Introduction
Neuroendocrine tumours (NETs) are slowly growing
tumours arising from neuroendocrine enterochromaffin (Kulchitsky)
cells, accounting for only 0.66% of all malignancies. NETs have an
increasing incidence and are most commonly found in the
gastrointestinal tract (55%) followed by the bronchopulmonary
system (30%) (1). NETs are
traditionally divided into subgroups based primarily on the
anatomical location from where they originate, i.e. the foregut
(bronchus, lung, stomach, pancreas, liver and first part of the
duodenum), the midgut (second part of the duodenum, jejenum, ileum,
appendix and proximal colon) and the hindgut (distal colon, rectum
and genitourinary tract) (2,3).
However, since the tumours in each group do not always share
similar clinical behaviour a new classification system is needed
for the future.
NETs most commonly metastasise to lymph nodes,
liver, lungs, peritoneum and pancreas (4). There is a tendency that orbital
metastases originate from primary gastrointestinal tract NETs,
whereas intraocular metastases arise from primary bronchial NETs
(5). Primary hepatic NETs are
extremely rare, and orbital metastases from NETs in general have
been reported only in a very few cases (5–9). We
present the first case of a primary hepatic NET with an orbital
metastasis including the genomic profiles of the two tumours.
Patients and methods
Case report
A 71-year-old female presented with impaired vision
and a visual field defect of her right eye. Six years earlier she
had been diagnosed with a large primary hepatic NET weighing 5.5 kg
and having the greatest diameter of 35 cm. The tumour was resected
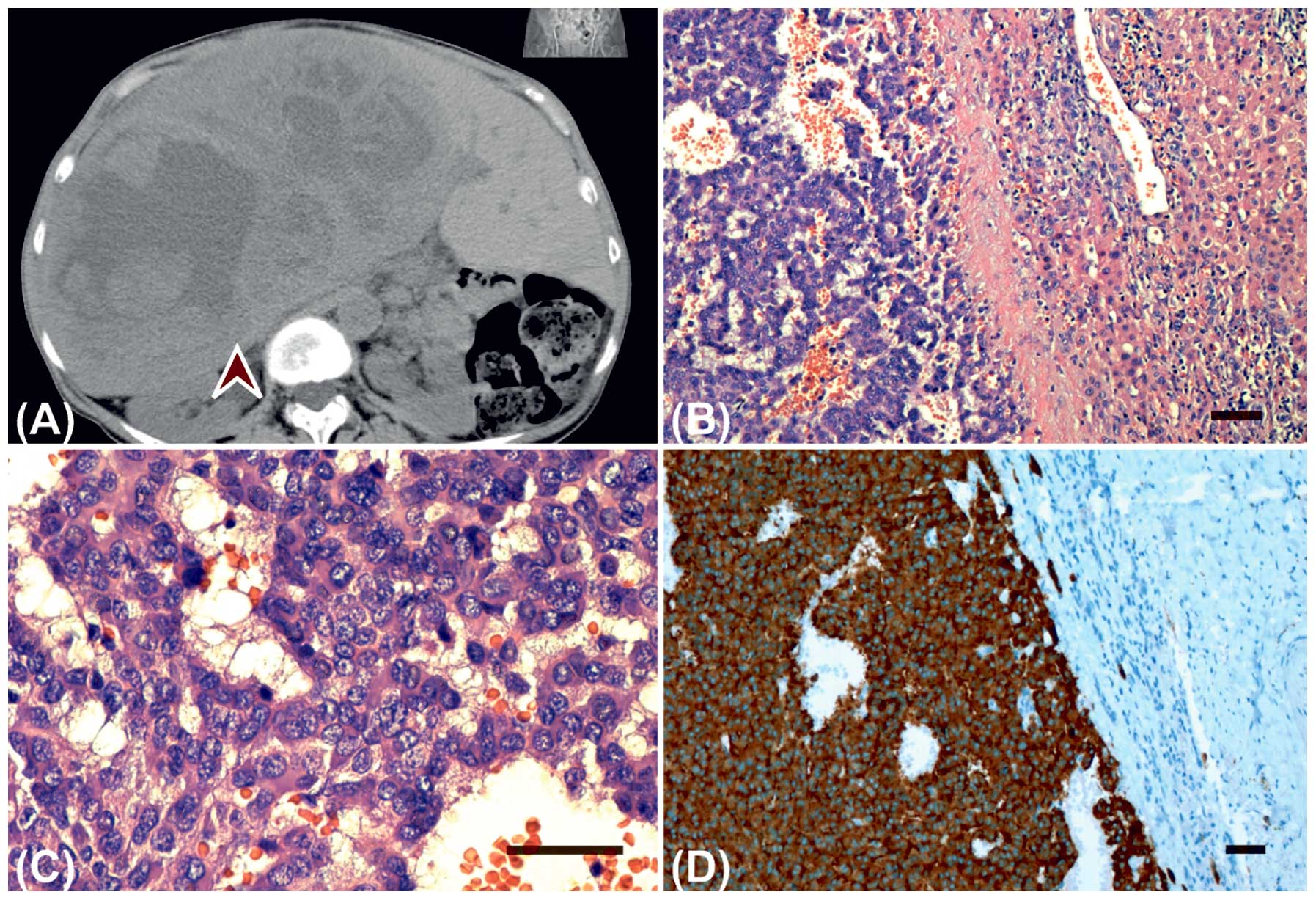
surgically (Fig. 1A).
