. Momentum is now gathering in the search for agents
with capacity to potentiate immune clearance of melanoma tumours
and to achieve tangible survival benefits (99). Parallel to the success of antibody
therapies, pathway inhibitor drugs are also making a positive
impact on disease management, despite drug resistance developing in
most patients within months of treatment.
Beyond clinical application of checkpoint blocking
antibodies, limitations of associated high toxicities and palpable
clinical benefits for only a proportion of patients, immunotherapy
of melanoma with antibodies now commands fresh consideration. The
next generation of antibodies may be designed to re-educate and
direct potent immune cells specifically against cancer cells. If
achieved, a new class of agents may realize the capabilities of
this modality to harness and potentially combine affinity and
specificity for targets with directing potent subsets of immune
effector cells against specific pathways or factors linked to
cancer. Targeting inhibitory receptors such as PD-1 and PD-L1, to
target tissue-resident antigen-educated T cell responses is likely
to yield clinical data in the near future, and promises to
potentiate T cell activation with potentially fewer toxic
side-effects. Focus has also centred on combinations of antibodies
with targeted pathway inhibitor drugs, other antibodies, vaccines
or radiotherapy.
Yet to be explored in melanoma are emerging
developments in antibody engineering including: bi-specific
antibodies, including those combining tumour and immunological
targets such as CD3 to focus T cells against tumours; toxin- or
cytokine-loaded antibodies; modified antibodies with engineered Fc
regions to enhance activatory effector functions (100). Numerous studies by us and others
point towards consideration of antibodies of classes such as IgA or
IgE with tissue immune surveillance functions that may be
well-suited to the phenotypes of tumour-resident effector cells
(101–107). Future studies may also provide
further insights into the pathways associated with immune cell
activation and re-direction in patient circulation and tumour
microenvironments (11,14,15,62,108).
Recent studies suggest the presence of an inflammatory B cell
infiltrate in melanoma and biased production of IgG4, an antibody
subclass with inefficient effector activities and with capacity to
block cytotoxic antibodies from attacking tumours (10,11,14,15,109,110). New understanding of immunological
mechanisms of response and strategies to counteract
immunosuppression in melanoma may lead to new therapeutic
interventions, but may also help identify predictive biomarkers for
patient stratification or for monitoring responses to treatments
(111). Clinically meaningful
advances in the treatment of this challenging disease may now be in
sight.
Acknowledgements
The authors acknowledge support from Cancer Research
UK (C30122/A11527); KCL Experimental Cancer Medicine Centre,
jointly funded by Cancer Research UK, the National Institute for
Health Research, Welsh Assembly Government, HSC R&D Office for
Northern Ireland and Chief Scientist Office, Scotland
(C10355/A15587); CR UK/EPSRC/MRC/NIHR KCL/UCL Comprehensive Cancer
Imaging Centre (C1519/A10331); The British Skin Foundation (S633);
Academy of Medical Sciences; Dermatrust. The research was supported
by the National Institute for Health Research (NIHR) Biomedical
Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust
and King’s College London. The views expressed are those of the
author(s) and not necessarily those of the NHS, the NIHR or the
Department of Health.
References
|
1
|
Balch CM, Gershenwald JE, Soong SJ, et al:
Final version of 2009 AJCC melanoma staging and classification. J
Clin Oncol. 27:6199–6206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gerner RE, Moore GE and Dickey C:
Combination chemotherapy in disseminated melanoma and other solid
tumors in adults. Oncology. 31:22–30. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Atkins MB: Cytokine-based therapy and
biochemotherapy for advanced melanoma. Clin Cancer Res.
12:2353s–2358s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ballantyne AD and Garnock-Jones KP:
Dabrafenib: first global approval. Drugs. 73:1367–1376. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bollag G, Tsai J, Zhang J, et al:
Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat
Rev Drug Discov. 11:873–886. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wright CJ and McCormack PL: Trametinib:
first global approval. Drugs. 73:1245–1254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sznol M: Advances in the treatment of
metastatic melanoma: new immunomodulatory agents. Semin Oncol.
