Introduction
Breast cancer has become the first common malignancy
in women in developed and developing countries. Approximately 1.3
million women are diagnosed with breast cancer annually worldwide
(1). Surgery is the important
therapeutic method of breast cancer. Chemotherapy, radiotherapy and
endocrine therapy are also important in breast cancer. Although
comprehensive therapy has been previously employed, approximately
0.5 million women patients succumb to breast cancer annually due to
recurrence, metastasis and resistance to therapy (2). Therefore, more effective therapeutic
strategies are required to improve treatment outcomes for breast
cancer patients.
Tumor cells do not exist in isolation during disease
progression. The occurrence of an intense fibro-inflammatory
reaction involving immune cells (3)
and cancer-associated fibroblasts (4,5) is a
prominent pathologic feature of breast cancer (6). The cooperative interactions among
tumor cells and reactive stroma strongly contribute to cancer
development and progression (7,8).
Cancer-associated fibroblasts (CAFs) have been indicated as the
main cell component of the tumor microenvironment involved in
cancer initiation, invasion and metastasis (9,10). In
breast malignancies, CAFs exert a pivotal role in tumor progression
and resistance to therapeutics through multiple mechanisms,
including the stimulation of new blood vessels (11), mainly generated by a hypoxic tumor
microenvironment (12–14). The mechanisms of cell sensing and
adaptation to stressful environments are activated within the
hypoxic tumor mass, leading to the growth and aggressiveness of
malignant cells (15). Despite this
finding, issues relating to maintenance of the tumor fibrotic
microenvironment during disease development remain to be
addressed.
Solid tumors often experience low oxygen tension
environments, which is predominantly caused by abnormal vasculature
formation of the rapidly growing tumor mass. Tumor hypoxia is
associated with enhanced tumor invasiveness, angiogenesis, and
distant metastasis (16–18). GPER and HIF-1α are recruited to the
HRE site located within the VEGF promoter region and cooperatively
act as a functional complex for the transcription of VEGF. Recent
studies have shown that hypoxia induced GPER expression in breast
cancer fibroblasts, and the cross-talk between HIF-1α and GPER
regulates the expression of the migratory factor CTGF (19).
We investigated the role of GPER in CAFs and
examined the effect of GPER silencing on hypoxia-driven breast
cancer progression. We found that GPER knockdown in CAFs suppressed
hypoxia-induced CAF activation and breast cancer cell invasion
through the inhibition of CTGF expression.
Materials and methods
Materials
The antibodies used in this study included
polyclonal rabbit anti-human HIF-1α (Bioworld, St. Louis Park, MN,
USA), polyclonal rabbit anti-human CTGF (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), polyclonal rabbit anti-human anti-GPER
(Santa Cruz Biotechnology, Inc.), monoclonal mouse anti-human MMP-9
(Santa Cruz Biotechnology, Inc.), polyclonal rabbit anti-human uPA
(Bioworld), monoclonal mouse anti-human α-SMA (Sigma, St. Louis,
MO, USA), monoclonal mouse anti-human cytokeratin14 (Sigma) and
monoclonal mouse anti-human β-actin (Santa Cruz Biotechnology,
Inc.).
Cell cultures
CAFs were extracted from invasive mammary ductal
carcinomas obtained from mastectomies as previously described
(20). These tissues were obtained
from the Department of Cancer Center at the First Affiliated
Hospital of Xi’an Jiaotong University. Signed informed consent from
all the patients was obtained. The study protocol and consent forms
were approved by the Ethics and Indications Committee of the First
Affiliated Hospital of Medical College, Xi’an Jiaotong University,
China. In particular, tissues obtained were cut into smaller
sections (1–2 mm diameter), placed in digestion solution [400 IU
collagenase, 100 IU hyaluronidase and 10% FBS (HyClone, Logan, UT,
USA), containing antibiotics and antimycotics solution] and
incubated overnight at 37°C. The cells were then separated by
differential centrifugation at 90 × g for 2 min.
The supernatant containing fibroblasts was
centrifuged at 485 × g for 8 min, the pellet obtained was suspended
in fibroblast growth medium (Medium 199 and Ham’s F12 mixed 1:1 and
supplemented with 10% FBS and 1% penicillin) and cultured at 37°C,
5% CO2. CAFs were then expanded into two 15-cm Petri
dishes and stored as cells passaged for 2–3 population doublings
within a total 7–10 days after tissue dissociation. CAFs were
passaged for up to five population doublings for subsequent
experiments to minimize clonal selection and culture stress, which
could occur during extended tissue culture. Primary cell cultures
of breast CAFs were characterized by immunofluorescence. Briefly,
the cells were incubated with anti-α-SMA and anti-cytokeratin14
(Fig. 1A), all antibodies were from
Sigma. MDA-MB-231 human breast cancer cells were purchased from the
Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and
were cultured at 37°C with 5% CO2 and 95% air in L-15
(Sigma) containing 10% heat-inactivated fetal bovine serum (FBS)
(HyClone) plus 100 μg/ml ampicillin and 100 μg/ml streptomycin.
Activation of CAFs
CAFs were cultured in regular growth medium to 80%
confluence. The cells were incubated under normoxic or hypoxic (3%
O2) conditions in fresh serum-free medium for 12 h prior
to the collection of conditioned media (CM).
Western blot analysis
CAFs or MDA-MB-231 cells (1×106) grown
under our experimental conditions were lysed for 20 min on ice in
300 μl of RIPA lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl,
1% Triton X-100, 2 mM EDTA, 1 mM sodium orthovanadate, 1 mM
phenylmethanesulfonyl-fluoride, 10 μg/ml aprotinin, 10 μg/ml
leupeptin]. Total proteins (100 μg) were loaded onto SDS-PAGE gels,
separated, and transferred onto PVDF membranes (Roche, Penzberg,
Germany). The membranes were blocked with 5% non-fat dry milk in
TBST [10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.05% Tween-20] and
were subsequently incubated with primary antibodies overnight at
4°C. After 5 washes of 10 min each in TBST, the membranes were
incubated with HRP-conjugated secondary antibodies (1:5000, Santa
Cruz Biotechnology, Inc.) for 2 h and subsequently washed again.
The peroxidase reaction was performed using an enhanced
chemiluminescence detection system to visualize the immunoreactive
bands.
Cell invasion assay
A chamber-based invasion assay (Millipore,
Billerica, MA, USA) was performed to evaluate breast cancer cell
invasion. Briefly, the upper surface of the membrane was coated
with matrigel (BD Biosciences, Franklin Lakes, NJ, USA). MDA-MB-231
cells (1×105) were resuspended in the upper chamber in
serum-free medium and allowed to migrate towards a serum gradient
(10%) in the lower chamber for 24 h. The medium was aspirated from
the inside of the insert, and the non-invasive cells on the upper
side were removed by scraping with a cotton swab. The membrane was
fixed with 4% paraformaldehyde and stained with crystal violet. The
number of migrating cells was counted on each membrane in 10 random
fields and photographed at a magnification of ×100. The values
reported were the averages of triplicate experiments.
