Introduction
Burkitt’s lymphoma (BL) is a malignancy of the human
lymphatic system that was first described in children in Central
Africa by the surgeon Denis Parsons Burkitt in 1956. The
pathogenesis of both the endemic and the sporadic types of BL are
closely related to impaired immunity and Epstein-Barr virus (EBV)
infection (1,2). In addition to EBV, BL is also
associated with a translocation involving the c-Myc and IgH genes,
t(8;14)(q24;q32) (3). The first
human BL cell line, Raji, was established in 1963 and was found to
integrate the EBV into its genome, leading to the discovery and
isolation of this virus (4). EBV
infection has been reported to contribute to BL development by
increasing the lymphomagenic potential of c-Myc
translocation-positive cells (5).
Moreover, Lassoued et al suggested that the EBV infection of
B lymphocytes and epithelial cells leads to oxidative stress that
facilitates viral transformation (6). Cerimele et al also demonstrated
that EBV-positive BL cells exhibit higher levels of reactive oxygen
species (ROS) than EBV-negative BL cells and that latent membrane
protein 1 (LMP1) is a major inducer of ROS (7). LMP1 is known to resemble CD40 and
function as a constitutively active tumor necrosis factor receptor
(TNFR) (8). Ha et al showed
that the ligation and activation of CD40 produces ROS by activating
the NAD(P)H oxidase (NOX) regulatory subunit p40phox
(9). The NOX family is an important
intrinsic source of ROS that consists of seven catalytic enzymes
(NOX1-5 and DUOX 1-2) and six regulatory subunits (10). Using NAD(P)H as a substrate, NOX
catalyzes the conversion of oxygen to superoxide.
In cancer cells, ROS stimulate oncogenic
transformation, cell proliferation, and mitochondrial malfunction
(11–14), and the redox status alters the
sensitivity of cancer cells to anticancer agents (15). In BL cells, an amonafide analog was
demonstrated to actively inhibit cell proliferation and induce
apoptosis via an ROS-mediated mitochondrial pathway (16). Rituximab was found to elevate ROS
generation and sensitize BL cells to X-irradiation (17). Since multiple effective and
clinically used chemotherapy agents induce excess ROS accumulation
to increase their anticancer activities (18,19),
most researchers have focused on developing ROS-stimulating
compounds to improve anticancer treatment outcomes (15,20).
However, less attention has been paid to the anticancer effects of
ROS-suppressing compounds.
Scientists believe that ROS are important
carcinogens (21) and that
ROS-suppressing compounds can be used as chemoprevention drugs to
reverse ROS-induced carcinogenesis (22). For example, polyphenols have been
reported to target cyclooxygenase-2 (COX-2) and scavenge ROS in
tumor cells, indicating the antitumorigenic potentials of these
compounds (23). In addition to the
polyphenol compounds derived from phytonutrients, microalgal
products have also exhibited chemopreventative activities in cancer
cells (24). The chemical compound
dibenziodolium chloride (DPI), which is a potent inhibitor of NOX,
is thought to significantly suppress ROS levels, and Yamaura et
al reported that inhibition of NOX activity by DPI arrests
melanoma cells at the G2-M phase of the cell cycle (25). Although anti-ROS compounds have been
proven to prevent the development of various types of cancers,
there are limited reports of their destruction of cancer cells.
In the present study, we found that EBV-encoded LMP1
was responsible for the activation of NOX and the accumulation of
cellular ROS in Raji BL cells. Inhibition of EBV-activated NOX by
DPI not only suppressed cellular ROS levels but also led to lactate
accumulation, which first arrested the Raji cells at the G2-M phase
of the cell cycle and subsequently resulted in significant
apoptosis. The obvious decreases in the expression levels of c-Myc
and Cdc25A within 6 h of DPI treatment suggest that c-Myc and
Cdc25A may be responsible for DPI-induced cell cycle arrest in Raji
BL cells. In this case, the ability of EBV infection to activate
NOX and stimulate ROS generation represents a novel therapeutic
target for BL treatment. Due to its ability to inhibit NOX-ROS-cell
cycle progression, it is possible to use DPI to suppress the
proliferation of BL cells.
Materials and methods
Materials
CM-H2DCF-DA was purchased from
Invitrogen-Molecular Probes (Carlsbad, CA, USA). DPI was purchased
from Sigma-Aldrich (St. Louis, MO, USA), dissolved in dimethyl
sulfoxide (DMSO) and freshly diluted in culture media before use.
The final DMSO concentration was <0.1% (v/v).
Cell lines
Raji cells, which were derived from a
well-established human BL cell line with latent EBV infection, JM1
cells (an acute lymphoblastic leukemia B cell line) and NALM 16
cells (a human hematopoietic B cell line) were maintained in our
laboratory in RPMI-1640 medium (Gibco-BRL, Gaithersburg, MD, USA)
supplemented with 10% heat-inactivated fetal bovine serum (Thermo
Scientific, HyClone, Logan, UT, USA). Cells were incubated in a
humidified, 5% CO2 atmosphere at 37°C.
Flow cytometry
Cellular ROS concentrations were measured by
incubating control or drug-treated Raji, JM1 and NALM16 cells with
1 μM CM-H2DCF-DA for 60 min followed by flow cytometric
analysis using a FACSCalibur equipped with CellQuest Pro software.
CM-H2DCF-DA is a fluorescent probe with relative
specificity for hydrogen peroxide (41). To determine the effects of the drug
on the cell cycle, propidium iodide (PI) staining after 75% alcohol
fixation was used, followed by flow cytometric analysis. Cell death
was determined by flow cytometry after the cells were double
stained with Annexin V-FITC and PI using an assay kit from BD
Pharmingen (San Diego, CA, USA) in a routine manner.
Examination of cellular lactate
generation
To analyze cellular lactate production, cells in the
exponential growth phase were incubated with fresh medium or medium
containing DPI for 24 h. Aliquots of culture medium were then
removed for analysis of lactate concentrations using a portable
Accutrend lactate analyzer with a linear range of standard lactate
concentrations, according to the manufacturer’s recommendations
(Roche, Mannheim, Germany).
Assays for DPI cytotoxicity
Cell viability was assessed using MTS assays. Raji
cells were plated in 96-well culture clusters (Costa, Cambridge,
MA, USA) at densities of 20,000–30,000 cells/ml, and serial
dilutions of DPI were prepared at concentrations of 2.5–20 μM using
a stock solution. All experimental concentrations were prepared in
triplicate. The percent absorbance of the DPI-treated cells
relative to that of the DMSO-treated control cells (<0.1% DMSO)
was plotted as a linear function of the DPI concentration. The
antiproliferative effect of DPI on the Raji cells was measured as
the percentage of viable cells relative to the DMSO-treated control
cells.