39:192–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oble DA, Loewe R, Yu P and Mihm MC Jr:
Focus on TILs: prognostic significance of tumor infiltrating
lymphocytes in human melanoma. Cancer Immun. 9:32009.PubMed/NCBI
|
|
9
|
Shimanovsky A, Jethava A and Dasanu CA:
Immune alterations in malignant melanoma and current immunotherapy
concepts. Expert Opin Biol Ther. 13:1413–1427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cipponi A, Mercier M, Seremet T, et al:
Neogenesis of lymphoid structures and antibody responses occur in
human melanoma metastases. Cancer Res. 72:3997–4007. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gilbert AE, Karagiannis P, Dodev T, et al:
Monitoring the systemic human memory B cell compartment of melanoma
patients for anti-tumor IgG antibodies. PLoS One. 6:e193302011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lacy KE, Karagiannis SN and Nestle FO:
Advances in the treatment of melanoma. Clin Med. 12:168–171. 2012.
View Article : Google Scholar
|
|
13
|
Fujimura T, Ring S, Umansky V, Mahnke K
and Enk AH: Regulatory T cells stimulate B7-H1 expression in
myeloid-derived suppressor cells in ret melanomas. J Invest
Dermatol. 132:1239–1246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karagiannis P, Gilbert AE, Nestle FO and
Karagiannis SN: IgG4 antibodies and cancer-associated inflammation:
insights into a novel mechanism of immune escape. Oncoimmunology.
2:e248892013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karagiannis P, Gilbert AE, Josephs DH, et
al: IgG4 subclass antibodies impair antitumor immunity in melanoma.
J Clin Invest. 123:1457–1474. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang T, Ge Y, Xiao M, et al:
Melanoma-derived conditioned media efficiently induce the
differentiation of monocytes to macrophages that display a highly
invasive gene signature. Pigment Cell Melanoma Res. 25:493–505.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Atkins MB, Lotze MT, Dutcher JP, et al:
High-dose recombinant interleukin 2 therapy for patients with
metastatic melanoma: analysis of 270 patients treated between 1985
and 1993. J Clin Oncol. 17:2105–2116. 1999.PubMed/NCBI
|
|
18
|
Hauschild A: Adjuvant interferon alfa for
melanoma: new evidence-based treatment recommendations? Curr Oncol.
16:3–6. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Petrella T, Quirt I, Verma S, Haynes AE,
Charette M, Bak K, et al: Single-agent interleukin-2 in the
treatment of metastatic melanoma: a systematic review. Cancer Treat
Rev. 33:484–496. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sasse AD, Sasse EC, Clark LG, Ulloa L and
Clark OA: Chemoimmunotherapy versus chemotherapy for metastatic
malignant melanoma. Cochrane Database Syst Rev.
CD0054132007.PubMed/NCBI
|
|
21
|
Borden EC: Interferons: pleiotropic
cellular modulators. Clin Immunol Immunopathol. 62:S18–24. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bart RS, Porzio NR, Kopf AW, Vilcek JT,
Cheng EH and Farcet Y: Inhibition of growth of B16 murine malignant
melanoma by exogenous interferon. Cancer Res. 40:614–619.
1980.PubMed/NCBI
|
|
23
|
Kirkwood JM, Strawderman MH, Ernstoff MS,
Smith TJ, Borden EC and Blum RH: Interferon alfa-2b adjuvant
therapy of high-risk resected cutaneous melanoma: the Eastern
Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 14:7–17.
