Introduction
Breast cancer is one of the most common cancer among
women aged between 50 and 70. About one million new cases of breast
cancers are diagnosed each year worldwide (1). The best lines of defense, radiation
therapy and chemotherapy, are unsatisfactory due to the untoward
side effects on healthy cells and the problem of drug resistance.
Therefore, the development of new therapeutic drugs for breast
cancer is clinically important.
Apoptotic (type Ι) and autophagic cell death (type
II) are two common forms of programmed cell death (PCD) (2). Autophagy is a degradative process that
is characterized by the sequestration of cytoplasmic components in
double-membrane vesicles which fuse with lysosomes to form
autolysosomes (3). Autophagy plays
an important role in cellular homeostasis by acting as a
housekeeper to clear damaged organelles such as broken
mitochondria, to remove misfolded proteins and to eliminate
intracellular pathogens (4). Aside
from its basic role in the turnover of proteins and organelles,
autophagy also shows multiple physiological and pathophysiological
functions in cell differentiation, immune defense and cell death
(5). Autophagy has been implicated
to play a role in cancer, cardiomyopathy and neurodegenerative
diseases (6,7).
Bcl-2 adenovirus E1B 19-kDa-interacting protein 3
(BNIP3), a member of the ‘BH3-only’ subfamily of pro-apoptotic
Bcl-2 family proteins which induces cell death, has a key role in
the pathogenesis of many diseases, such as cancer (8). BNIP3 is primarily localized to the
mitochondria with the N-terminus oriented into the cytoplasm and
the C-terminus inside the mitochondria, and has been documented to
be the key regulator of mitochondrial permeability transition pore
(mPTP) and mitochondrial dysfunction (9). Previous research has shown that
overexpression of BNIP3 induces cell death accompanied by rapid and
strong mitochondrial dysfunction caused by the alteration of
mitochondrial transmembrane potential (ΔΨm) and the opening of mPTP
(10,11), which could lead to the production of
reactive oxygen species (ROS). Recently, BNIP3 is known as a potent
inducer of autophagy in many different cell types (9). As BNIP3 may represent a potential
therapeutic target in cancer, it is important to understand how
BNIP3 regulates mitochondrial function and autophagy.
Silibinin is a type of flavonoid which is the major
bioactive component of silymarin isolated from milk thistle
(Silybum marianum). It has been used as a traditional
medicine specifically as a hepatoprotective drug in Europe and Asia
and shows obvious clinical curative effects for jaundice, hepatitis
and gallbladder disease (12).
Recently, many studies have shown that silibinin has anticancer
activities in various types of cancer cells, such as colon,
prostate, skin and glioma cancer cells. In these studies, silibinin
caused cell death through inhibition of proliferation, activation
of the MAPK pathway and induction of inflammation and apoptosis
(13,14). However, the mechanisms underlying
the antitumor effect of silibinin have not been clearly elucidated.
Thus, the present study was designed to elucidate the effect of
silibinin on the human MCF7 breast cancer cell line and to
determine its underlying mechanisms. Here, we provide evidence that
autophagy plays an important role in silibinin-induced cell death.
Furthermore, we showed that autophagic cell death induced by
silibinin was accompanied by ROS-dependent alteration of ΔΨm and
loss of ATP production that involved BNIP3.
Materials and methods
Reagents
Silibinin,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
N-acetylcysteine (NAC), ascorbic acid (AA), 3-methyladenine (3-MA),
bafilomycin-A1 (Baf-A1), mouse anti-human BNIP3 and rabbit
anti-human light chain 3 (LC3) were all obtained from
Sigma-Aldrich. Antibodies for rabbit anti-human Bcl-2, β-actin,
rabbit anti-human Atg12 and rabbit anti-human Beclin-1 were all
purchased from Cell Signaling Technology (Beverly, MA, USA).
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazol
ylcarbocyanine iodide (JC-1) and dihydroethidium (DHE) were
obtained from Molecular Probes. BNIP3 siRNA was purchased from
RiboBio Co. (guangdong, China).
Cell culture
MCF7 cells were cultured in RPMI-1640 media
supplemented with 10% fetal calf serum, 100 u/ml penicillin, and
100 μg/ml streptomycin. Cells were incubated at 37°C in a
humidified atmosphere of 5% CO2 and 95% air.
Serum-starved cells were cultured as above without 10% fetal calf
serum for 24 h.