Visual acuity at referral was 0.3 of the right eye
and 0.8 of the left eye. A 2-mm protrusion of the right eye as well
as restricted ocular motility at superior and temporal gaze were
observed. Furthermore, a visual field defect was present in the
periphery of all 4 quadrants. Computed tomography scan (CT) and
contrast enhanced magnetic resonance imaging (MRI) scan
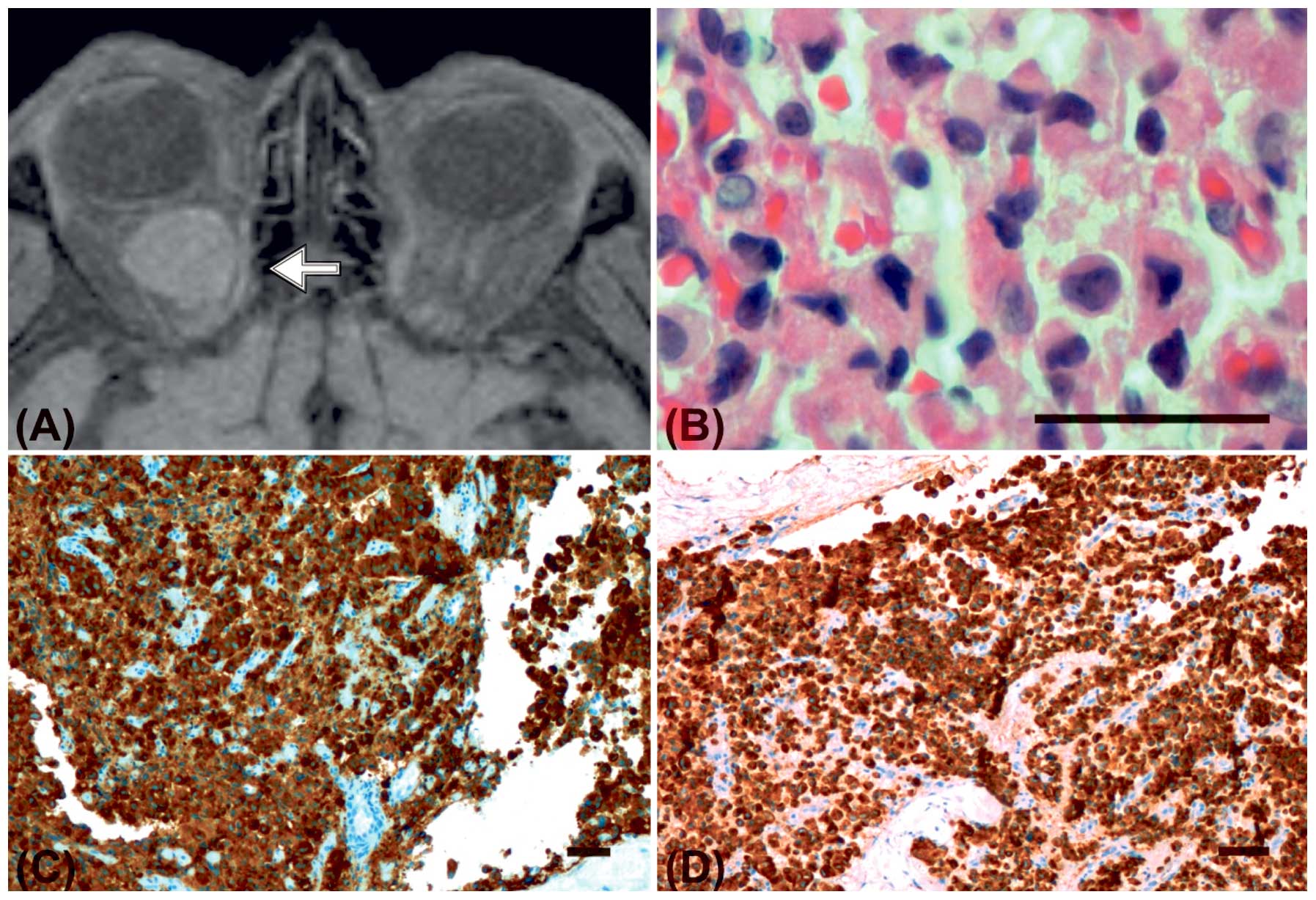
demonstrated a retrobulbar, intraconal mass measuring 2.2×1.8×1.5
cm with contrast uptake (Fig. 2A).