NOX activity assay
DPI is widely used as an inhibitor of flavoenzymes,
particularly NOX. To determine cellular NOX activity, 10 μM DPI was
added 4 h before the cells were harvested. The control and the
DPI-treated NP69 and NP69-LMP1 cells were lysed in hypotonic
phosphate buffer containing protease inhibitors, disrupted by
sonication, and centrifuged for 10 min at 1,500 rpm. The
supernatant containing the cytosolic and mitochondrial fractions
was further ultracentrifuged at 100,000 × g for 30 min at 4°C. The
resulting pellet, which contained the membranous fractions of the
cytosol and mitochondria, was resuspended in buffer B [50 mM Tris
(pH 7.5), 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail
(one tablet for 10 ml of buffer)] and used for the assay. The
samples were adjusted to the same protein concentration (1 μg/μl).
Then, 5-μl aliquots of the samples were incubated with 94 μl of
phosphate buffer (50 mM K2HPO4, 1 mM EGTA,
and 150 mM sucrose) and with 1 μl of 3-NAD(P)H (4 mM) for 15 min.
Before measurements were performed, 2.5 μl of lucigenin (2 mM) was
added, and the lucigenin-derived chemiluminescence of the cell
homogenates was assessed over 1 min in a 20/20n tube
luminometer (Turner Biosystems, Sunnyvale, CA, USA).
Small interfering RNAs and
transfection
The siRNA pools for LMP1 and the non-targeting
siRNAs were purchased from Thermo Scientific Dharmacon.
Lipofectamine™ 2000 transfection reagent (Invitrogen) was used to
introduce the siRNA (final concentration of 50 nM) or plasmids [the
ratio of DNA (μg) to lipid reagent (μl) was 1 to 2.5] into the
cells.
Reverse transcriptase PCR and qPCR assays
for LMP1
Total RNA was isolated from cells using an RNeasy
Mini kit (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions, and cDNA was synthesized as previously
described (4). Thirty-five cycles
of PCR were performed, and the primer sequences used to amplify
LMP1 were as follows: forward, 5′-CGTTATGAGTGACTGGACTGGA-3′ and
reverse, 5′-TGAACAGCACAATTCCAAGG-3′. Quantifications of LMP1 mRNA
expression in the NPC and non-cancer cells were performed by
amplifications using SYBR-Green. The absolute threshold cycle
values (Ct values) of LMP1 were determined using SDS software v.2.1
(Applied Biosystems). LMP1 mRNA expression was analyzed by
determining its Ct values relative to those of β-actin.
Immunoblot analysis
Protein analyses by immunoblotting were performed as
previously described (42) using
primary antibodies against c-Myc (sc-764; Santa Cruz Biotechnology,
Santa Cruz, CA, USA), Cdh1 (ab3242; Abcam), Chk1 (#2345; Cell
Signaling), Cdc25A (sc-97; Santa Cruz Biotechnology), DDB1 (#5428;
Cell Signaling), CDK2 (sc-163), cyclin E (sc-481) (both from Santa
Cruz Biotechnology) and GAPDH (ab9489; Abcam). Total cell lysates
were harvested, subjected to 12% SDS-polyacrylamide gel
electrophoresis and then transferred to polyvinylidene difluoride
membranes (Roche, Basel, Switzerland). Immunoblotting involved
incubation with the primary antibodies followed by the addition of
secondary antibodies conjugated to horseradish peroxidase (Cell
Signaling) to facilitate detection. Subsequently, enhanced
chemiluminescence reagent (Cell Signaling) was added to develop the
blots.
Statistical analysis
All analyses for comparing the significance of the
measured levels were performed using the one-way ANOVA test with
SPSS 19.0 software.
Results
Accumulation of cellular ROS in Raji BL
cells is triggered by EBV-encoded LMP1
BL is unique among the various types of B cell
malignancies since its carcinogenesis is closely associated with
EBV infection. EBV-encoded LMP1 is predominantly expressed in B
cell lymphocytes latently infected with EBV. The virus-encoded
nuclear protein 2 (EBNA2) and its downstream factor LMP1 have been
reported to facilitate ROS production in malignant B cells
(7). The NOX family proteins
consume NAD(P)H and transfer electrons across biological membranes
to generate superoxide and ROS (26). Thus, we hypothesized that LMP1 may
be able to stimulate ROS generation by activating NOX in BL cells.
To test our hypothesis, we first analyzed ROS and NOX activity
levels in Raji cells (from a well-established BL cell line with
latent EBV infection) and in two other EBV-negative, malignant B
cell lines, JM1 (an acute lymphoblastic leukemia B cell line) and
NALM 16 (a human hematopoietic B cell line). As illustrated in
Fig. 1A and B, the Raji cells
exhibited a much higher basal ROS level (9.7- and 35.9-fold) and
significantly higher NOX activity (2.9- and 10.5-fold) compared
with the EBV-negative B cell lines NALM16 and JM1, respectively
(p<0.01). Next, we detected EBV-encoded LMP1 mRNA levels by
RT-PCR and qPCR. As shown in Fig. 1C
and D, the expression of LMP1 mRNA was much higher in the Raji
BL cells than in the JM1 and NALM16 cells. To identify the effects
of LMP1 on NOX activation in the Raji BL cells, we knocked down
LMP1 expression using RNAi and then analyzed NOX activity using
lucigenin and a luminometer. As shown in Fig. 1E, inhibition of LMP1 expression by
siRNA markedly suppressed NOX activity, suggesting that EBV LMP1
may be responsible for activating NOX enzyme activity as well as
the generation of excessive ROS in the EBV-positive Raji BL
cells.