Reverse-transcription quantitative PCR
assay (RT-qPCR)
Total RNAs were extracted from CAFs or breast cancer
cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and
reverse transcription was performed using the PrimeScript RT
Reagent kit (Takara, Dalian, China) according to the manufacturer’s
instructions. Real-time experiments were carried out using the iQ5
Multicolor Real-time PCR Detection System (Bio-Rad, Hercules, CA,
USA) and a SYBR-Green PCR kit (Takara). The following PCR program
was used: denaturation at 95°C for 30 sec, followed by 40 cycles
consisting of denaturation at 95°C for 5 sec, annealing at 60°C for
30 sec, and extension at 72°C for 30 sec. A melting curve analysis
was applied to assess the specificity of the amplified PCR
products. The PCR primer sequences used were: HIF-1α 5′-AAG
TCTAGGGATGCAGCA-3′ (forward) and 5′-CAAGATCA CCAGCATCATG-3′
(reverse), GPER 5′-ACACACCTG GGTGGACACAA-3′ (forward) and
5′-GGAGCCAGAAG CCACATCTG-3′ (reverse), VEGF 5′-TGCAGATTATGCG
GATCAAACC-3′ (forward) and 5′-TGCATTCACATTTGT TGTGCTGTAG-3′
(reverse), CTGF 5′-ACCTGTGGGATG GGCATCT-3′ (forward) and
5′-CAGGCGGCTCTG CTTCTCTA-3′ (reverse), IL-6 5′-AGTTCCTGCAGTCCAG
CCTGAG-3′ (forward) and 5′-TCAAACTGCATAGCCACTT TCC-3′ (reverse),
GAPDH 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and
5′-TCCACCACCCTGTTGCTGAT-3′ (reverse), The amount of each target
gene was quantified by the comparative C(T) method using GAPDH as
the normalization control (21).
Enzyme-linked immunosorbent assay
(ELISA)
The cells were conditioned in serum-free medium for
24 h. The culture media were then collected and centrifuged at
1,500 rpm for 5 min to remove particles. The supernatants were then
frozen at −80°C until use. The production of CTGF, IL-6, and VEGF
in the supernatants of CAFs was assessed by ELISA using a
commercially available ELISA kit (R&D Systems, Minneapolis, MN,
USA) according to the manufacturer’s instructions.
Immunofluorescence microscopy
After the designated treatment, CAFs were fixed with
4% paraformaldehyde for 10 min at room temperature, permeabilized
in 0.5% Triton X-100 for 10 min, and blocked in 1% BSA for 1 h.
Fixed cells were then incubated with mouse anti-human-α-SMA
antibodies (1:100) or mouse anti-human-cytokeratin14 antibodies
(1:100) at 4°C overnight. The cells were washed and incubated with
goat anti-mouse dyelight 594 (red) IgG antibody (Qenshare
Biological Inc., Xi’an, China) at 1:200 dilution for 60 min. Nuclei
were stained with DAPI for 5 min. The cells were visualized by a
fluorescent microscope (Observer A1, Carl Zeiss Microscopy GmbH,
Germany) using appropriate excitation and emission spectra at a
magnification of ×400.
RNA interference
siRNA against GPER (5′-CUGACACC GUCGACCAGGATT-3′,
5′-UCCUGGUCGACGGUGUC GTT-3′), siRNA against CTGF
(5′-AGAAUAUGAUGUUCA UCAATT-3′, 5′-UUGAUGAACAUCAUAUUCUTT-3′), and a
negative control siRNA (5′-UUCUCCGAACGUGUCAC GUTT-3′,
5′-ACGUGACACGUUCGGAGAATT-3′) were obtained from GenePharm
(Shanghai, China). Cells (2×105 per well) were seeded in
six-well plates and transfected with 100 nM siRNA using
Lipofectamine RNAiMAX reagent (Invitrogen) according to the
manufacturer’s instructions. After 48-h transfection, the cells
were used for subsequent experiments.
Statistical analysis
The data are presented as the means ± SD from at
least three independent experiments. Statistical analysis of the
data was performed using Student’s t-test using SPSS software
(version 13.0; SPSS, Chicago, IL, USA). P<0.05 was considered
statistically significant.
Results
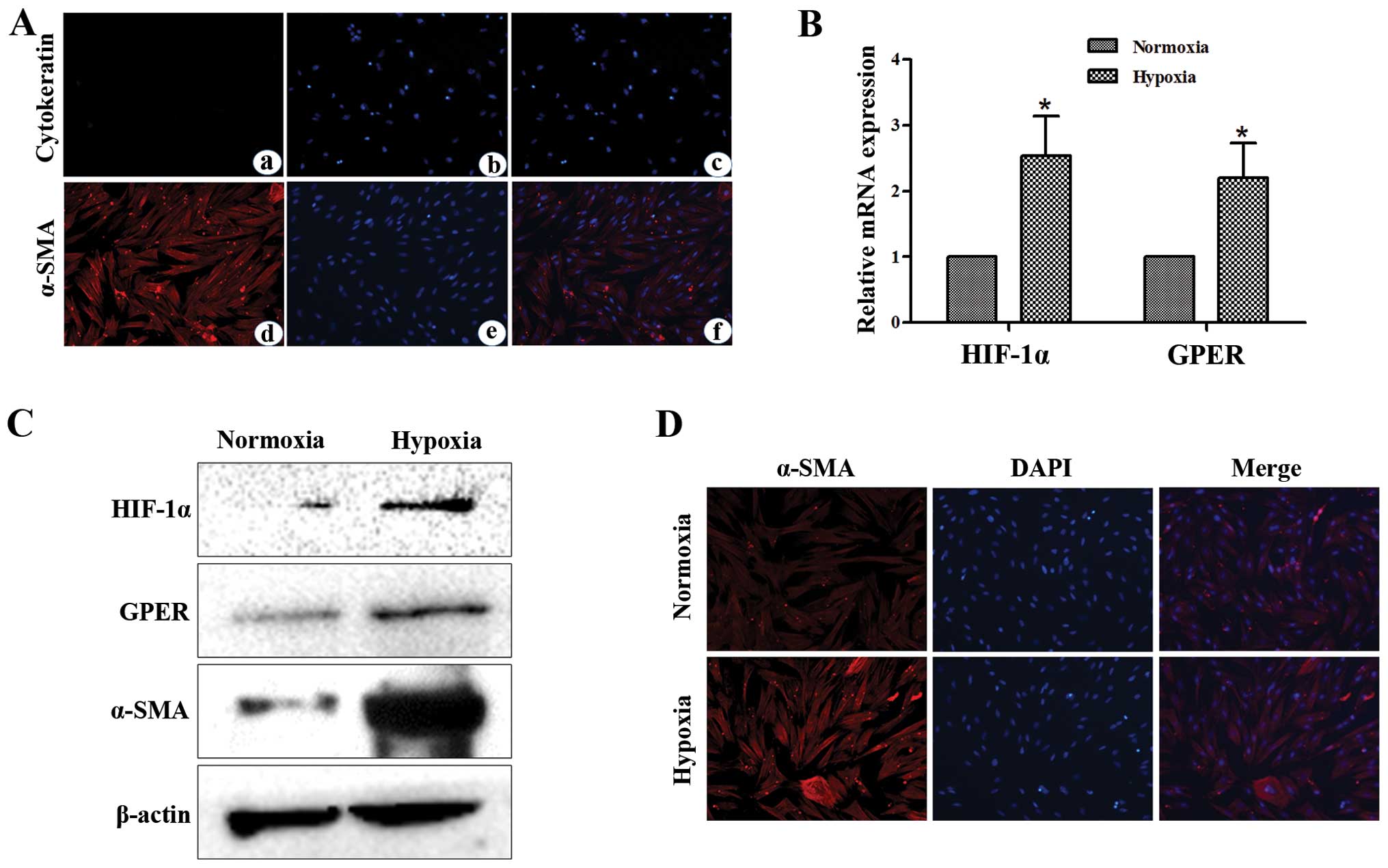
Hypoxia induces HIF-1α, GPER and α-SMA
expression in CAFs
Primary cell culture of breast CAFs was
characterized by immunofluorescence. Briefly, the cells were
incubated with human anti-α-SMA and human anti-cytokeratin
(Fig. 1A). To provide insight into
the response to hypoxia in the main components of the tumor
microenvironment such as CAFs, we showed that hypoxia induced the
mRNA expression of HIF-1α and its target gene GPER, as
ascertained by qPCR (Fig. 1B). The
induction of HIF-1α and GPER mRNA expression was paralleled by
increased protein levels of these factors in CAFs exposed to a
low-oxygen tension (3% O2) for 12 h (Fig. 1C). Furthermore, we observed that
hypoxia increased activation of CAFs, as revealed by α-SMA
expression (Fig. 1C and D).