1996.PubMed/NCBI
|
|
24
|
Wheatley K, Ives N, Hancock B, Gore M,
Eggermont A and Suciu S: Does adjuvant interferon-alpha for
high-risk melanoma provide a worthwhile benefit? A meta-analysis of
the randomised trials. Cancer Treat Rev. 29:241–252. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hillner BE: Cost-effectiveness assessment
of interferon alfa-2b as adjuvant therapy of high-risk resected
cutaneous melanoma. Eur J Cancer. 34:S18–S21. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eggermont AM, Suciu S, Santinami M, et al:
Adjuvant therapy with pegylated interferon alfa-2b versus
observation alone in resected stage III melanoma: final results of
EORTC 18991, a randomised phase III trial. Lancet. 372:117–126.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheever MA and Higano CS: PROVENGE
(Sipuleucel-T) in prostate cancer: the first FDA-approved
therapeutic cancer vaccine. Clin Cancer Res. 17:3520–3526. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zikich D, Schachter J and Besser MJ:
Immunotherapy for the management of advanced melanoma: the next
steps. Am J Clin Dermatol. 14:261–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dannull J, Haley NR, Archer G, et al:
Melanoma immunotherapy using mature DCs expressing the constitutive
proteasome. J Clin Invest. 123:3135–3145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan J, Ku GY, Gallardo HF, et al: Safety
and immunogenicity of a human and mouse gp100 DNA vaccine in a
phase I trial of patients with melanoma. Cancer Immun.
9:52009.PubMed/NCBI
|
|
31
|
Reichert JM and Dhimolea E: The future of
antibodies as cancer drugs. Drug Discov Today. 17:954–963. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Azijli K, Stelloo E, Peters GJ and van den
Eertwegh AJ: New developments in the treatment of metastatic
melanoma: immune checkpoint inhibitors and targeted therapies.
Anticancer Res. 34:1493–1505. 2014.PubMed/NCBI
|
|
33
|
Price MA, Colvin Wanshura LE, Yang J, et
al: CSPG4, a potential therapeutic target, facilitates malignant
progression of melanoma. Pigment Cell Melanoma Res. 24:1148–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Osada T, Wang Y, et al: CSPG4
protein as a new target for the antibody-based immunotherapy of
triple-negative breast cancer. J Natl Cancer Inst. 102:1496–1512.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J, Price MA, Neudauer CL, et al:
Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK
activation by distinct mechanisms. J Cell Biol. 165:881–891. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang J, Price MA, Li GY, et al: Melanoma
proteoglycan modifies gene expression to stimulate tumor cell
motility, growth, and epithelial-to-mesenchymal transition. Cancer
Res. 69:7538–7547. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chekenya M, Krakstad C, Svendsen A, et al:
The progenitor cell marker NG2/MPG promotes chemoresistance by
activation of integrin-dependent PI3K/Akt signaling. Oncogene.
27:5182–5194. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maciag PC, Seavey MM, Pan ZK, Ferrone S
and Paterson Y: Cancer immunotherapy targeting the high molecular
weight melanoma-associated antigen protein results in a broad
antitumor response and reduction of pericytes in the tumor
vasculature. Cancer Res. 68:8066–8075. 2008. View Article : Google Scholar
|
|
39
|
Kusama M, Kageshita T, Chen ZJ and Ferrone
S: Characterization of syngeneic antiidiotypic monoclonal
antibodies to murine anti-human high molecular weight
melanoma-associated antigen monoclonal antibodies. J Immunol.
143:3844–3852. 1989.
|
|
40
|
Chen ZJ, Yang H, Kageshita T and Ferrone
S: Human high-molecular-weight melanoma-associated antigen mimicry
by mouse antiidiotypic monoclonal antibody TK7-371. Cancer Res.
51:4790–4797. 1991.PubMed/NCBI
|
|
41
|
Mittelman A, Chen ZJ, Yang H, Wong GY and
Ferrone S: Human high molecular weight melanoma-associated antigen
(HMW-MAA) mimicry by mouse anti-idiotypic monoclonal antibody
MK2-23: induction of humoral anti-HMW-MAA immunity and prolongation
of survival in patients with stage IV melanoma. Proc Natl Acad Sci
USA. 89:466–470. 1992. View Article : Google Scholar
|
|
42
|
Mittelman A, Chen GZ, Wong GY, Liu C,
Hirai S and Ferrone S: Human high molecular weight-melanoma
associated antigen mimicry by mouse anti-idiotypic monoclonal
antibody MK2-23: modulation of the immunogenicity in patients with
malignant melanoma. Clin Cancer Res. 1:705–713. 1995.PubMed/NCBI
|
|
43
|
Mittelman A, Wang X, Matsumoto K and
Ferrone S: Antiantiidiotypic response and clinical course of the
disease in patients with malignant melanoma immunized with mouse
antiidiotypic monoclonal antibody MK2-23. Hybridoma. 14:175–181.