Measurement of cell viability
Cell growth was measured using the MTT assay. Cells
were seeded in a 96-multiwell plate at a concentration of
5×103 cells/well. After overnight incubation, the medium
was replaced with fresh medium containing silibinin at various
concentrations and incubation was carried out for 24 or 48 h. MTT
(0.5 mg/ml) was added during the last 4 h of incubation. In each
well, the absorbance was measured at 570 nm. For the trypan blue
assay, cells were seeded in a 6-multiwell plate at a concentration
of 5×105 cells/well in RPMI-1640 media with/without 10%
fetal calf serum. After 24 h, 100 μM of silibinin was added
and incubation was carried out for 2, 6, 12 or 24 h. Cells were
harvested and stained with 0.4% trypan blue solution for 10 min.
Non-viable cells were stained blue, and the ratio of blue cells to
total cells was recognized as the cell death rate.
Measurement of ROS
Cells were seeded in a 6-multiwell plate at a
concentration of 5×105 cells/well. After overnight
incubation, the medium was replaced with a fresh medium containing
silibinin at various concentrations and incubation was carried out
for 24 h. Next, the cells were harvested and incubation was carried
out with 5 μM DHE in PBS for 20 min at 37°C. Then the cells
were washed with PBS twice before FACS analysis.
Measurement of ΔΨm
Cells were seeded in a 6-multiwell plate at a
concentration of 5×105 cells/well. After overnight
incubation, the medium was replaced with fresh medium containing
silibinin at various concentrations and incubation was carried out
for 24 h. Next, the cells were harvested and incubated with 5
μM JC-1 in PBS for 20 min at 37°C. Then the cells were
washed with PBS twice before FACS analysis.
ATP measurement
Cells were seeded in a 6-multiwell plate at a
concentration of 5×105 cells/well. After overnight
incubation, the medium was replaced with fresh medium containing
silib-inin at various concentrations and incubation was carried out
for 24 h. Next, the cells were harvested and lysed in 1% NP-40 and
when the ATP in the lysates and luciferin/luciferase enzyme complex
combined, a reaction which produces light occurs. The relative
light units were detected by the DTX 880 Multimode Detector
(Beckman Coulter).
RNA interference
The siRNA sequences were: BNIP3-siRNA sense,
5′-CACGAGCGUCAUGAAGAAAUU-3′ starting at nucleotide 439 from the Aug
start codon of human BNIP3 coding sequence (GenBank™ accession no.
MM 004052) and BNIP3-siRNA antisense, 5′-UUUCUUCAUGACGCUCGUGUU-3′.
siRNA (100 nM) was transfected into MCF7 cells using Lipofectamine™
RNAiMax (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions for ~48 h.
Quantitative real-time fluorescence
polymerase chain reaction (qRT-PCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen) from MCF7 cells following treatment with 100 μM
silibinin or other reagents. cDNA was synthesized using ReverTra
Ace® qPCR RT kit (Toyobo, Japan) according to the
manufacturer’s instructions. qRT-PCR reactions were run on an
Applied Biosystems 7500 Real-Time PCR system machine. Gene
expression levels in all samples were examined using SYBR-Green
(Takara, Japan) according to the manufacturer’s instructions. The
sequences were as follows: BNIP3 sense, 5′-GTTCCAGCCTCGGTTTCT-3′
and antisense, 5′-AGCCCTGTTGGTATCTTGTG-3′; GAPDH sense,
5′-TGCCAAATATGATGACATCAAGAA-3′ and antisense,
5′-GGAGTGGGTGTCGCTGTTG-3′. After correcting the levels of
expression of each mRNA to GAPDH, the relative levels of
transcripts in the MCF7 cells were calculated using the
2−ΔΔCt method (15).
Western blot analysis
Cells were extracted in lysis buffer (Cell Signaling
Technology) and the lysates were separated on a 12.5% SDS-PAGE
using the SDS-PAGE system as described (16). Proteins were then transferred to a
PVDF membrane (Millipore, Billerica, MA, USA) in transfer buffer.
After blocking with 5% non-fat dried milk for 2 h, the membrane was
incubated with the primary antibodies overnight at 4°C. Then the
immunoreactive bands were visualized by enhanced chemiluminescence
using HRP-conjugated secondary antibodies and blots were developed
by an enhanced chemiluminescence detection system
(Amersham-Pharmacia Biotech).