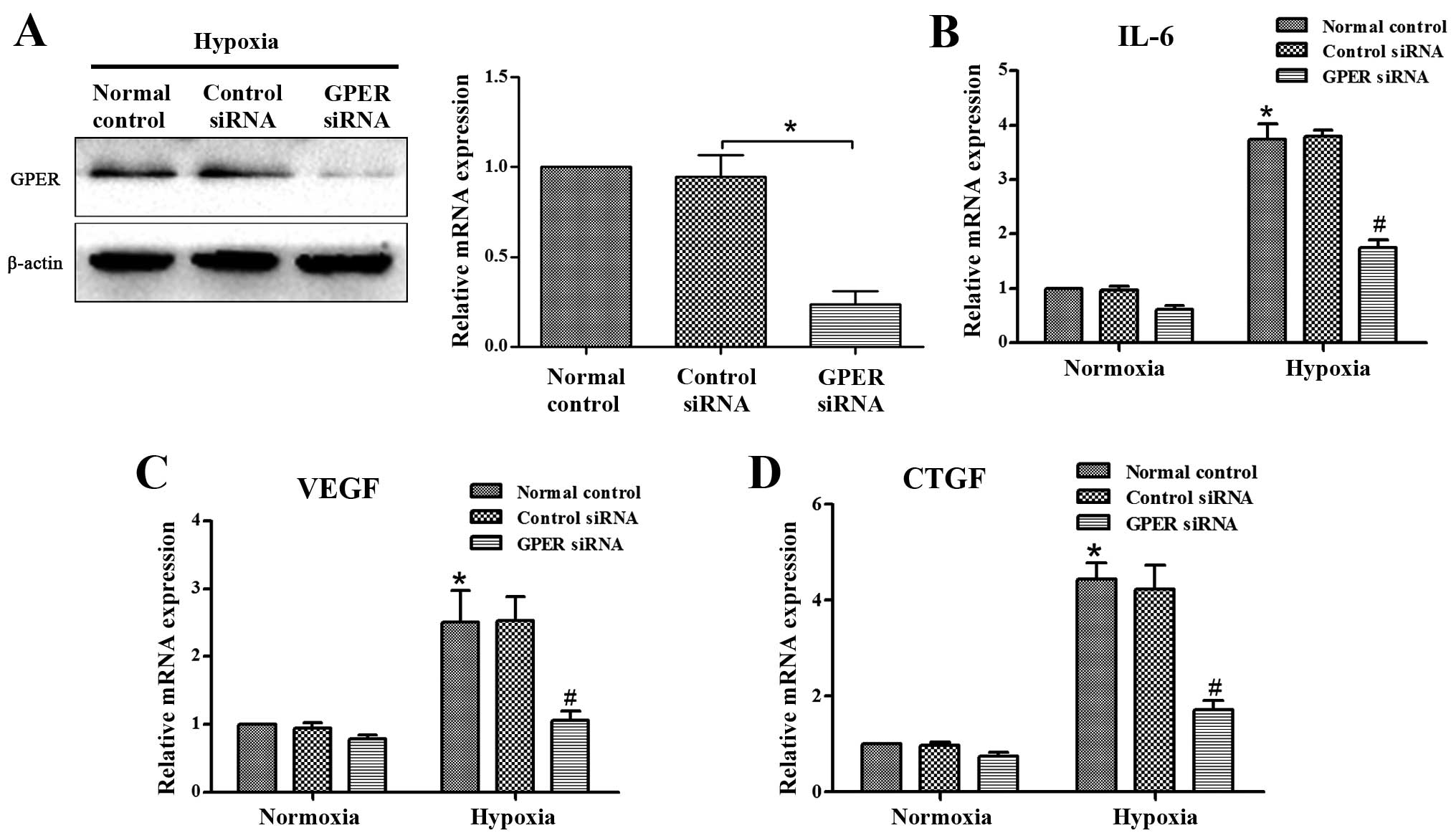
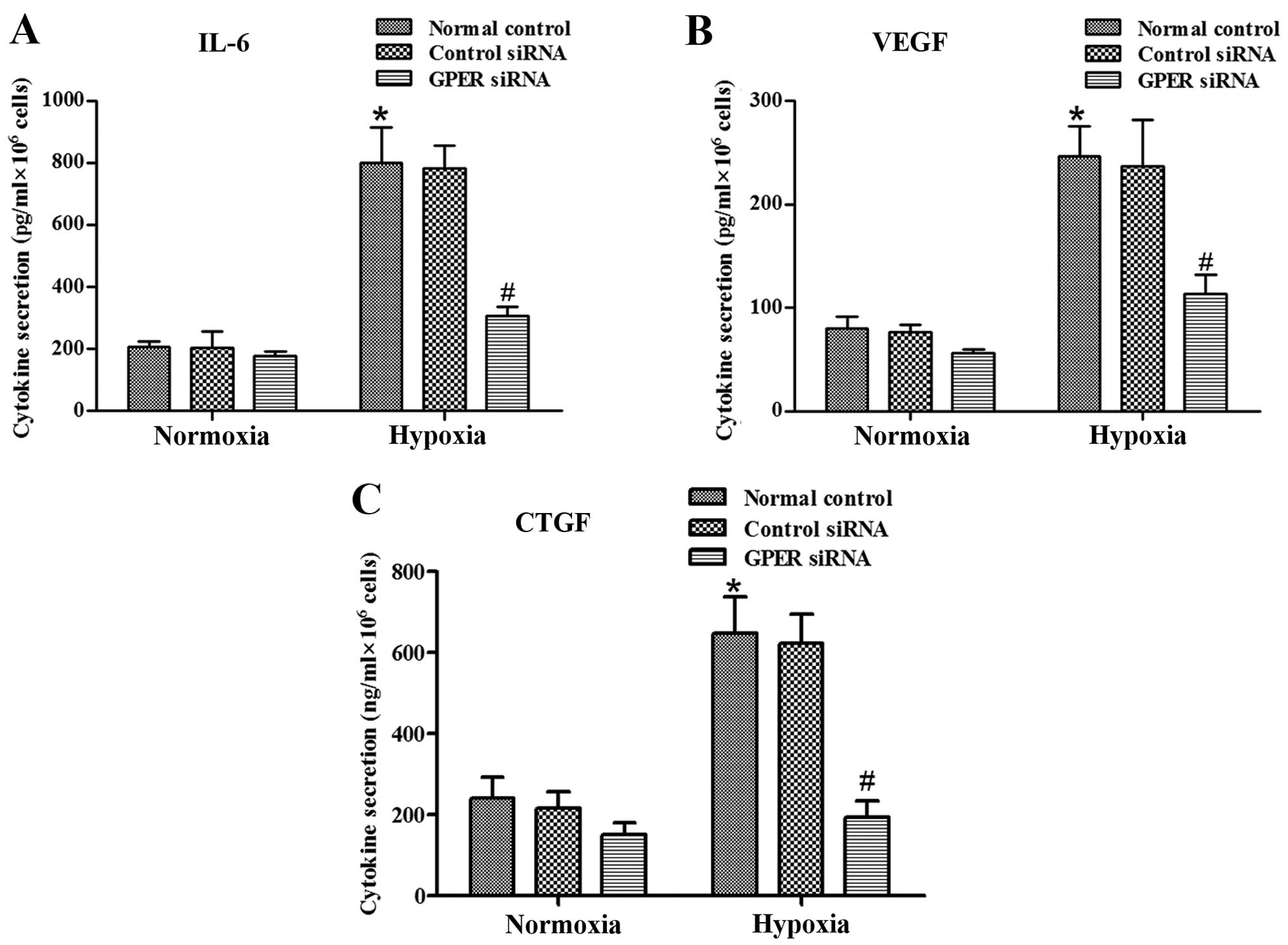
GPER silencing abrogates
hypoxia-activated IL-6, VEGF and CTGF secretion in CAFs
Previous results from other laboratories indicated
that the activated stroma secretes large amounts of IL-6, VEGF and
CTGF, leading to a significant increase in the invasive capacity of
the surrounding tumor cells (22–24).
To verify whether hypoxia-activated CAFs overexpress these soluble
growth factors and cytokines, we performed RT-qPCR and ELISA to
quantify IL-6, VEGF and CTGF expression. As shown in Fig. 2B–D and Fig. 3A–C, CAFs cultured under hypoxic
conditions exhibit higher levels of IL-6, VEGF and CTGF
transcription and secretion. These factors are known to be involved
in modulating the response of tumor cells to activated CAFs. GPER
was involved in hypoxia-induced VEGF expression in breast cancer
CAFs (19). We investigated the
role of GPER in these hypoxia-induced effects, and found that GPER
was knocked down by siRNA (Fig.
2A). The data showed that silencing of GPER abrogated the
hypoxia-induced overexpression of these factors in CAFs (Fig. 2B–D and Fig. 3A–C).