Somatostatin receptor scintigraphy (octreoscan) showed increased
uptake in the cranium, neck, mediastinum, right upper arm, abdomen
and between the right kidney and the liver. A biopsy from the right
orbit via the upper eyelid was performed. Ptosis of the right eye
was permanently present after the procedure. The patient underwent
30 Gy radiation to the right orbit, but the tumour did not respond.
Fifteen months later, the orbital tumour was surgically excised en
bloc. It was adherent to the superior rectus muscle and the levator
muscle of the eyelid. Vision declined to 0.17 at the right eye.
Metastases to the cerebrum developed 20 months later and they were
surgically removed. Metastasis to the sacral bone developed 8 years
after initial diagnosis of the hepatic neuroendocrine carcinoma and
was treated with 20 Gy radiation. When the neuroendocrine carcinoma
had started to spread 8 years after the initial diagnosis,
IntronA®, Sandostatin®, Nexavar®
and Dotatoc® were instituted. Twelve years after
diagnosis of the primary hepatic neuroendocrine carcinoma, the
patient was treated with Temodal, but due to progression of the
liver metastases, the treatment was changed to etoposide
monotherapy. This was, however, also stopped due to the continuous
progression of the liver metastases and increasing renal
failure.
At the last follow-up time (15 years after the
diagnosis of hepatic NET) an MRI scan revealed a recurrent mass
measuring 2×1.5×1.5 cm in the right orbit. The patient is still
alive and receives palliative treatment. She has no light
perception in the right eye and only 0.4 in the left eye.
Histopathology
The orbital biopsy and the liver specimen were
formalin-fixed and paraffin-embedded (FFPE), and 4-μm sections were
mounted on glass slides. The sections were stained with
haematoxylin and eosin (H&E) and periodic acid-Schiff (PAS).
Immunohistochemical reactions were performed using the
streptavidin-biotin method. Antibodies against CD31, CD34, CD56,
pan-cytokeratin (clone AE1/AE3), CK7, chromogranin A, Ki-67 (clone
MIB-1), neuron-specific enolase (NSE), S100, serotonin,
somatostatin, synaptophysin, thyroid transcription factor (TTF-1)
and vimentin (all from Dako Inc., Copenhagen, Denmark) were
applied. HepPar-1 antibody, polyclonal antibodies against CEA (both
from Dako) and monoclonal antibodies against arginase-1 (Abcam,
Cambridge, UK) were applied to rule out that the tumour was a
hepatocellular carcinoma with neuroendocrine dedifferentiation.
Appropriate controls were performed.
ArrayCGH
Genomic DNA was isolated from the FFPE tumour
tissues from the primary tumour and the metastatic lesion using the
DNeasy® Blood and Tissue kit (Qiagen GmbH, Hilden,
Germany). ArrayCGH analysis was subsequently performed using the
human genome CGH microarray 244K oligonucleotide array (G4411B;
Agilent Technologies Inc., Palo Alto, CA, USA). The arrayCGH
experiments were performed essentially as previously described and
as recommended by the manufacturer (10,11).
Slides were scanned on an Agilent High-Resolution C Microarray
Scanner, followed by data extraction and normalization using
Feature Extraction v.10.7.1 (Agilent Technologies) with linear
normalization (protocol CGH_107_Sep09). Data analysis was carried
out using Nexus Copy Number Software® Discovery Edition
v. 6.0 (BioDiscovery Inc., El Segundo, CA, USA) as previously
described (11). Each aberration
was checked manually to confirm the accuracy of the call. Regions
partially or completely covered by a previously reported copy
number variation (Database of Genomic Variants; http://dgvbeta.tcag.ca/dgv/app/home?ref=NCBI36/hg18)
were excluded from the analysis.