1995. View Article : Google Scholar
|
|
44
|
Murray JL, Gillogly M, Kawano K, et al:
Fine specificity of high molecular weight-melanoma-associated
antigen-specific cytotoxic T lymphocytes elicited by anti-idiotypic
monoclonal antibodies in patients with melanoma. Cancer Res.
64:5481–5488. 2004. View Article : Google Scholar
|
|
45
|
Torisu-Itakura H, Schoellhammer HF, Sim
MS, et al: Redirected lysis of human melanoma cells by a
MCSP/CD3-bispecific BiTE antibody that engages patient-derived T
cells. J Immunother. 34:597–605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rybczynska AA, Dierckx RA, Ishiwata K,
Elsinga PH and van Waarde A: Cytotoxicity of sigma-receptor ligands
is associated with major changes of cellular metabolism and
complete occupancy of the sigma-2 subpopulation. J Nucl Med.
49:2049–2056. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
de Bruyn M, Rybczynska AA, Wei Y, et al:
Melanoma-associated Chondroitin Sulfate Proteoglycan
(MCSP)-targeted delivery of soluble TRAIL potently inhibits
melanoma outgrowth in vitro and in vivo. Mol Cancer. 9:3012010.
|
|
48
|
Geldres C, Savoldo B, Hoyos V, et al: T
lymphocytes redirected against the chondroitin sulfate
proteoglycan-4 control the growth of multiple solid tumors both in
vitro and in vivo. Clin Cancer Res. 20:962–971. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mehnert JM, McCarthy MM, Jilaveanu L, et
al: Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3
in melanoma tissue microarrays. Hum Pathol. 41:375–384. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Spinella F, Caprara V, Cianfrocca R, et
al: The interplay between hypoxia, endothelial and melanoma cells
regulates vascularization and cell motility through endothelin-1
and vascular endothelial growth factor. Carcinogenesis. 35:840–848.
2014. View Article : Google Scholar
|
|
51
|
Hsu JY and Wakelee HA: Monoclonal
antibodies targeting vascular endothelial growth factor: current
status and future challenges in cancer therapy. BioDrugs.
23:289–304. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Molhoek KR, Griesemann H, Shu J,
Gershenwald JE, Brautigan DL and Slingluff CL Jr: Human melanoma
cytolysis by combined inhibition of mammalian target of rapamycin
and vascular endothelial growth factor/vascular endothelial growth
factor receptor-2. Cancer Res. 68:4392–4397. 2008. View Article : Google Scholar
|
|
53
|
Del Vecchio M, Mortarini R, Canova S, et
al: Bevacizumab plus fotemustine as first-line treatment in
metastatic melanoma patients: clinical activity and modulation of
angiogenesis and lymphangiogenesis factors. Clin Cancer Res.
16:5862–5872. 2010.PubMed/NCBI
|
|
54
|
Varker KA, Biber JE, Kefauver C, et al: A
randomized phase 2 trial of bevacizumab with or without daily
low-dose interferon alfa-2b in metastatic malignant melanoma. Ann
Surg Oncol. 14:2367–2376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guenterberg KD, Grignol VP, Relekar KV, et
al: A pilot study of bevacizumab and interferon-alpha2b in ocular
melanoma. Am J Clin Oncol. 34:87–91. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Corrie PG, Marshall A, Dunn JA, et al:
Adjuvant bevacizumab in patients with melanoma at high risk of
recurrence (AVAST-M): preplanned interim results from a
multicentre, open-label, randomised controlled phase 3 study.