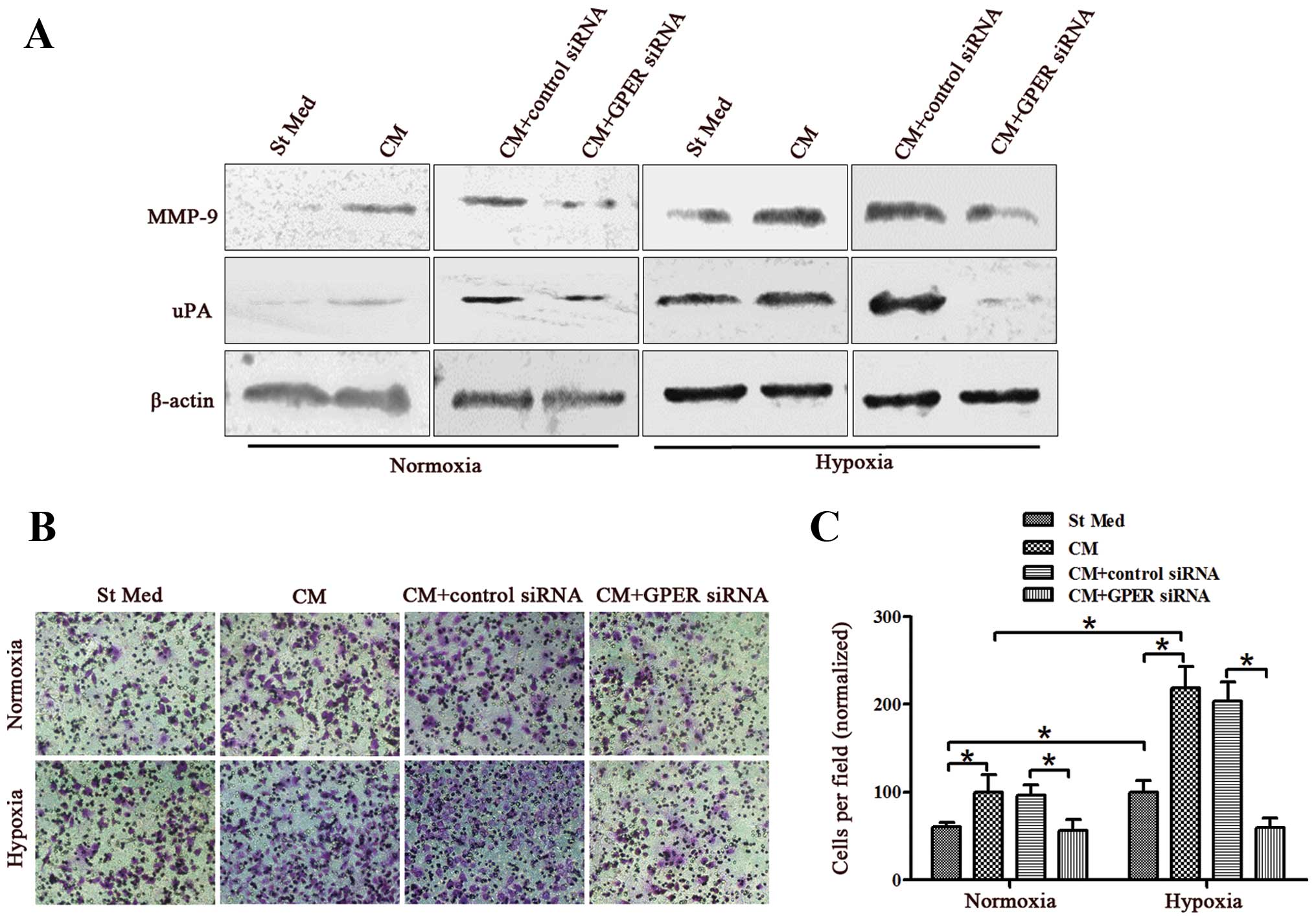
Knockdown of GPER in CAFs suppresses
breast cancer cell invasion induced by CAF conditioned media under
hypoxic conditions
It has been demonstrated that high levels of GPER
expression in cancer cells are linked to enhanced invasive
potential (25,26). Thus, we investigated whether GPER
derived from stromal components also influenced the behavior of
tumor cells. We examined whether media from CAFs cultured under
hypoxic conditions promoted the metastatic potential of cancer
cells (using MDA-MB-231 cells derived from human primary breast
adenocarcinoma). We treated MDA-MB-231 cells with conditioned media
(CM) from CAFs activated by hypoxia with or without GPER silencing,
and assayed their ability to express invasion-associated enzymes
(e.g., MMP-9 or uPA) and to invade through a reconstituted matrigel
barrier. The results revealed that CM from CAFs significantly
increased the MMP-9 and uPA levels of breast cancer cells under
either normoxic or hypoxic conditions (Fig. 4A). Moreover, CM from CAFs was mildly
active in promoting the invasiveness of breast cancer cells under
the two conditions (Fig. 4B and C).