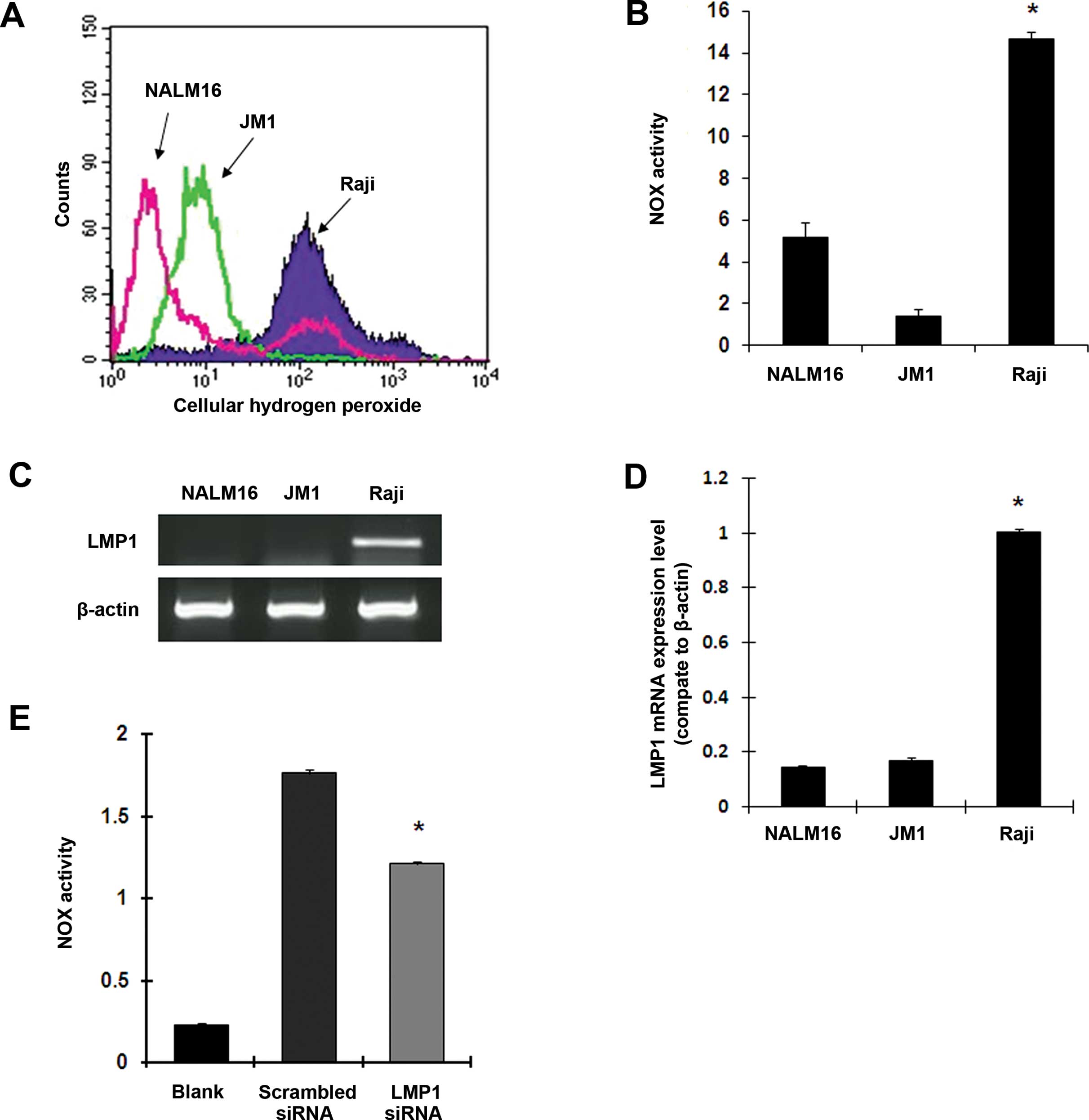 | Figure 1Epstein-Barr virus-encoded LMP1
upregulates NOX activity and ROS levels in Raji BL cells. (A)
Comparison of cellular ROS concentrations in Raji, JM1 and NALM16
cells using flow cytometry and H2DCF-DA. Each histogram
is representative of three experiments (p<0.001, Raji vs. JM1
cells). (B) Comparison of NOX activity in Raji, JM1 and NALM16
cells, measured using a luminometer and lucigenin in the presence
of NAD(P)H (mean ± SD of three experiments; *p<0.01).
(C) mRNA expression of LMP1 in NALM16, JM1 and Raji cells, measured
by RT-PCR assays. β-actin served as an internal control. (D)
Quantitative real-time PCR was performed to quantify mRNA
expression of LMP1 in Raji, JM1 and NALM16 cells (mean ± SD of
three experiments; *p<0.001). (E) The effects of LMP1
siRNA on NOX activity in Raji cells were measured using a
luminometer and lucigenin in the presence of NAD(P)H (mean ± SD of
three experiments; *p<0.01). LMP1, latent membrane
protein 1; NOX, NAD(P)H oxidase; ROS, reactive oxygen species; BL,
Burkitt’s lymphoma. |
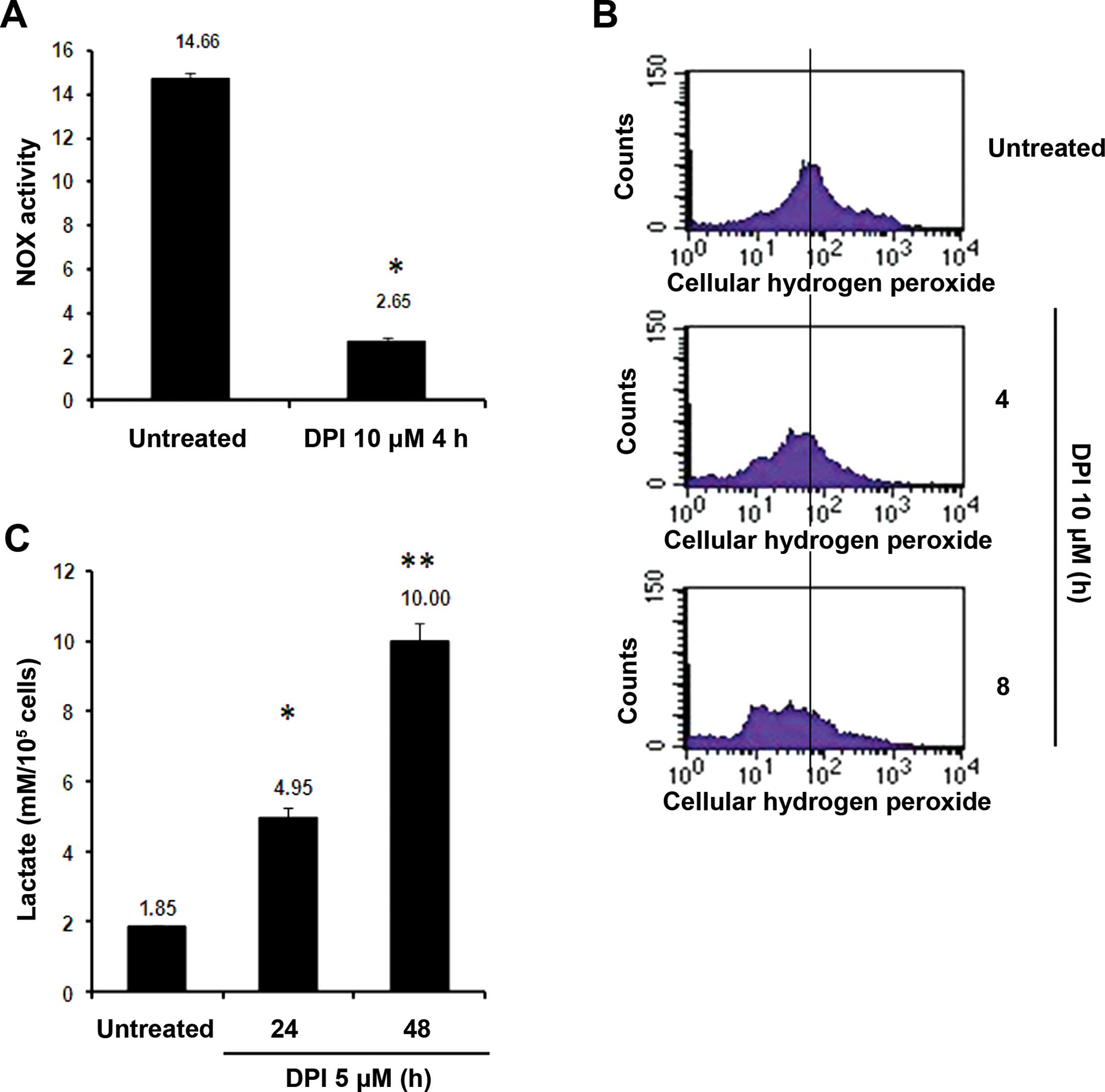
Inhibition of NOX activity by DPI
suppresses cellular ROS accumulation and stimulates lactate
generation
Over the past decade, most studies have focused on
evaluating ROS-stimulated compounds, investigating their effects on
the destruction of cancer cells since excessive ROS accumulation is
thought to result in the apoptosis of these cells (15,20).