Results
Histopathology
A biopsy from the orbital mass contained nests of
pleomorphic tumour cells (Fig. 2B),
that diffusely infiltrated the orbital connective tissue and fat.
The tumour cells were intermediate-sized and polyhedral with
eosinophilic cytoplasm with perinuclear eosinophilic inclusions and
rounded to oval nuclei with finely granular chromatin. There were
no areas of necrosis, and mitoses were infrequent. The orbital
tumour cells demonstrated positive immunoreactivity for
synaptophysin (Fig. 2C), CD56,
cytokeratin (AE1/AE3) (Fig. 2D),
vimentin, and NSE. Scattered cells were positive for somatostatin,
whereas staining for chromogranin was negative. Stainings for
hepatocytes (arginase-1, polyclonal CEA and HepPar1) were negative.
The average proliferation index (Ki-67) was <10%.
The histopatological assessment of the resected
liver tumour revealed a trabecularly arranged pattern consisting of
relatively uniform cells predominantly placed in anastomosing,
irregular cell strands. The stroma was highly vascular. The tumour
cells contained round or oval nuclei, abundant pink-coloured and
granular cytoplasm, and infrequent mitoses (Fig. 1B and 1C). Immunohistochemistry of
the liver tumour was consistent with the immunoprofile of the
orbital specimen (Fig. 1D). These
findings were in agreement with the diagnosis of a primary
low-grade hepatic NET with an orbital metastasis.
Genomic imbalances
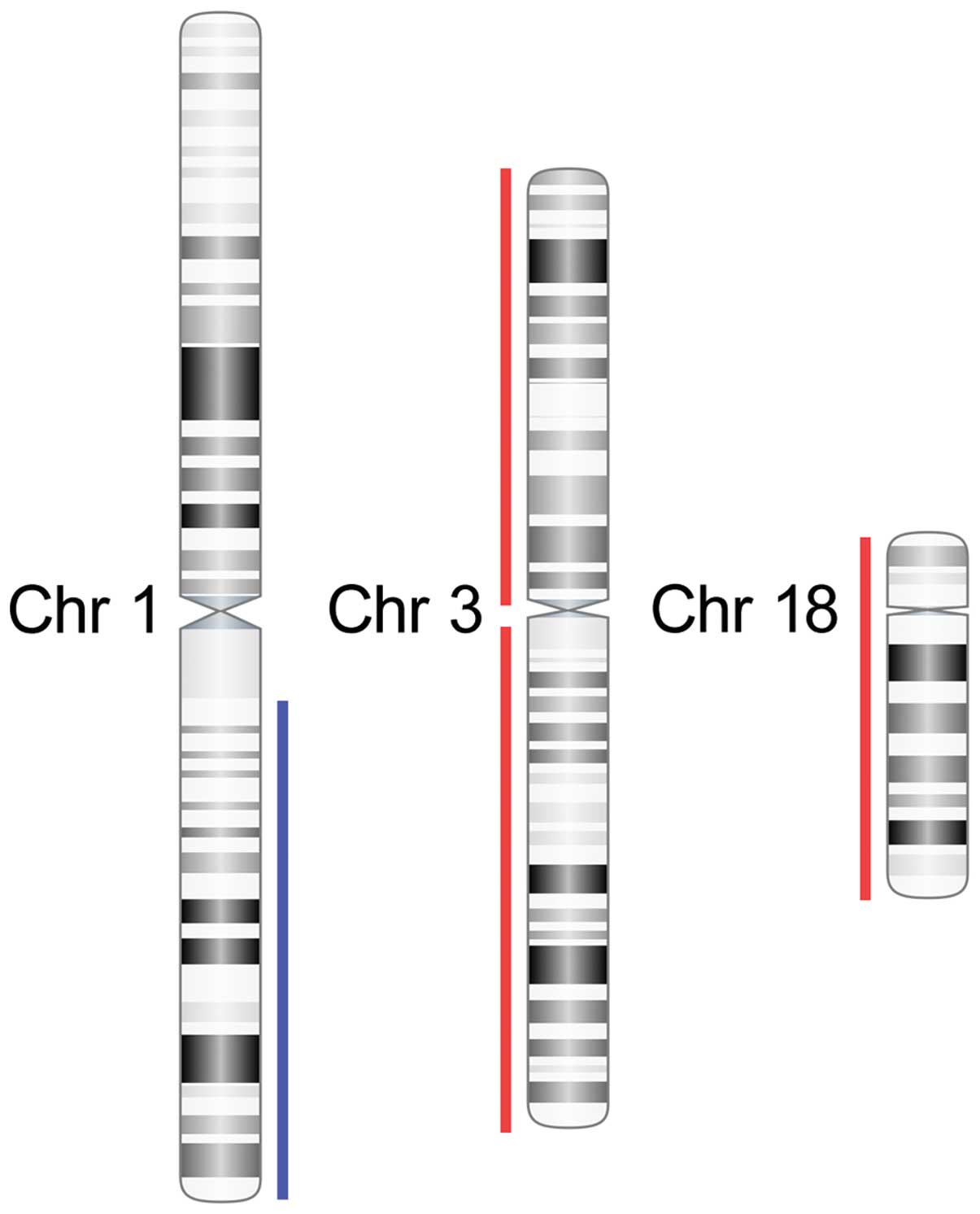
Genome-wide arrayCGH analysis of the primary tumour
using 244K arrays revealed a quiet genome with few copy number
alterations. Three major aberrations were detected including gain
of one copy of the long arm of chromosome 1 (1q21.1-qter) and
losses of one copy each of chromosomes 3 and 18 (Fig. 3). No segmental aberrations,
high-copy gains/amplifications or homozygous deletions were
detected.
The DNA isolated from the orbital metastasis was
degraded and yielded array data of poorer quality. However,
analysis of this array using less stringent criteria for aberration
calls confirmed the presence of the same aberrations as in the
liver tumour as well as gain of 2q24.2-qter and loss of one copy of
the long arm of chromosome 10 (data not shown).
Discussion
We here present the first reported case of an
orbital metastasis with origin from a primary hepatic
neuroendocrine carcinoma including genetic analysis of both