Exposure of CAFs to hypoxia during their activation enhances their
ability to affect breast cancer motility, leading to a 1.7-fold
(normoxia) or 2.2-fold (hypoxia) increase in invasiveness (Fig. 4B and C). However, GPER knockdown
eliminated the effects of activated CAFs and hypoxia on breast
cancer invasiveness (Fig. 4). These
findings suggested that CAFs are sensitive to hypoxia, which
enhances their promotion of breast cancer invasiveness.
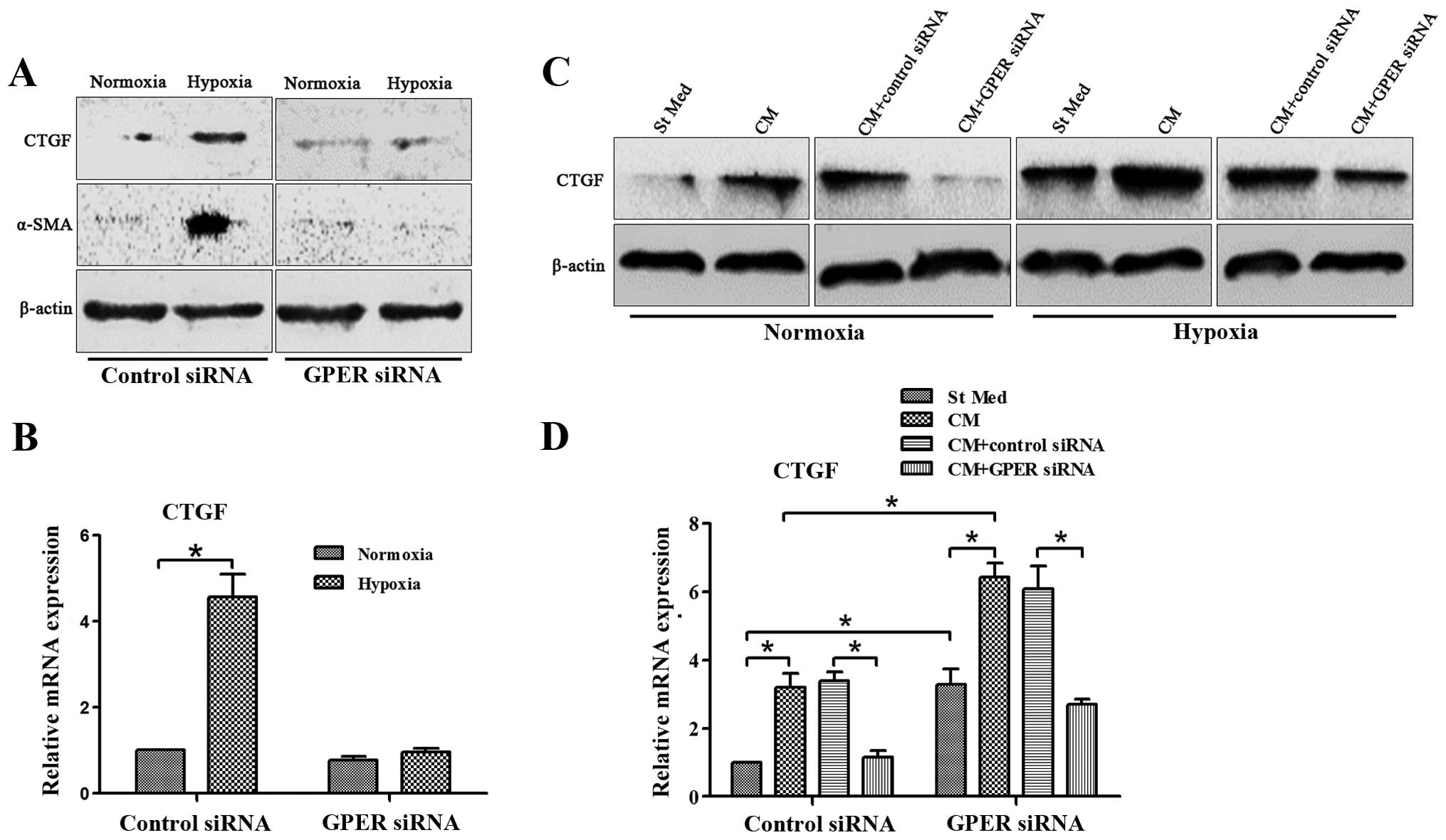
GPER silencing in CAFs inhibits
hypoxia-increased CTGF expression in CAFs and breast cancer cells
cultured with CM from CAFs
In tumor cells, CTGF has been reported to regulate
growth, migration, invasion, and angiogenesis (27–29).
We investigated whether CTGF is involved in the hypoxia-driven
programs by GPER. We therefore analyzed CTGF expression in CAFs and
breast cancer cells under normoxic and hypoxic conditions. In CAFs
exposed to hypoxia, there was a significant increase in CTGF
expression, whereas CTGF exhibited a low expression under normoxic
conditions (Fig. 5A and B).