Statistical analysis
Data (mean ± SEM) were statistically analyzed using
one-way analysis of variance (ANOVA) followed by Dunnett’s test.
P<0.05 was considered to indicate a statistically significant
result.
Results
Silibinin induces autophagic cell
death
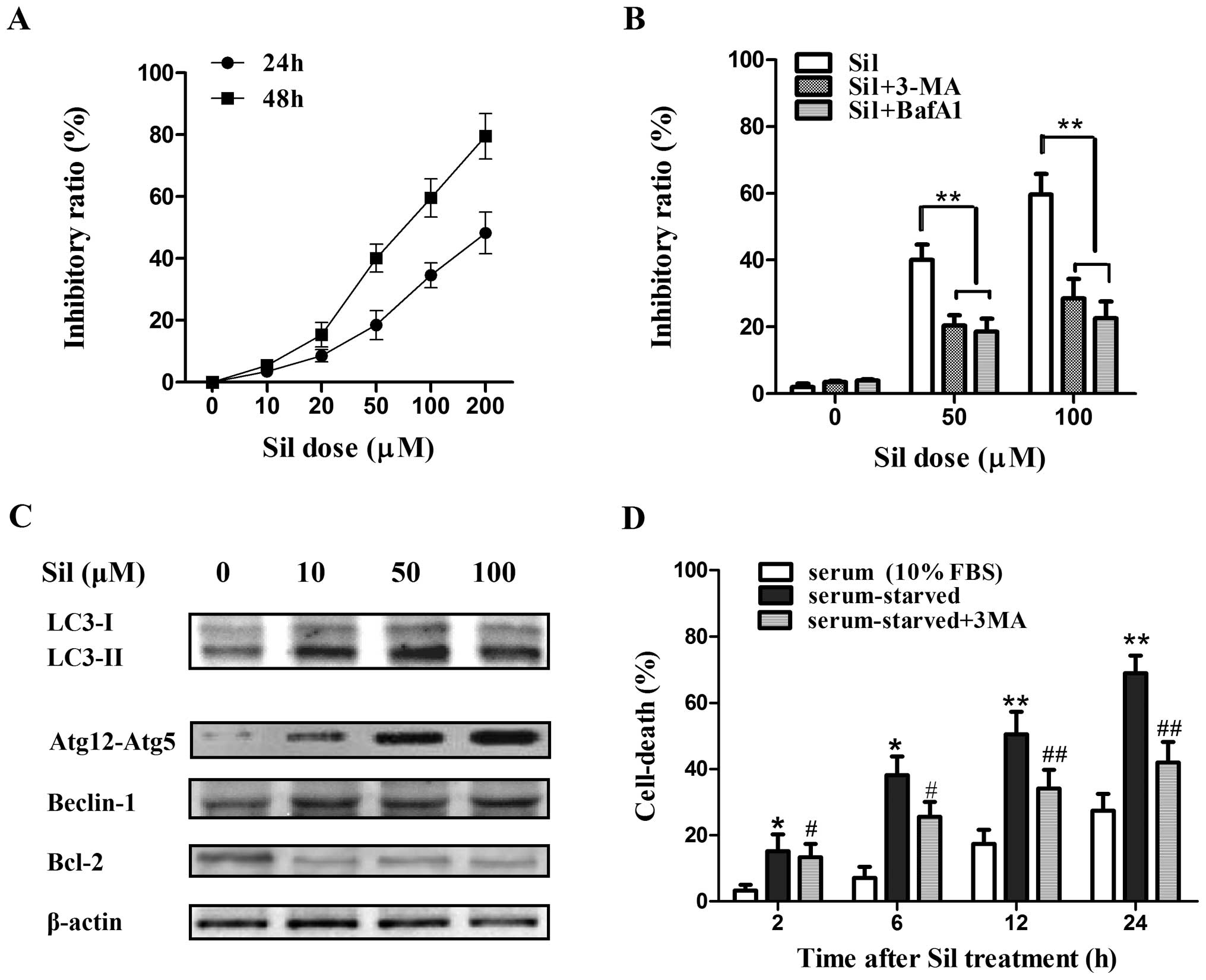
First, we assessed the efficacy of silibinin against
the cell viability of MCF7 human breast cancer cells. The cells
were exposed to various concentrations of silibinin for 24 and 48
h, and the cell viability was measured by MTT assay. Silibinin
caused a marked increase in cell death in a time- and
concentration-dependent manner (Fig.