However, little attention has been paid to the application of novel
compounds that suppress cellular ROS levels in anti-cancer
treatments. In the present study, we found that the
small-molecular-weight compound DPI was able to effectively inhibit
NOX activity (by ~82%, Fig. 2A) in
the Raji cells. Since the aforementioned findings suggest that
LMP1-activated NOX is responsible for stimulating ROS generation in
Raji BL cells, we suspected that DPI may suppress cellular ROS
levels in Raji cells by inhibiting NOX activity. As shown in
Fig. 2B, flow cytometry indicated
that 5 μM DPI effectively suppressed ROS accumulation in the Raji
cells by blocking NOX activity, causing ~35 and 57% decreases in
cellular ROS concentrations at 4 and 8 h, respectively. Notably, in
addition to suppressing ROS levels, the DPI treatment was able to
markedly elevate lactate production in the Raji cells. As shown in
Fig. 2C, treatment of Raji cells
with 5 μM DPI caused significant increases in lactate levels by
2.6- and 5.4-fold at 24 and 48 h, respectively. These increases may
have occurred due to upregulation of lactate dehydrogenase (LDH)
activity by inhibition of NOX since NOX and LDH utilize the same
substrate, NAD(P)H, to generate ROS and lactate.
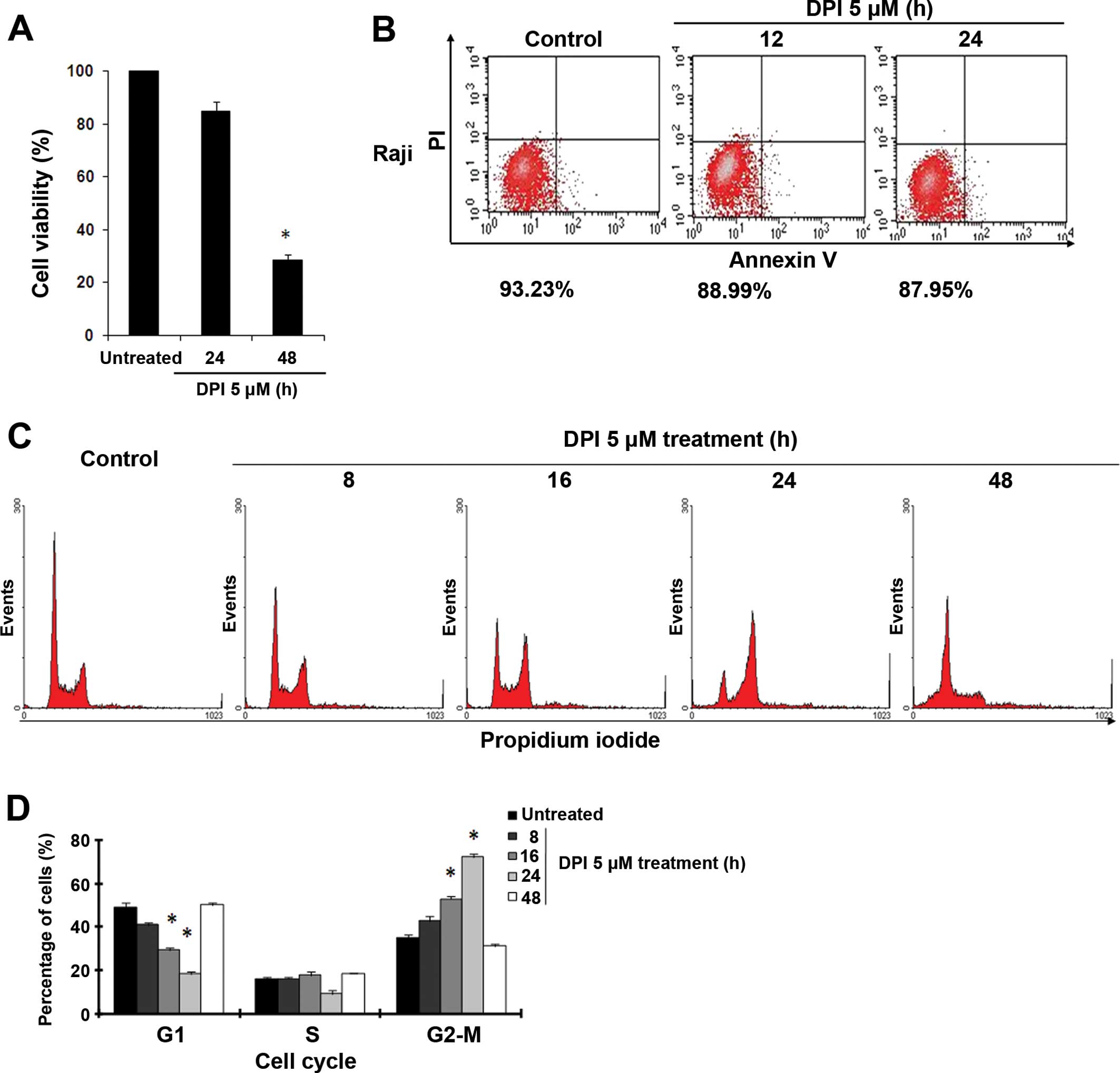
DPI treatment arrests Raji cells at the
G2-M phase of the cell cycle
Lactate accumulation can be harmful to cancer cells
and cause irreversible damage, suggesting that a hyperacidic
environment and ROS inhibition may mediate the anticancer effects
of DPI. As shown in Fig. 3A, DPI
effectively inhibited proliferation of the Raji cells at 48 h, and
~72% of their capacity for proliferation was inhibited at 5 μM DPI.