MDA-MB-231 cells were treated with CM from CAFs activated by
hypoxia for 24 h under normoxic and hypoxic conditions. Hypoxia
significantly increased CTGF expression in MDA-MB-231 cells, and
treatment with CM from CAFs greatly increased this effect.
Moreover, CM from CAFs was able to upregulate CTGF expression in
MDA-MB-231 cells even under normoxic conditions (Fig. 5C and D). However, GPER silencing in
CAFs abrogated CTGF expression in CAFs and breast cancer cells
cultured with CM from CAFs under normoxic and hypoxic conditions
(Fig. 5). Moreover, GPER knockdown
eliminated CAF activation induced by hypoxia (Fig. 5A).
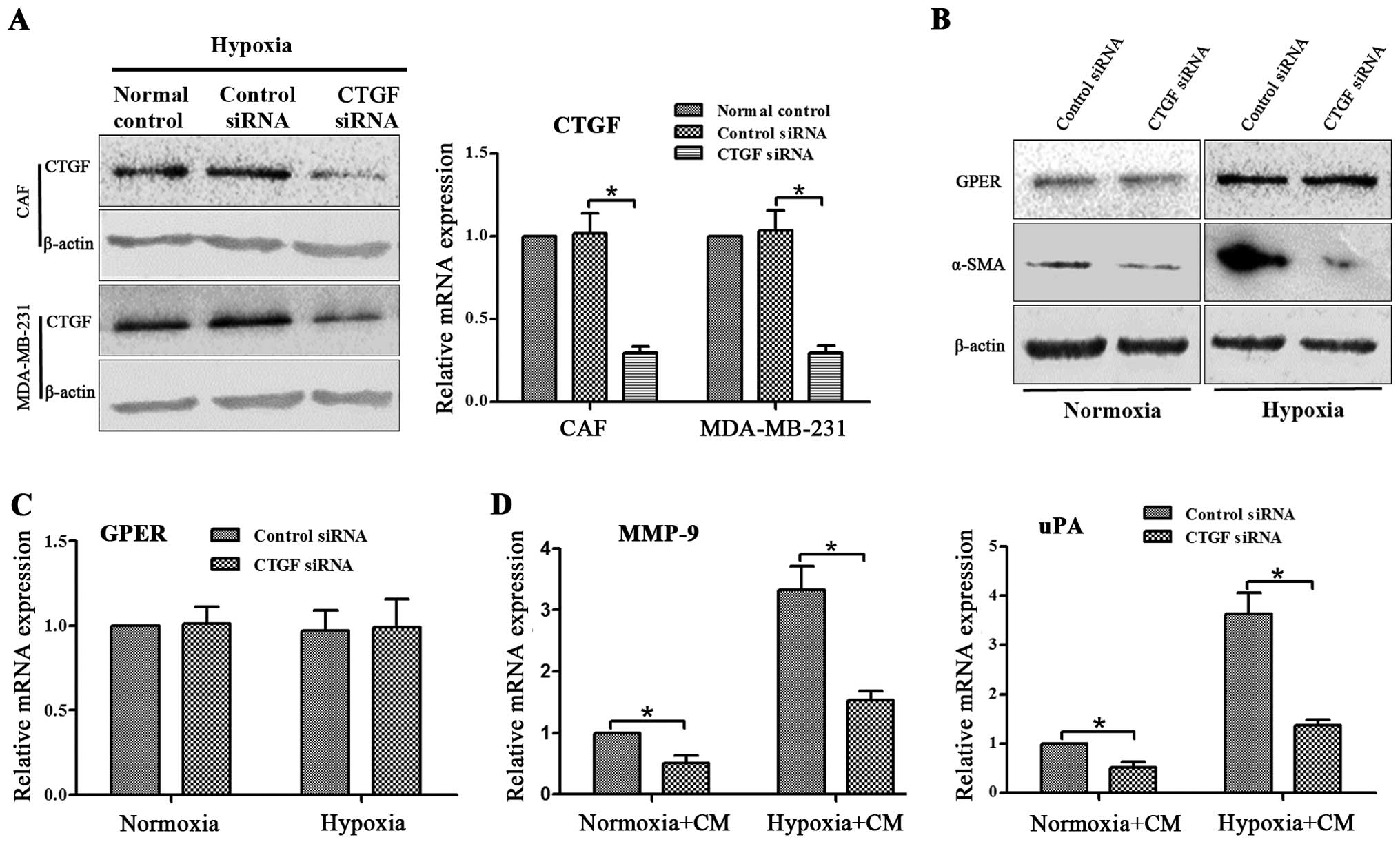
CTGF is responsible for the observed
effects of GPER on CAF activation and breast cancer invasion
Since GPER knockdown in CAFs may eliminate CTGF
upregulation under hypoxia exposure in CAFs and MDA-MB-231 cells,
we investigated whether CTGF is responsible for the observed
effects of GPER on activation of CAFs and breast cancer invasion.
CTGF siRNA was applied to the knockdown of CTGF expression in CAFs
and MDA-MB-231 cells. Since the expression level of CTGF in CAFs
under normoxic conditions was extremely low, we detected the
interference efficiency of CTGF siRNA in CAFs and MDA-MB-231 cells
under hypoxic conditions (Fig. 6A).
The GPER and α-SMA expression in CAFs and the MMP-9 and uPA
expression in MDA-MB-231 cells were then examined. CTGF siRNA
significantly suppressed α-SMA expression in CAFs under normoxic
and hypoxic conditions (Fig. 6B).
However, GPER expression was not affected by CTGF siRNA (Fig. 6B and C). Moreover, knockdown of CTGF
in MDA-MB-231 cells decreased the MMP-9 and uPA expression of
MDA-MB-231 cells cultured with CM from hypoxia-activated CAFs under
the two conditions (Fig. 6D). Since
CTGF siRNA did not influence GPER expression in CAFs, and GPER
knockdown downregulated CTGF expression, these data indicated that
CTGF is a downstream gene of GPER, and is responsible for
the observed effects of GPER on CAFs activation and breast cancer
invasion.
Discussion
The results of this study are consistent with a
mandatory role for some components of the tumor microenvironment,
i.e., CAFs and hypoxia, in the progression of breast cancer towards
an aggressive phenotype. We provide evidence that i) stromal
reactivity depends on hypoxia (particularly on its associated GPER
expression); ii) hypoxia and activated CAFs exhibit synergy in
promoting breast cancer invasiveness, increasing CTGF
expression.