1A). Then two autophagy inhibitors, 3-MA (which blocks the
initial stages of autophagy by inhibiting class III PI3 kinase) and
Baf-A1 (which blocks autophagosome-lysosome fusion by inhibiting
the vacuolar H+-ATPase), were further used to
investigate whether silibinin-induced cell death is attributed to
autophagy. MCF7 cells were treated with 5 mM 3-MA or 50 nM Baf-A1
for 2 h prior to 24 h of silibinin treatment (100 μM). 3-MA
and Baf-A1 pretreatment both greatly abrogated silibinin-induced
cell death (Fig. 1B), indicating
that autophagy contributed to the cell death in the MCF7 cells by
silibinin. Next, to further confirm the induction of autophagy by
silibinin, a set of autophagy-related factors including LC3-I and
LC3-II, Atg12-Atg5 formation, and Beclin-1 in the MCF7 cells after
treatment with silibinin (0–100 μM) for 24 h were
investigated by western blot analysis. Notably, the conversion of
LC3-I to LC3-II (LC3-II/LC3-I ratio), an established indicator of
autophagy, was greatly enhanced by silibinin (Fig. 1C). The formation of LC3-II is known
to depend on the Atg12-Atg5 conjugate, and Atg12-Atg5 formation was
significantly elevated (Fig. 1C),
confirming the activated autophagy by silibinin. Moreover, the
expression of Beclin-1, which plays a vital role in the regulation
of early stages of autophagosome formation, was increased although
the increase was not as marked as the level of LC3-II or Atg12-Atg5
formation. Recently, Bcl-2 was reported to act as an anti-autophagy
protein via its inhibitory interaction with Beclin-1 (17). As expected, an obvious decrease in
the level of Bcl-2 was observed (Fig.
1C). Notably, MCF7 cells treated with silibinin (100 μM)
under serum-starved conditions underwent an immediate increase in
cell death at 2 h, which occurred earlier than that under