However, both cell viability assays and Annexin V/PI flow
cytometric analyses showed that 24 h of incubation with DPI (5 μM)
did not cause obvious cell apoptosis in the Raji cells (Fig. 3A and B). Instead of undergoing
apoptosis, many of the cells were arrested at the G2-M phase of the
cell cycle within 24 h of DPI treatment (Fig. 3C). Compared to the 34% of untreated
Raji cells at the G2-M phase, 43, 53 and 72% of these cells were
arrested at the G2-M phase after DPI treatments for 8, 16 and 24 h,
respectively, as quantified by flow cytometry using PI as a
fluorescent probe (Fig. 3C and D).
When we prolonged the DPI incubation time to 48 h, this G2-M phase
arrest disappeared, and large numbers of Raji cells were arrested
at the sub-G1 phase, indicating the occurrence of apoptotic cell
death (Fig. 3C and D). These data
suggest that the anticancer activity mediated by DPI first resulted
in cell cycle arrest during the G2-M phase, which may have led to
apoptotic cell death.
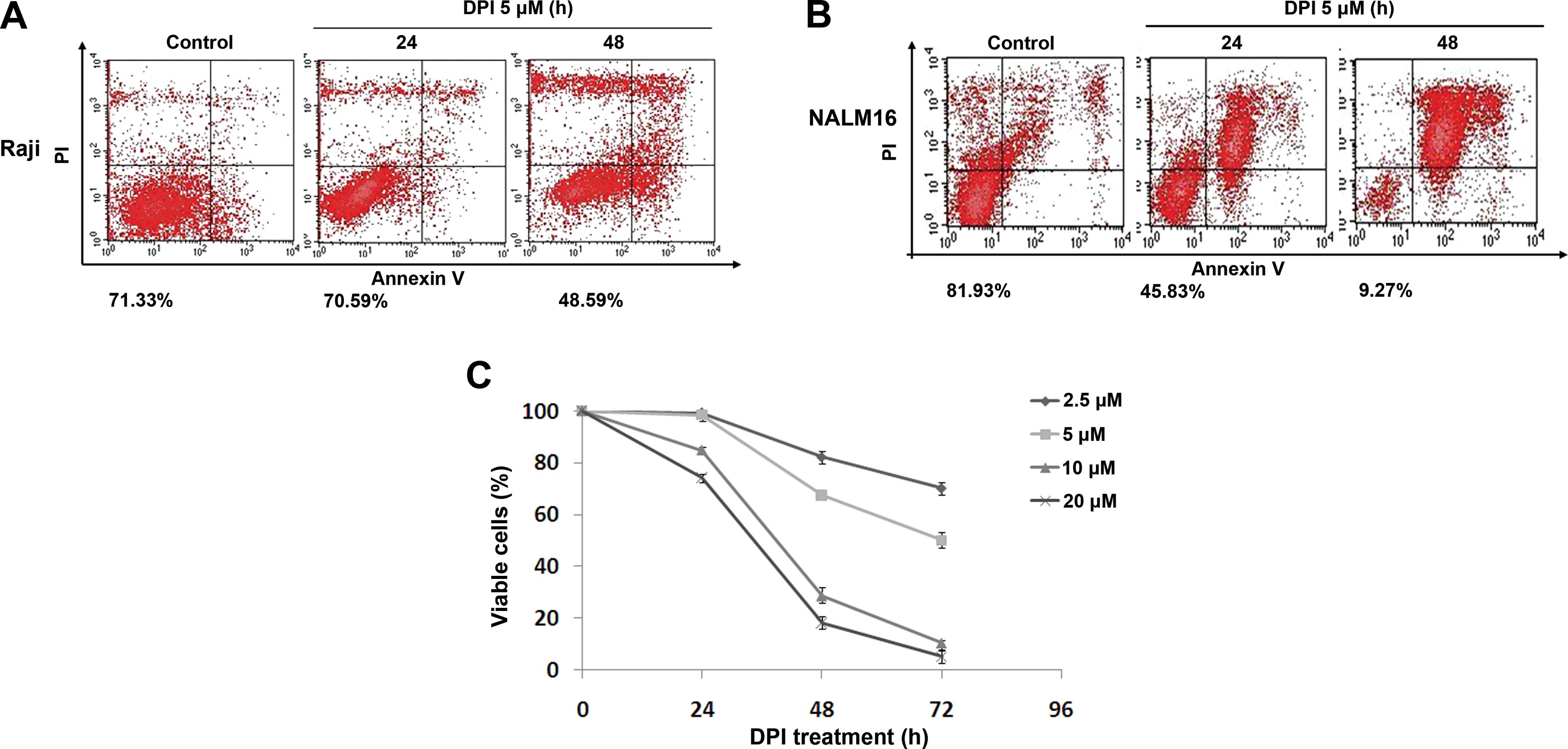
DPI treatment induces apoptosis in Raji
cells
Since the aforementioned flow cytometry data
indicated that the apoptotic cell death was the result of
DPI-induced cell cycle arrest, we further examined the cell-killing
effects of DPI on Raji BL cells. Annexin V/PI flow cytometry assays
were adopted to evaluate the cytotoxicity of DPI in the Raji cells.
As shown in Fig. 4A and B, compared
to the NALM16 cells, which exhibited obvious cell death at both 24
and 48 h after the 5-μM DPI treatment, Raji cells only exhibited
significant apoptotic cell death at 48 h after the 5-μM DPI
treatment, implying a delayed response of these cells to
DPI-induced cell cycle arrest. In other words, the DPI-induced
destruction of Raji cells may have been caused by the effects of
DPI on the cell cycle. Subsequent MTS assays were performed to
assess cell viability after DPI treatment at a series of
concentrations ranging from 2.5 to 20 μM. As illustrated in
Fig. 4C, the DPI treatment induced
time- and dose-dependent cell death in Raji cells, and a
significant decrease in cell viability was first observed at 48 h
after treatment. Based on these data, we believe that DPI is a
potent killer of Raji BL cells and may represent an improved, novel
treatment strategy for the treatment of BL patients.