tumours. Primary hepatic NETs are extremely rare with only 90–100
cases reported to date (6), and the
orbit is an extremely rare anatomical site for metastasis from NETs
with less than 70 reported cases in the literature (5,7–9). The
5-year survival rate for hepatic neuroendocrine carcinoma is
reported to be only 18% (2). In the
present case, the patient is still alive 15 years after being
diagnosed with the liver tumour, but only after intense surgical
and oncological treatment. The ‘golden standard’ of treating
orbital carcinoid is biopsy followed by local radiotherapy and
additional systemic chemotherapy (5). In the present case, the orbital tumour
was removed surgically, since the tumour did not respond adequately
to radiotherapy and systemic chemotherapy and is also at present
showing evidence of local recurrence.
Using arrayCGH we demonstrated that both the primary
hepatic NET and the orbital metastasis had gain of one copy of 1q
and losses of one copy each of chromosomes 3 and 18 (Fig. 3). Loss of chromosome 18 is a common
feature in gastrointestinal NETs and has been reported in 43–88% of
tumours (12,13). Deletion of 18q21, the location of
the tumour suppressor genes DCC (deleted in colorectal carcinoma)
and DPC4/SMAD4 (involved in transcriptional regulation), may play a
role in the pathogenesis of gastrointestinal and pancreatic
endocrine tumours (13). Loss of
chromosome 3 is only found in approximately 25% of gastrointestinal
NETs (12). In contrast, loss of
chromosome 3p has been reported as the most common alteration in
NETs of the lung (14). A high
frequency of loss of heterozygosity (LOH) at a locus proximal to
the von Hippel-Lindau (VHL) gene on 3p has been shown to be
necessary for malignant conversion of pancreatic islet cell
tumours, and LOH at 3q (15) is
reported in 50% of sporadic pancreatic endocrine tumours associated
with hepatic metastases (12).
Hepatocellular carcinomas (HCC) metastasising to the
orbit are extremely rare and only 17 cases have been reported in
the English language literature (16). In Europe, the incidence of HCC is in
the range 3–12 of 100,000 individuals, whereas it exceeds 30 of
100,000 in East Asia (17). Common
genetic alterations in HCC are mutations in TP53 and CTNNB1 (the
gene for β-catenin) and amplification of genes in 1q21-q22
(18). In contrast, the observed
alterations involving chromosomes 3 and 18 in the present case are
uncommon in HCC (18).