Lancet Oncol. 15:620–630. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shrimali RK, Yu Z, Theoret MR, Chinnasamy
D, Restifo NP and Rosenberg SA: Antiangiogenic agents can increase
lymphocyte infiltration into tumor and enhance the effectiveness of
adoptive immunotherapy of cancer. Cancer Res. 70:6171–6180. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Perez DG, Suman VJ, Fitch TR, et al: Phase
2 trial of carboplatin, weekly paclitaxel, and biweekly bevacizumab
in patients with unresectable stage IV melanoma: a North Central
Cancer Treatment Group study, N047A. Cancer. 115:119–127. 2009.
View Article : Google Scholar
|
|
59
|
Perez EA, Hillman DW, Dentchev T, et al:
North Central Cancer Treatment Group (NCCTG) N0432: phase II trial
of docetaxel with capecitabine and bevacizumab as first-line
chemotherapy for patients with metastatic breast cancer. Ann Oncol.
21:269–274. 2010. View Article : Google Scholar
|
|
60
|
Allison JP, Chambers C, Hurwitz A, et al:
A role for CTLA-4 mediated inhibitory signals in peripheral T cell
tolerance? Novartis Found Symp. 215:92–98. 1998.PubMed/NCBI
|
|
61
|
Linsley PS, Brady W, Urnes M, Grosmaire
LS, Damle NK and Ledbetter JA: CTLA-4 is a second receptor for the
B cell activation antigen B7. J Exp Med. 174:561–569. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Simpson TR, Li F, Montalvo-Ortiz W, et al:
Fc-dependent depletion of tumor-infiltrating regulatory T cells
co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J
Exp Med. 210:1695–1710. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bulliard Y, Jolicoeur R, Windman M, et al:
Activating Fc γ receptors contribute to the antitumor activities of
immunoregulatory receptor-targeting antibodies. J Exp Med.
210:1685–1693. 2013.
|
|
64
|
Friedline RH, Brown DS, Nguyen H, et al:
CD4+regulatory T cells require CTLA-4 for the
maintenance of systemic tolerance. J Exp Med. 206:421–434.
2009.PubMed/NCBI
|
|
65
|
Phan GQ, Yang JC, Sherry RM, et al: Cancer
regression and autoimmunity induced by cytotoxic T
lymphocyte-associated antigen 4 blockade in patients with
metastatic melanoma. Proc Natl Acad Sci USA. 100:8372–8377. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Weber JS, O’Day S, Urba W, et al: Phase
I/II study of ipilimumab for patients with metastatic melanoma. J
Clin Oncol. 26:5950–5956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ribas A, Camacho LH, Lopez-Berestein G, et
al: Antitumor activity in melanoma and anti-self responses in a
phase I trial with the anti-cytotoxic T lymphocyte-associated
antigen 4 monoclonal antibody CP-675,206. J Clin Oncol.
23:8968–8977. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Robert C, Thomas L, Bondarenko I, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hersh EM, O’Day SJ, Powderly J, et al: A
phase II multicenter study of ipilimumab with or without
dacarbazine in chemotherapy-naive patients with advanced melanoma.
Invest New Drugs. 29:489–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hodi FS, O’Day SJ, McDermott DF, et al:
Improved survival with ipilimumab in patients with metastatic
melanoma. New Engl J Med. 363:711–723. 2010. View Article : Google Scholar
|
|
71
|
Callahan MK, Postow MA and Wolchok JD:
Immunomodulatory therapy for melanoma: ipilimumab and beyond. Clin
Dermatol. 31:191–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Postow MA, Callahan MK and Wolchok JD: The
antitumor immunity of ipilimumab: (T-cell) memories to last a
lifetime? Clin Cancer Res. 18:1821–1823. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Della Vittoria Scarpati G, Fusciello C,
Perri F, et al: Ipilimumab in the treatment of metastatic melanoma:
management of adverse events. Onco Targets Ther. 7:203–209.