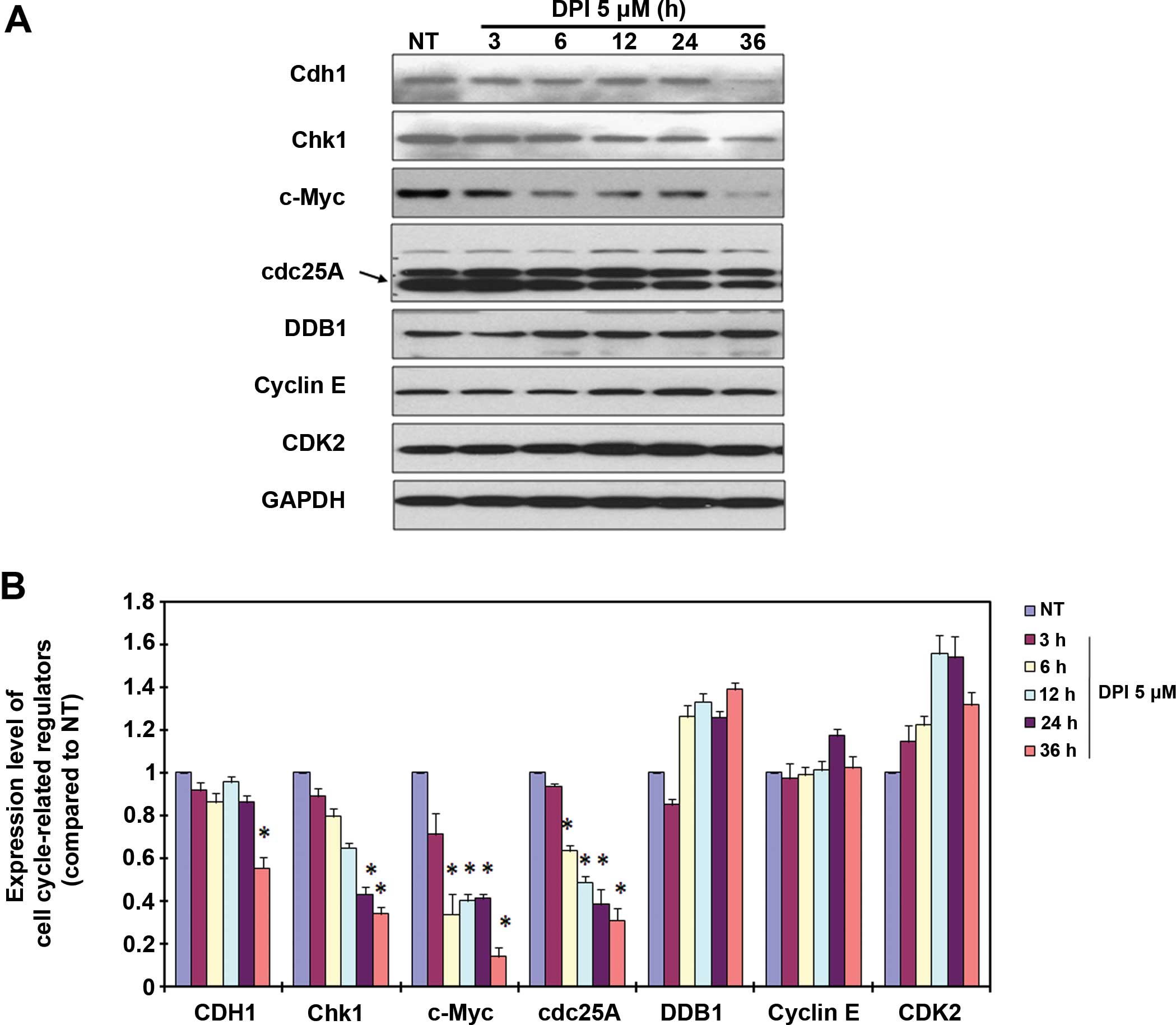
Cell cycle regulators are involved in
DPI-induced cell cycle arrest
By inhibiting NOX activity, DPI arrested the Raji
cells at the G2-M phase of the cell cycle and effectively induced
apoptotic cell death. To further investigate the related molecular
mechanism, a panel of cell cycle regulators was analyzed by
immunoblotting following treatments for 3–36 h. The data indicated
that the cell cycle regulators Cdh1, Chk1, c-Myc and Cdc25 were
modified by the 5-μM DPI treatment (Fig. 5A and B). Among these four proteins,
c-Myc and Cdc25 were significantly downregulated at 6 h after
treatment, Chk1 was significantly downregulated at 12 h after
treatment, and Cdh1 was only obviously downregulated at 36 h after
treatment. Based on these modifications of the cell cycle
regulators, we suspect that c-Myc and Cdc25 are quite possibly
involved in DPI-induced G2-M cell cycle arrest in the Raji BL
cells.
Discussion
The incidence of BL, which is a highly aggressive
form of B lymphoma that remains problematic for children in endemic
areas, has increased dramatically worldwide due to its association
with immunodeficiency (27). EBV,
which is a frequently occurring virus found in all human
populations, is strongly linked to the pathogenesis of BL. While
most studies have focused on the activities of cancer-related
viruses in stimulating the oncogenic signaling pathways of host
cells, few investigations have evaluated their roles in modifying
the redox status of tumor cells. Recent studies have shown that the
hepatitis B virus X protein (HBx) is involved in viral metabolism
and ROS accumulation in liver disease (28). Human T-cell leukemia virus type 1
(HTLV-1) has also been demonstrated to stimulate ROS generation
under glucose deprivation (29).
Previous studies have reported that EBV-positive BL cells express
high levels of mitogen-activated protein kinase (MAPK) and have
high ROS concentrations compared with EBV-negative malignant B
cells (7). In the present study,
the role of EBV in ROS stimulation in BL cells and its underlying
applications for clinical use were investigated. Following latent
EBV infection and LMP1 expression, EBV-positive BL cells (Raji
cells) exhibited much higher NOX activity and ROS levels than two
EBV-negative malignant B cell lines (JM1 and NALM16). By inhibiting
NOX activity, DPI was a potent suppressor of cellular ROS levels,
which resulted in lactate accumulation that initially caused
significant cell cycle arrest during the G2-M phase and
subsequently led to substantial apoptosis of Raji BL cells.
Moreover, we suggest that c-Myc and Cdc25A may contribute to
DPI-induced cell cycle arrest and the subsequent apoptosis.
EBV infection is known to have three latency
programs (latency I, II and III), each of which leads to the
production of a distinct panel of viral proteins and RNAs (30). Consistent with a previous report
that emphasized the role of EBNA2 and its downstream factor LMP1 in
ROS induction in BL cells with EBV type III latent infections
(7), our findings suggest an
important role of LMP1 in activating NOX and modulating ROS
generation in Raji BL cells. When we knocked down LMP1 expression
by RNAi, NOX enzyme activity was suppressed.
The NOX family consists of a group of enzymes that
utilize molecular oxygen to generate ROS. Since activated NOX
enzymes in cancer cells consume the substrate NAD(P)H, which is
also used as a substrate by LDH to produce lactate, some reports
have indicated that NOX is also involved in modulating glycolysis
(31,32). Since the activation of NOX enzymes
is always linked with inflammation and malignant disorders, it can
be targeted to treat these diseases (33,34).