serum-sufficient (10% FBS) conditions, and the autophagy inhibitor
3-MA partially inhibited cell death by silibilin under this
circumstance (Fig. 1D), suggesting
that the MCF7 cells were more sensitive to silibinin-induced
autophagic cell death under the starvation condition.
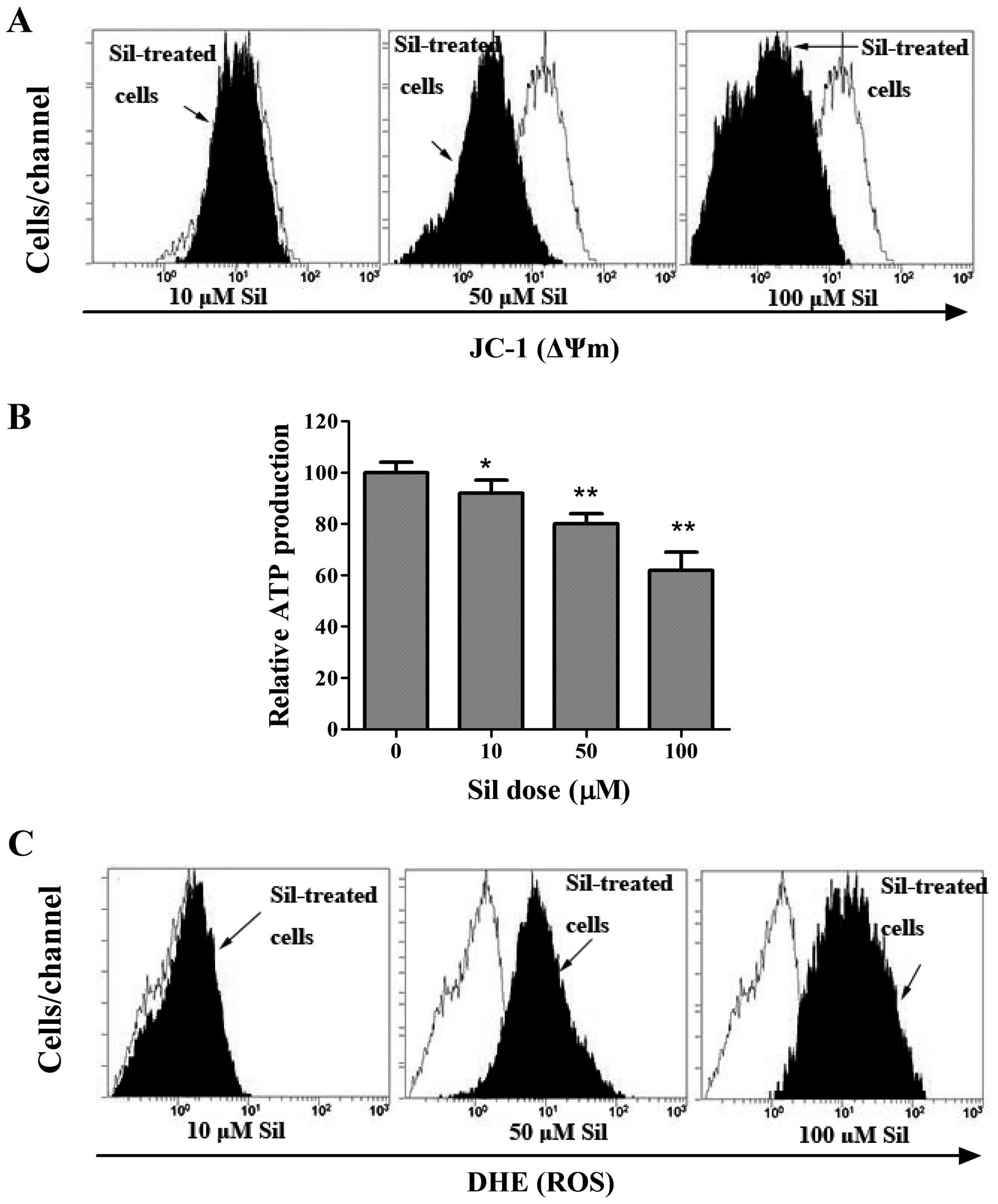
Silibinin causes ROS generation and
mitochondrial dysfunction
To elucidate the mechanism of the induction of
autophagic cell death by silibinin, we next investigated the
mitochondrial activities during silibinin treatment. ΔΨm, as an
important parameter of mitochondrial function, is often employed as
an indicator of cellular viability. Thus, we adopted a
potential-dependent fluorescent carbocyanine and lipophilic dye
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocy-anine
iodide (JC-1), which accumulates in the mitochondria, to indicate
the dissipation of ΔΨm. As shown in Fig. 2A, in the control cells, the bright
red fluorescence generated by JC-1 was indicative of high membrane
potential, whereas silibinin caused a dose-dependent reduction in
ΔΨm, as indicated by a shift in the fluorescence peak to the left.
Since ΔΨm is known to be essential for various functions including
the production of ATP via oxidative phosphorylation (18), we sought to determine whether loss
of ΔΨm caused changes in ATP content. As expected, cellular ATP
levels progressively declined in the silibinin-treated MCF7 cells
(Fig. 2B), suggesting mitochondrial
dysfunction. The generation of ROS is known to be induced by the
opening of the mPTP (19). Thereby,
the fluorogenic probe, DHE, was used to assess the effect of
silibinin on ROS generation. Compared to the control cells, the
cells stained with DHE showed a progressively enhanced ROS level by
exposure to increasing concentrations of silibinin (Fig. 2C).