2014.PubMed/NCBI
|
|
74
|
Ribas A, Kefford R, Marshall MA, et al:
Phase III randomized clinical trial comparing tremelimumab with
standard-of-care chemotherapy in patients with advanced melanoma. J
Clin Oncol. 31:616–622. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Iwai Y, Ishida M, Tanaka Y, Okazaki T,
Honjo T and Minato N: Involvement of PD-L1 on tumor cells in the
escape from host immune system and tumor immunotherapy by PD-L1
blockade. Proc Natl Acad Sci USA. 99:12293–12297. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hino R, Kabashima K, Kato Y, et al: Tumor
cell expression of programmed cell death-1 ligand 1 is a prognostic
factor for malignant melanoma. Cancer. 116:1757–1766. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ahmadzadeh M, Johnson LA, Heemskerk B, et
al: Tumor antigen-specific CD8 T cells infiltrating the tumor
express high levels of PD-1 and are functionally impaired. Blood.
114:1537–1544. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Brahmer JR, Drake CG, Wollner I, et al:
Phase I study of single-agent anti-programmed death-1 (MDX-1106) in
refractory solid tumors: safety, clinical activity,
pharmacodynamics, and immunologic correlates. J Clin Oncol.
28:3167–3175. 2010. View Article : Google Scholar
|
|
79
|
Topalian SL, Sznol M, McDermott DF, et al:
Survival, durable tumor remission, and long-term safety in patients
with advanced melanoma receiving nivolumab. J Clin Oncol.
32:1020–1030. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lipson EJ, Sharfman WH, Drake CG, et al:
Durable cancer regression off-treatment and effective reinduction
therapy with an anti-PD-1 antibody. Clin Cancer Res. 19:462–468.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hamid O, Robert C, Daud A, et al: Safety
and tumor responses with lambrolizumab (anti-PD-1) in melanoma. New
Engl J Med. 369:134–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Brahmer JR, Tykodi SS, Chow LQ, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. New Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yao X, Ahmadzadeh M, Lu YC, et al: Levels
of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively
associated with clinical response to adoptive immunotherapy of
human cancer. Blood. 119:5688–5696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
De Panfilis G, Campanini N, Santini M, et
al: Phase- and stage-related proportions of T cells bearing the
transcription factor FOXP3 infiltrate primary melanoma. J Invest
Dermatol. 128:676–684. 2008.PubMed/NCBI
|
|
85
|
Miracco C, Mourmouras V, Biagioli M, et
al: Utility of tumour-infiltrating
CD25+FOXP3+regulatory T cell evaluation in
predicting local recurrence in vertical growth phase cutaneous
melanoma. Oncol Rep. 18:1115–1122. 2007.PubMed/NCBI
|
|
86
|
Agius E, Lacy KE, Vukmanovic-Stejic M, et
al: Decreased TNF-alpha synthesis by macrophages restricts
cutaneous immunosurveillance by memory CD4+T cells
during aging. J Exp Med. 206:1929–1940. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jacobs JF, Punt CJ, Lesterhuis WJ, et al:
Dendritic cell vaccination in combination with anti-CD25 monoclonal
antibody treatment: a phase III study in metastatic melanoma
patients. Clin Cancer Res. 16:5067–5078. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Curran MA, Montalvo W, Yagita H and
Allison JP: PD-1 and CTLA-4 combination blockade expands
infiltrating T cells and reduces regulatory T and myeloid cells
within B16 melanoma tumors. Proc Natl Acad Sci USA. 107:4275–4280.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wolchok JD, Kluger H, Callahan MK, et al:
Nivolumab plus ipilimumab in advanced melanoma. New Engl J Med.
369:122–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ly LV, Sluijter M, van der Burg SH, Jager
MJ and van Hall T: Effective cooperation of monoclonal antibody and
peptide vaccine for the treatment of mouse melanoma. J Immunol.
190:489–496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Alderson KL, Luangrath M, Elsenheimer MM,
et al: Enhancement of the anti-melanoma response of Hu14.18K322A by
αCD40 + CpG. Cancer Immunol Immunother. 62:665–675. 2013.PubMed/NCBI
|
|
92
|
Flaherty KT: Dividing and conquering:
controlling advanced melanoma by targeting oncogene-defined
subsets. Clin Exp Metastasis. 29:841–846. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chapman PB, Hauschild A, Robert C, et al:
Updated overall survival (OS) results for BRIM-3, a phase III
randomized, open-label, multicenter trial comparing BRAF inhibitor
vemurafenib (vem) with dacarbazine (DTIC) in previously untreated
patients with BRAFV600E-mutated melanoma. J Clin Oncol.