Breast tumors are characterized by an extensive
desmoplastic stroma, abundantly populated by fibroblasts, and CAFs
were shown to support the growth of mammary tumors (30). Accumulating evidence indicates that
tumor desmoplasia plays a central role in disease progression and
that activated CAFs are responsible for the excess matrix
production. The mechanisms underlying the interplay between tumor
and stroma are complex. Various growth factors, such as
transforming growth factor (TGF)-α, TGF-β, insulin-like growth
factor (IGF)-I, IGF-II and platelet-derived growth factor (PDGF),
have been identified. These growth factors secreted by cancer cells
and can stimulate stromal cells (31–33),
which mediate effects on tumor growth, invasion, metastasis, and
resistance to chemotherapy. It is therefore conceivable that the
different stromal frameworks they encounter in this pathway grossly
affect their behavior and their terminal differentiation. This
result is consistent with our observation that MDA-MB-231 cells
sense activated stromal CAFs with a clear increase in their
invasiveness. This behavior of breast cancer cells in response to
their activated CAF counterparts is common among other tumors, such
as melanoma, pancreatic carcinoma, and prostate carcinoma, the
motility and aggressiveness of which is enhanced following contact
with CAFs (24,34–36).
In addition, activated stromal prostate fibroblasts induce
stem-like characteristics in carcinoma cells, thereby strengthening
the effect of these fibroblasts on metastatic tumor growth
(35,37,38).
Solid tumors often experience low-oxygen tension,
which is predominantly caused by abnormal vasculature formation in
the rapidly growing tumor mass. Our data indicate that hypoxia
activated CAFs and elicited the secretion of key cytokines such as
VEGF, IL-6, and CTGF, which are known to exert angiogenic and
inflammatory functions. Tumor hypoxia is recognized as a key factor
in tumor progression in several cancer models, as it is correlated
with de novo angiogenesis and with profound changes in tumor
metabolism as well as achievement of motile behavior (39,40).
These events synergistically facilitate the metastatic spread of
aggressive cells. Recent studies on breast cancer have shown that
GPER is an HIF-1-regulated gene, which contributes to adaptation to
a low-oxygen environment in breast cancer cells and in
cardiomyocytes (41).
Our results suggest that CAFs also sense hypoxia
through GPER upregulation, as GPER siRNA knockdown efficiently
abolishes CAF activation and the effects of CAFs on breast cancer
cells. As expected, GPER knockdown abolished the expression of
VEGF, IL-6, and CTGF induced by activated CAFs, suggesting a key
role for hypoxia-driven GPER expression in the regulation of
angiogenic and inflammatory responses during breast cancer
progression. Recchia et al recently reported that hypoxia
leads to the upregulation of CTGF (41), which is a target gene of HIF-1α
(41,42) and GPER (19,43).
We also showed that CTGF participates in hypoxia-driven
GPER-induced effects on CAFs and breast cancer cells.
VEGF and CTGF, which are involved in angiogenesis
and invasion of cancer and endothelial cells, and IL-6, which is
involved in the organization of the pro-inflammatory response, have
already been reported to be under the transcriptional control of
HIF-1 (44,45). We have shown that in breast cancer,
the secretion of these cytokines by activated CAFs is dependent on
concomitant exposure to hypoxia. These results indicate that
activated CAFs exposed to hypoxia are active players in attracting
breast cancer cells to different locations. Active factors in this
chemoattraction include CTGF, VEGF, and IL-6, confirming their
pleiotropic role in breast cancer progression. Thus, the
surrounding stroma, with intralesional hypoxic areas, may play a
role in attracting metastatic breast cancer cells from the primary
lesions, thereby facilitating satellite metastases.
GPER and HIF-1α are recruited to the HRE site
located within the VEGF promoter region and cooperatively act as a
functional complex for the transcription of VEGF (19). The present results show that GPER
knockdown abrogated hypoxia-driven CAF activation. Moreover, GPER
silencing inhibited breast cancer cell invasion induced by CAF CM,
and abolished hypoxia-activated CTGF, VEGF, and IL-6 secretion in
CAFs. Additionally, GPER knockdown suppressed hypoxia-enhanced CTGF
expression in CAFs and breast cancer cells cultured with CM from
CAFs. However, siRNA-mediated downregulation of CTGF abolished the
effects of GPER silencing on inhibiting CAF activation and breast
cancer invasion. These data indicate that GPER silencing has a
protective effect against hypoxia in the breast tumor-stromal
interaction, which is associated with its ability to ameliorate
CTGF upregulation.
Acknowledgements
This study is supported by the National Natural
Science Foundations of China (NSFC) (Nos. 31201060/C0709,
30973175/H1621, 81172490/H1621), Program for New Century Excellent
Talents in University (NCET-12-0440), Scientific and Technological
Research Foundation of Shaanxi Province (Nos. 2012K13-01-06,
2007K09-09), Project sponsored by Scientific Research Foundation
for the Returned overseas Chinese Scholars of State Education
Ministry (0601-18920006), Research Foundation of Health Department
of Shaan’xi Province (No. 2010D41), Qing Nian Jiao Shi Gen Zong Ji
Hua of Xi’an Jiaotong University (‘The Fundamental Research Funds
for the Central Universities’) (J.R., 2012), Program for Changjiang
Scholars and Innovative Research Team in University (PCSIRT:1171),
and the Research Fundation of Xi’an Jiao Tong University of China
(J.R.).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Youlden DR, Cramb SM, Dunn NA, Muller JM,
Pyke CM and Baade PD: The descriptive epidemiology of female breast
cancer: an international comparison of screening, incidence,
survival and mortality. Cancer Epidemiol. 36:237–248. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clark CE, Hingorani SR, Mick R, Combs C,
Tuveson DA and Vonderheide RH: Dynamics of the immune reaction to
pancreatic cancer from inception to invasion. Cancer Res.
67:9518–9527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yen TW, Aardal NP, Bronner MP, Thorning
DR, Savard CE, Lee SP and Bell RH Jr: Myofibroblasts are
responsible for the desmoplastic reaction surrounding human
pancreatic carcinomas. Surgery. 131:129–134. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hwang RF, Moore T, Arumugam T,
Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB and Logsdon CD:
Cancer-associated stromal fibroblasts promote pancreatic tumor
progression. Cancer Res. 68:918–926. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neesse A, Michl P, Frese KK, et al:
Stromal biology and therapy in pancreatic cancer. Gut. 60:861–868.
2011. View Article : Google Scholar
|
|
7
|
Polyak K and Kalluri R: The role of the
microenvironment in mammary gland development and cancer. Cold
Spring Harb Perspect Biol. 2:a0032442010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bhowmick NA, Neilson EG and Moses HL:
Stromal fibroblasts in cancer initiation and progression. Nature.
432:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gaggioli C, Hooper S, Hidalgo-Carcedo C,
Grosse R, Marshall JF, Harrington K and Sahai E: Fibroblast-led
collective invasion of carcinoma cells with differing roles for
RhoGTPases in leading and following cells. Nat Cell Biol.
9:1392–1400. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shimoda M, Mellody KT and Orimo A:
Carcinoma-associated fibroblasts are a rate-limiting determinant
for tumour progression. Semin Cell Dev Biol. 21:19–25. 2010.