DPI is a well-established inhibitor of NOX enzymes. In pancreatic
adenocarcinoma cells, DPI potently inhibits ROS production and
induces apoptosis (35). In chronic
myelogenous leukemia (CML) cells, both DPI and NOX RNAi suppress
BCR-ABL-induced cell proliferation and reduce cellular growth
(36). In addition to DPI, multiple
other compounds, such as plumbagin, celastrol and phenothiazines,
have been shown to effectively target NOX (37–39).
Here, we demonstrated that inhibition of NOX by DPI not only
suppressed ROS levels yet also elevated lactate production.
Excessive lactate concentrations can lead to lactic acidosis,
causing great harm to cells. In this manner, DPI induced a marked
G2-M phase cell cycle arrest that eventually led to irreversible
apoptosis in Raji BL cells.
The underlying mechanisms of NOX in cell cycle
progression have rarely been investigated, but some information can
be obtained from recent publications. ROS produced by NOX enzymes
can influence cell cycle progression by regulating phosphorylation
and ubiquitination of cyclin-dependent kinases (CDKs) and cell
cycle regulators (40). The cell
cycle progression induced by platelet-derived growth factor (PDGF)
is partially mediated by NOX1-induced ROS production (26), and NOX4 has been associated with
G2-M phase cell cycle progression in melanoma cells (25). In these reports, Cdc25
phosphorylation and the ubiquitination of CDK inhibitor proteins
(CKIs) have been suggested to be involved in NOX-mediated cell
cycle progression. In our system, CDK2 expression seemed to be
elevated after DPI treatment, possibly due to ubiquitination of
CKIs. Consistent with previous findings, Cdc25 expression was
altered by DPI treatment. More importantly, c-Myc, which is a vital
oncoprotein in BL cells, was significantly downregulated by DPI,
strongly implying its potential use for the treatment of BL.
In summary, the present study linked EBV infection
with NOX-activated ROS generation in BL cells, suggesting that NOX
activation is a potential target for development of anti-cancer
treatment strategies for BL patients. DPI, which is a specific
inhibitor of NOX enzymes, was demonstrated to be capable of
decreasing cellular ROS concentrations and of stimulating excess
lactate generation, resulting in a G2-M phase cell cycle arrest and
eventually leading to significant apoptosis in Raji cells.
Moreover, the involved cell cycle regulators were likely c-Myc and
Cdc25. Therefore, we propose that EBV-induced NOX activation and
ROS generation are potent targets for BL treatment. By halting cell
cycle progression and inducing apoptosis, DPI demonstrated the
ability to kill Raji BL cells, indicating that it may be valuable
for development as an effective anti-BL treatment.
References
|
1
|
Vereide DT, Seto E, Chiu YF, Hayes M,
Tagawa T, Grundhoff A, Hammerschmidt W and Sugden B: Epstein-Barr
virus maintains lymphomas via its miRNAs. Oncogene. 33:1258–1264.
2014. View Article : Google Scholar
|
|
2
|
Bornkamm GW: Epstein-Barr virus and the
pathogenesis of Burkitt’s lymphoma: more questions than answers.
Int J Cancer. 124:1745–1755. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Molyneux EM, Rochford R, Griffin B, Newton
R, Jackson G, Menon G, Harrison CJ, Israels T and Bailey S:
Burkitt’s lymphoma. Lancet. 379:1234–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Drexler HG and Minowada J: History and
classification of human leukemia-lymphoma cell lines. Leuk
Lymphoma. 31:305–316. 1998.PubMed/NCBI
|
|
5
|
Bieging KT, Amick AC and Longnecker R:
Epstein-Barr virus LMP2A bypasses p53 inactivation in a MYC model
of lymphomagenesis. Proc Natl Acad Sci USA. 106:17945–17950. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lassoued S, Ben Ameur R, Ayadi W, Gargouri
B, Ben Mansour R and Attia H: Epstein-Barr virus induces an
oxidative stress during the early stages of infection in B
lymphocytes, epithelial, and lymphoblastoid cell lines. Mol Cell
Biochem. 313:179–186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cerimele F, Battle T, Lynch R, Frank DA,
Murad E, Cohen C, Macaron N, Sixbey J, Smith K, Watnick RS,
Eliopoulos A, Shehata B and Arbiser JL: Reactive oxygen signaling
and MAPK activation distinguish Epstein-Barr Virus (EBV)-positive
versus EBV-negative Burkitt’s lymphoma. Proc Natl Acad Sci USA.
102:175–179. 2005. View Article : Google Scholar
|
|
8
|
Zheng H, Li LL, Hu DS, Deng XY and Cao Y:
Role of Epstein-Barr virus encoded latent membrane protein 1 in the
carcinogenesis of nasopharyngeal carcinoma. Cell Mol Immunol.
4:185–196. 2007.PubMed/NCBI
|
|
9
|
Ha YJ and Lee JR: Role of TNF
receptor-associated factor 3 in the CD40 signaling by production of
reactive oxygen species through association with
p40phox, a cytosolic subunit of nicotinamide adenine
dinucleotide phosphate oxidase. J Immunol. 172:231–239. 2004.
View Article : Google Scholar
|
|
10
|
Lambeth JD, Kawahara T and Diebold B:
Regulation of Nox and Duox enzymatic activity and expression. Free
Radic Biol Med. 43:319–331. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Devi GS, Prasad MH, Saraswathi I, Raghu D,
Rao DN and Reddy PP: Free radicals antioxidant enzymes and lipid
peroxidation in different types of leukemias. Clin Chim Acta.
293:53–62. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park SY, Chang I, Kim JY, Kang SW, Park
SH, Singh K and Lee MS: Resistance of mitochondrial DNA-depleted
cells against cell death: role of mitochondrial superoxide
dismutase. J Biol Chem. 279:7512–7520. 2004. View Article : Google Scholar
|
|
13
|
Behrend L, Henderson G and Zwacka RM:
Reactive oxygen species in oncogenic transformation. Biochem Soc
Trans. 31:1441–1444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roth D, Krammer PH and Gulow K: Dynamin
related protein 1-dependent mitochondrial fission regulates
oxidative signalling in T cells. FEBS Lett. 588:1749–1754. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trachootham D, Zhou Y, Zhang H, Demizu Y,
Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J and
Huang P: Selective killing of oncogenically transformed cells
through a ROS-mediated mechanism by β-phenylethyl isothiocyanate.