85022012.
|
|
94
|
Hauschild A, Grob JJ, Demidov LV, et al:
Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre,
open-label, phase 3 randomised controlled trial. Lancet.
380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Knight DA, Ngiow SF, Li M, et al: Host
immunity contributes to the anti-melanoma activity of BRAF
inhibitors. J Clin Invest. 123:1371–1381. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ngiow SF, Knight DA, Ribas A, McArthur GA
and Smyth MJ: BRAF-targeted therapy and immune responses to
melanoma. Oncoimmunology. 2:e244622013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ascierto PA, Simeone E, Giannarelli D,
Grimaldi AM, Romano A and Mozzillo N: Sequencing of BRAF inhibitors
and ipilimumab in patients with metastatic melanoma: a possible
algorithm for clinical use. J Transl Med. 10:1072012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ribas A, Hodi FS, Callahan M, Konto C and
Wolchok J: Hepatotoxicity with combination of vemurafenib and
ipilimumab. N Engl J Med. 368:1365–1366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Culos KA and Cuellar S: Novel targets in
the treatment of advanced melanoma: new first-line treatment
options. Ann Pharmacother. 47:519–526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Woof JM: Insights from Fc receptor
biology: a route to improved antibody reagents. MAbs. 4:291–293.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Bossi G, Buisson S, Oates J, Jakobsen BK
and Hassan NJ: ImmTAC-redirected tumour cell killing induces and
potentiates antigen cross-presentation by dendritic cells. Cancer
Immunol Immunother. 63:437–448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
McCormack E, Adams KJ, Hassan NJ, et al:
Bi-specific TCR-anti CD3 redirected T-cell targeting of NY-ESO-1-
and LAGE-1-positive tumors. Cancer Immunol Immunother. 62:773–785.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Karagiannis SN, Josephs DH, Karagiannis P,
et al: Recombinant IgE antibodies for passive immunotherapy of
solid tumours: from concept towards clinical application. Cancer
Immunol Immunother. 61:1547–1564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Josephs DH, Spicer JF, Karagiannis P,
Gould HJ and Karagiannis SN: IgE immunotherapy: a novel concept
with promise for the treatment of cancer. MAbs. 6:54–72. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Boross P, Lohse S, Nederend M, et al: IgA
EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med.
5:1213–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lohse S, Brunke C, Derer S, et al:
Characterization of a mutated IgA2 antibody of the m(1) allotype
against the epidermal growth factor receptor for the recruitment of
monocytes and macrophages. J Biol Chem. 287:25139–25150. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lohse S, Derer S, Beyer T, et al:
Recombinant dimeric IgA antibodies against the epidermal growth
factor receptor mediate effective tumor cell killing. J Immunol.
186:3770–3778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Peggs KS, Quezada SA, Chambers CA, Korman
AJ and Allison JP: Blockade of CTLA-4 on both effector and
regulatory T cell compartments contributes to the antitumor
activity of anti-CTLA-4 antibodies. J Exp Med. 206:1717–1725. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Silina K, Rulle U, Kalnina Z and Line A:
Manipulation of tumour-infiltrating B cells and tertiary lymphoid
structures: a novel anti-cancer treatment avenue? Cancer Immunol
Immunother. Apr 3–2014.(Epub ahead of print).
|
|
110
|
Cipponi A, Wieers G, van Baren N and
Coulie PG: Tumor-infiltrating lymphocytes: apparently good for
melanoma patients. But why? Cancer Immunol Immunother.
60:1153–1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Tsoka S, Ainali C, Karagiannis P, et al:
Toward prediction of immune mechanisms and design of
immunotherapies in melanoma. Crit Rev Biomed Eng. 40:279–294. 2012.
View Article : Google Scholar : PubMed/NCBI
|