View Article : Google Scholar :
|
|
12
|
Liao D and Johnson RS: Hypoxia: a key
regulator of angiogenesis in cancer. Cancer Metastasis Rev.
26:281–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rapisarda A and Melillo G: Role of the
hypoxic tumor microenvironment in the resistance to anti-angiogenic
therapies. Drug Resist Updat. 12:74–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lundgren K, Holm C and Landberg G: Hypoxia
and breast cancer: prognostic and therapeutic implications. Cell
Mol Life Sci. 64:3233–3247. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michieli P: Hypoxia, angiogenesis and
cancer therapy: to breathe or not to breathe. Cell Cycle.
8:3291–3296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lei J, Ma J, Ma Q, et al: Hedgehog
signaling regulates hypoxia induced epithelial to mesenchymal
transition and invasion in pancreatic cancer cells via a
ligand-independent manner. Mol Cancer. 12:662013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lei J, Huo X, Duan W, et al: α-Mangostin
inhibits hypoxia-driven ROS-induced PSC activation and pancreatic
cancer cell invasion. Cancer Lett. 347:129–138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Francesco EM, Lappano R, Santolla MF,
Marsico S, Caruso A and Maggiolini M: HIF-1alpha/GPER signaling
mediates the expression of VEGF induced by hypoxia in breast cancer
associated fibroblasts (CAFs). Breast Cancer Res. 15:R642013.
View Article : Google Scholar
|
|
20
|
Madeo A and Maggiolini M: Nuclear
alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced
gene expression and migration in breast cancer-associated
fibroblasts. Cancer Res. 70:6036–6046. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arias-Pulido H, Chaher N, Gong Y, Qualls
C, Vargas J and Royce M: Tumor stromal vascular endothelial growth
factor A is predictive of poor outcome in inflammatory breast
cancer. BMC Cancer. 12:2982012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Staton CA, Reed MW and Brown NJ: A
critical analysis of current in vitro and in vivo angiogenesis
assays. Int J Exp Pathol. 90:195–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Comito G, Giannoni E, Di GP, Segura CP,
Gerlini G and Chiarugi P: Stromal fibroblasts synergize with
hypoxic oxidative stress to enhance melanoma aggressiveness. Cancer
Lett. 324:31–41. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu T, Liu M, Luo H, Wu C, Tang X, Tang S,
Hu P, Yan Y, Wang Z and Tu G: GPER mediates enhanced cell viability
and motility via non-genomic signaling induced by 17β-estradiol in
triple-negative breast cancer cells. J Steroid Biochem Mol Biol.
143:392–403. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang QF, Wu TT, Yang JY, Dong CR, Wang N,
Liu XH and Liu ZM: 17β-estradiol promotes the invasion and
migration of nuclear estrogen receptor-negative breast cancer cells
through cross-talk between GPER1 and CXCR1. J Steroid Biochem Mol
Biol. 138:314–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dhar A and Ray A: The CCN family proteins
in carcinogenesis. Exp Oncol. 32:2–9. 2010.PubMed/NCBI
|
|
28
|
Chu CY, Chang CC, Prakash E and Kuo ML:
Connective tissue growth factor (CTGF) and cancer progression. J
Biomed Sci. 15:675–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hall-Glenn F, De Young RA, Huang BL, et
al: CCN2/connective tissue growth factor is essential for pericyte
adhesion and endothelial basement membrane formation during
angiogenesis. PLoS One. 7:e305622012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ronnov-Jessen L and Petersen OW: Induction
of alpha-smooth muscle actin by transforming growth factor-beta 1
in quiescent human breast gland fibroblasts. Implications for
myofibroblast generation in breast neoplasia. Lab Invest.
68:696–707. 1993.PubMed/NCBI
|
|
32
|
Ellis MJ, Singer C, Hornby A, Rasmussen A
and Cullen KJ: Insulin-like growth factor mediated
stromal-epithelial interactions in human breast cancer. Breast
Cancer Res Treat. 31:249–261. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bronzert DA, Pantazis P, Antoniades HN,
Kasid A, Davidson N, Dickson RB and Lippman ME: Synthesis and
secretion of platelet-derived growth factor by human breast cancer
cell lines. Proc Natl Acad Sci USA. 84:5763–5767. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Taddei ML, Giannoni E, Raugei G, Scacco S,
Sardanelli AM, Papa S and Chiarugi P: Mitochondrial oxidative
stress due to complex I dysfunction promotes fibroblast activation
and melanoma cell invasiveness. J Signal Transduct.
2012:6845922012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Giannoni E, Bianchini F, Masieri L, Serni
S, Torre E, Calorini L and Chiarugi P: Reciprocal activation of
prostate cancer cells and cancer-associated fibroblasts stimulates
epithelial-mesenchymal transition and cancer stemness. Cancer Res.
70:6945–6956. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cirri P and Chiarugi P:
Cancer-associated-fibroblasts and tumour cells: a diabolic liaison
driving cancer progression. Cancer Metastasis Rev. 31:195–208.
2012. View Article : Google Scholar
|
|
37
|
Giannoni E, Bianchini F, Calorini L and
Chiarugi P: Cancer associated fibroblasts exploit reactive oxygen
species through a proinflammatory signature leading to epithelial
mesenchymal transition and stemness. Antioxid Redox Signal.
14:2361–2371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giaccia AJ and Schipani E: Role of
carcinoma-associated fibroblasts and hypoxia in tumor progression.
Curr Top Microbiol Immunol. 345:31–45. 2010.PubMed/NCBI
|
|
40
|
Melillo G: Targeting hypoxia cell
signaling for cancer therapy. Cancer Metastasis Rev. 26:341–352.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Recchia AG, De Francesco EM, Vivacqua A,
Sisci D, Panno ML, Ando S and Maggiolini M: The G protein-coupled
receptor 30 is up-regulated by hypoxia-inducible factor-1alpha
(HIF-1alpha) in breast cancer cells and cardiomyocytes. J Biol
Chem. 286:10773–10782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lappano R, Recchia AG, De Francesco EM,
Angelone T, Cerra MC, Picard D and Maggiolini M: The cholesterol
metabolite 25-hydroxycholesterol activates estrogen receptor
alpha-mediated signaling in cancer cells and in cardiomyocytes.
PLoS One. 6:e166312011. View Article : Google Scholar
|
|
43
|
Pandey DP, Lappano R, Albanito L, Madeo A,
Maggiolini M and Picard D: Estrogenic GPR30 signalling induces
proliferation and migration of breast cancer cells through CTGF.
EMBO J. 28:523–532. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Semenza GL: Oxygen homeostasis. Wiley
Interdiscip Rev Syst Biol Med. 2:336–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Youn SW, Lee SW, Lee J, et al: COMP-Ang1
stimulates HIF-1α-mediated SDF-1 overexpression and recovers
ischemic injury through BM-derived progenitor cell recruitment.
Blood. 117:4376–4386. 2011. View Article : Google Scholar : PubMed/NCBI
|