Cancer Cell. 10:241–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin B, Chen Z, Xu Y, Zhang H, Liu J and
Qian X: 7-b, a novel amonafide analogue, cause growth inhibition
and apoptosis in Raji cells via a ROS-mediated mitochondrial
pathway. Leuk Res. 35:646–656. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fengling M, Fenju L, Wanxin W, Lijia Z,
Jiandong T, Zu W, Xin Y and Qingxiang G: Rituximab sensitizes a
Burkitt lymphoma cell line to cell killing by X-irradiation. Radiat
Environ Biophys. 48:371–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuang Y, Balakrishnan K, Gandhi V and Peng
X: Hydrogen peroxide inducible DNA cross-linking agents: targeted
anti-cancer prodrugs. J Am Chem Soc. 133:19278–19281. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fang J, Nakamura H and Iyer AK:
Tumor-targeted induction of oxystress for cancer therapy. J Drug
Target. 15:475–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu ZY, Wang J, Cheng G, Zhu XF, Huang P,
Yang D and Zeng YX: Apogossypolone targets mitochondria and light
enhances its anticancer activity by stimulating generation of
singlet oxygen and reactive oxygen species. Chin J Cancer.
30:41–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Acharya A, Das I, Chandhok D and Saha T:
Redox regulation in cancer: a double-edged sword with therapeutic
potential. Oxid Med Cell Longev. 3:23–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee KW and Lee HJ: The roles of
polyphenols in cancer chemoprevention. Biofactors. 26:105–121.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roy P, George J, Srivastava S, Tyagi S and
Shukla Y: Inhibitory effects of tea polyphenols by targeting
cyclooxygenase-2 through regulation of nuclear factor kappa B, Akt
and p53 in rat mammary tumors. Invest New Drugs. 29:225–231. 2011.
View Article : Google Scholar
|
|
24
|
Talero E, Ávila-Roman J and Motilva V:
Chemoprevention with phytonutrients and microalgae products in
chronic inflammation and colon cancer. Curr Pharm Des.
18:3939–3965. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamaura M, Mitsushita J, Furuta S, Kiniwa
Y, Ashida A, Goto Y, Shang WH, Kubodera M, Kato M, Takata M, Saida
T and Kamata T: NADPH oxidase 4 contributes to transformation
phenotype of melanoma cells by regulating G2-M cell
cycle progression. Cancer Res. 69:2647–2654. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mbulaiteye SM, Anderson WF, Ferlay J,
Bhatia K, Chang C, Rosenberg PS, Devesa SS and Parkin DM:
Pediatric, elderly, and emerging adult-onset peaks in Burkitt’s
lymphoma incidence diagnosed in four continents, excluding Africa.
Am J Hematol. 87:573–578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gu JM, Lim SO, Oh SJ, Yoon SM, Seong JK
and Jung G: HBx modulates iron regulatory protein 1-mediated iron
metabolism via reactive oxygen species. Virus Res. 133:167–177.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Silic-Benussi M, Cavallari I, Vajente N,
Vidali S, Chieco-Bianchi L, Di Lisa F, Saggioro D, D’Agostino DM
and Ciminale V: Redox regulation of T-cell turnover by the p13
protein of human T-cell leukemia virus type 1: distinct effects in
primary versus transformed cells. Blood. 116:54–62. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hutzinger R, Feederle R, Mrazek J,
Schiefermeier N, Balwierz PJ, Zavolan M, Polacek N, Delecluse HJ
and Hüttenhofer A: Expression and processing of a small nucleolar
RNA from the Epstein-Barr virus genome. PLoS Pathog.
5:e10005472009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang
F, Feng L, Pelicano H, Wang H, Keating MJ, Liu J, McKeehan W, Wang
H, Luo Y and Huang P: Novel role of NOX in supporting aerobic
glycolysis in cancer cells with mitochondrial dysfunction and as a
potential target for cancer therapy. PLoS Biol. 10:e10013262012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prata C, Maraldi T, Fiorentini D, Zambonin
L, Hakim G and Landi L: Nox-generated ROS modulate glucose uptake
in a leukaemic cell line. Free Radic Res. 42:405–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han CY, Umemoto T, Omer M, Den Hartigh LJ,
Chiba T, LeBoeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED and
Chait A: NADPH oxidase-derived reactive oxygen species increases
expression of monocyte chemotactic factor genes in cultured
adipocytes. J Biol Chem. 287:10379–10393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Block K and Gorin Y: Aiding and abetting
roles of NOX oxidases in cellular transformation. Nat Rev Cancer.
12:627–637. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mochizuki T, Furuta S, Mitsushita J, Shang
WH, Ito M, Yokoo Y, Yamaura M, Ishizone S, Nakayama J, Konagai A,
Hirose K, Kiyosawa K and Kamata T: Inhibition of NADPH oxidase 4
activates apoptosis via the AKT/apoptosis signal-regulating kinase
1 pathway in pancreatic cancer PANC-1 cells. Oncogene.
25:3699–3707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Singh MM, Irwin ME, Gao Y, Ban K, Shi P,
Arlinghaus RB, Amin HM and Chandra J: Inhibition of the NADPH
oxidase regulates heme oxygenase 1 expression in chronic myeloid
leukemia. Cancer. 118:3433–3445. 2012. View Article : Google Scholar :
|
|
37
|
Ding Y, Chen ZJ, Liu S, Che D, Vetter M
and Chang CH: Inhibition of Nox-4 activity by plumbagin, a
plant-derived bioactive naphthoquinone. J Pharm Pharmacol.
57:111–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gianni D, Taulet N, Zhang H, Der
Mardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM
and Rosen H: A novel and specific NADPH oxidase-1 (Nox1)
small-molecule inhibitor blocks the formation of functional
invadopodia in human colon cancer cells. ACS Chem Biol. 5:981–993.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jaquet V, Marcoux J, Forest E, Leidal KG,
McCormick S, Westermaier Y, Perozzo R, Plastre O, Fioraso-Cartier
L, Diebold B, Scapozza L, Nauseef WM, Fieschi F, Krause KH and
Bedard K: NADPH oxidase (NOX) isoforms are inhibited by celastrol
with a dual mode of action. Br J Pharmacol. 164:507–520. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Verbon EH, Post JA and Boonstra J: The
influence of reactive oxygen species on cell cycle progression in
mammalian cells. Gene. 511:1–6. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pelicano H, Feng L, Zhou Y, Carew JS,
Hileman EO, Plunkett W, Keating MJ and Huang P: Inhibition of
mitochondrial respiration: a novel strategy to enhance drug-induced
apoptosis in human leukemia cells by a reactive oxygen
species-mediated mechanism. J Biol Chem. 278:37832–37839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guo C, Pan ZG, Li DJ, Yun JP, Zheng MZ, Hu
ZY, Cheng LZ and Zeng YX: The expression of p63 is associated with
the differential stage in nasopharyngeal carcinoma and EBV
infection. J Transl Med. 4:232006. View Article : Google Scholar : PubMed/NCBI
|