Taken together, the present hepatic NET and its
orbital metastasis have genetic features consistent with
gastrointestinal NETs. The mechanism for development of primary
hepatic NETs is unclear to date, while theories include origin from
the biliary system or from ectopic adrenal or pancreatic tissues in
the liver (19). The results of the
present study indicate that hepatic NETs exhibit genetic
similarities mainly to gastrointestinal NETs; however, further
studies are needed to clarify these issues. Genetic studies may
also provide new biomarkers that can form the basis for a new
classification system and serve as prognostic biomarkers.
References
|
1
|
Pinchot SN, Holen K, Sippel RS and Chen H:
Carcinoid Tumors. Oncologist. 13:1255–1269. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Williams ED and Sandler M: The
classification of carcinoid tumours. Lancet. 1:238–239. 1963.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Friedman JR and Kaestner KH: On the origin
of the liver. J Clin Invest. 121:4630–4633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Modlin IM, Lye KD and Kidd M: A 5-decade
analysis of 13,715 carcinoid tumors. Cancer. 97:934–959. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mehta JS, Abou-Rayyah Y and Rose GE:
Orbital carcinoid metastases. Ophthalmology. 113:466–472. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fenoglio LM, Severini S, Ferrigno D and
Gollè G: Primary hepatic carcinoid: A case report and literature
review. World J Gastroenterol. 15:2418–2422. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turaka K, Mashayekhi A and Shields Cl: A
case series of neuroendocrine (carcinoid) tumor metastasis to the
orbit. Oman J Ophthalmol. 4:125–128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gupta A, Chazen JL and Phillips CD:
Carcinoid tumor metastases to the extraocular muscles: MR imaging
and CT findings and review of the literature. AJNR Am J
Neuroradiol. 32:1208–1211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsuo T, Ichimura K, Tanaka T and
Takenaka T: Neuroendocrine tumor (carcinoid) metastatic to orbital
extraocular muscle: case report and literature review. Strabismus.
18:123–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barrett MT, Scheffer A, Ben-Dor A, Sampas
N, Lipson D, Kincaid R, Tsang P, Curry B, Baird K, Meltzer PS,
Yakhini Z, Bruhn L and Laderman S: Comparative genomic
hybridization using oligonucleotide microarrays and total genomic
DNA. Proc Natl Acad Sci USA. 101:17765–17770. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Persson F, Winnes M, Andrén Y, et al:
High-resolution array CGH analysis of salivary gland tumors reveals
fusion and amplification of the FGFR1 and PLAG1 genes in ring
chromosomes. Oncogene. 27:3072–3080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zikusoka MN, Kidd M, Eick G, Latich I and
Modlin IM: The molecular genetics of gastroenteropancreatic
neuroendocrine tumors. Cancer. 104:2292–2309. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andersson E, Swärd C, Stenman G, Ahlman H
and Nilsson O: High-resolution genomic profiling reveals gain of
chromosome 14 as a predictor of poor outcome in ileal carcinoids.
Endocr Relat Cancer. 16:953–966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leotlela PD, Jauch A, Holtgreve-Grez H and
Thakker RV: Genetics of neuroendocrine and carcinoid tumours.
Endocr Relat Cancer. 10:437–450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tönnies H, Toliat MR, Ramel C and Pape UF:
Analysis of sporadic neuroendocrine tumours of the enteropancreatic
system by comparative genomic hybridisation. Gut. 48:536–541.
2001.PubMed/NCBI
|
|
16
|
Guerriero S, Infante G and Giancipoli E:
Hepatocellular carcinoma metastasis to the orbit in a coinfected
HIV+ HBV+ patient previously treated with
orthotopic liver transplantation: a case report. Case Rep
Ophthalmol Med. 2011:5492702011.PubMed/NCBI
|
|
17
|
Hirunwiwatkul P, Tirakunwichcha S,
Meesuaypong P and Shuangshoti S: Orbital metastasis of
hepatocellular carcinoma. J Neuroophthalmol. 28:47–50. 2008.
View Article : Google Scholar
|
|
18
|
van Malenstein H, van Pelt J and Verslype
C: Molecular classification of hepatocellular carcinoma anno 2011.
Eur J Cancer. 47:1789–1797. 2011.PubMed/NCBI
|
|
19
|
Gao J, Hu Z, Wu J, Bai L and Chai X:
Primary hepatic carcinoid tumor. World J Surg Oncol. 9:1512011.
View Article : Google Scholar
|