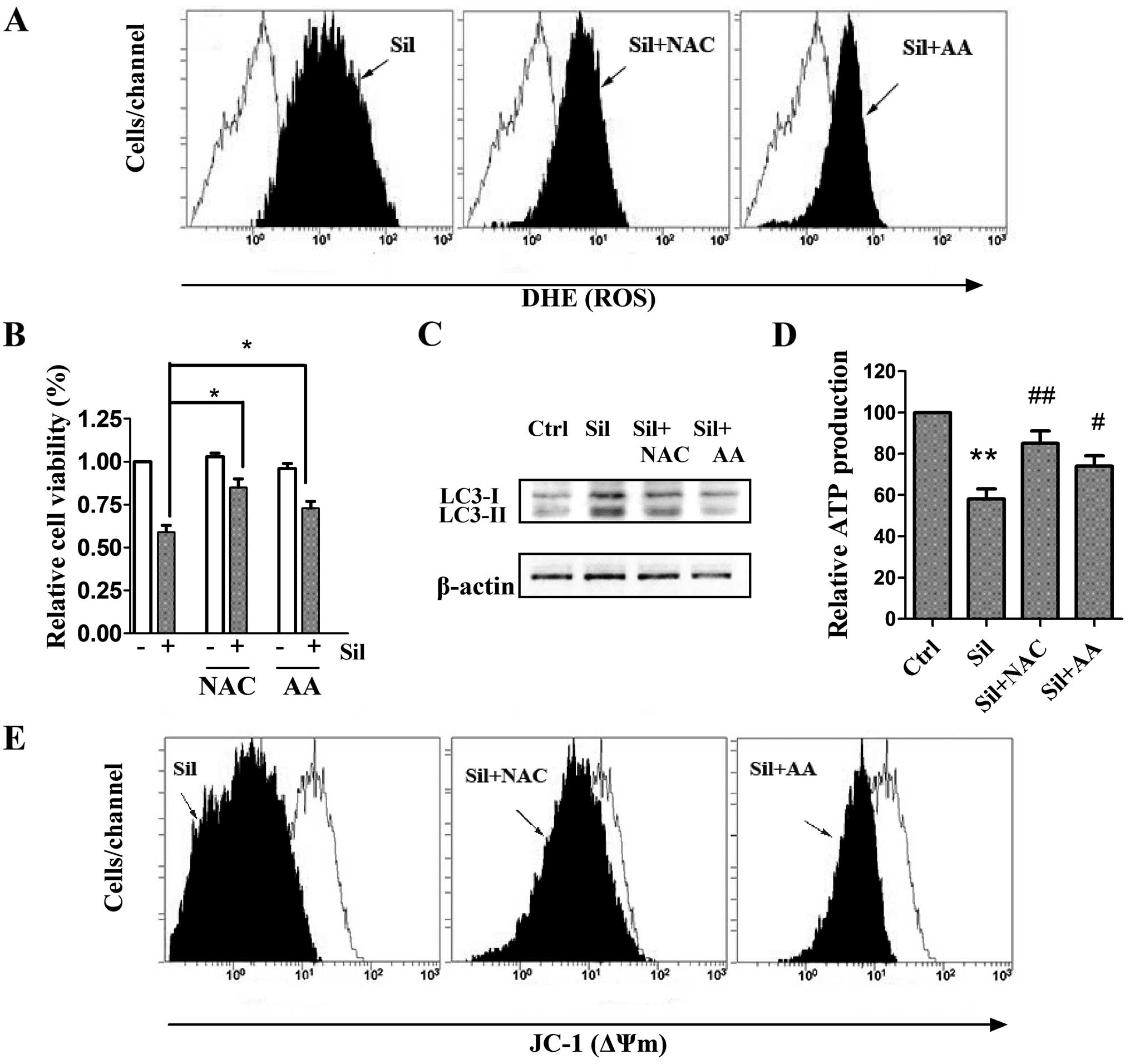
Antioxidants prevent silibinin-triggered
ROS generation as well as autophagic cell death, mitochondrial
dysfunction and ATP depletion
The pivotal role of ROS in mediating
silibinin-induced cell death has previously been revealed (20), and it has also been reported that
ROS could regulate autophagy in various cell models (21). Therefore, we investigated whether
ROS scavenging could inhibit silibinin-promoted autophagy by
adopting two antioxidants, NAC and AA, known as strong scavengers
of ROS. Cells were pretreated with 5 mM NAC and 500 μM AA
for 2 h and stimulated with silibinin (100 μM) for another
24 h. We observed that the antioxidants exerted little effect on
the intracellular ROS levels (data not shown). Notably,
pretreatment with both NAC and AA greatly inhibited the
silibinin-stimulated ROS generation (Fig. 3A). After verifying the inhibition of
ROS production by the antioxidants, the involvement of ROS in
silibinin-induced autophagic cell death and mitochondrial
dysfunction was then investigated. As shown in Fig. 3B and C, pre-incubation with NAC or
AA effectively prevented not only the loss of cell viability but
also the elevated autophagic LC3-II/LC3-I ratio, indicating that
autophagic cell death was ROS-dependent. Notably, pretreatment of
the antioxidants effectively recovered ATP levels and ΔΨm during
silibinin treatment (Fig. 3D and
E). These results indicate that the ROS generation may be a
cause of silibinin-triggered autophagic cell death and
mitochondrial dysfunction as well as ATP depletion.
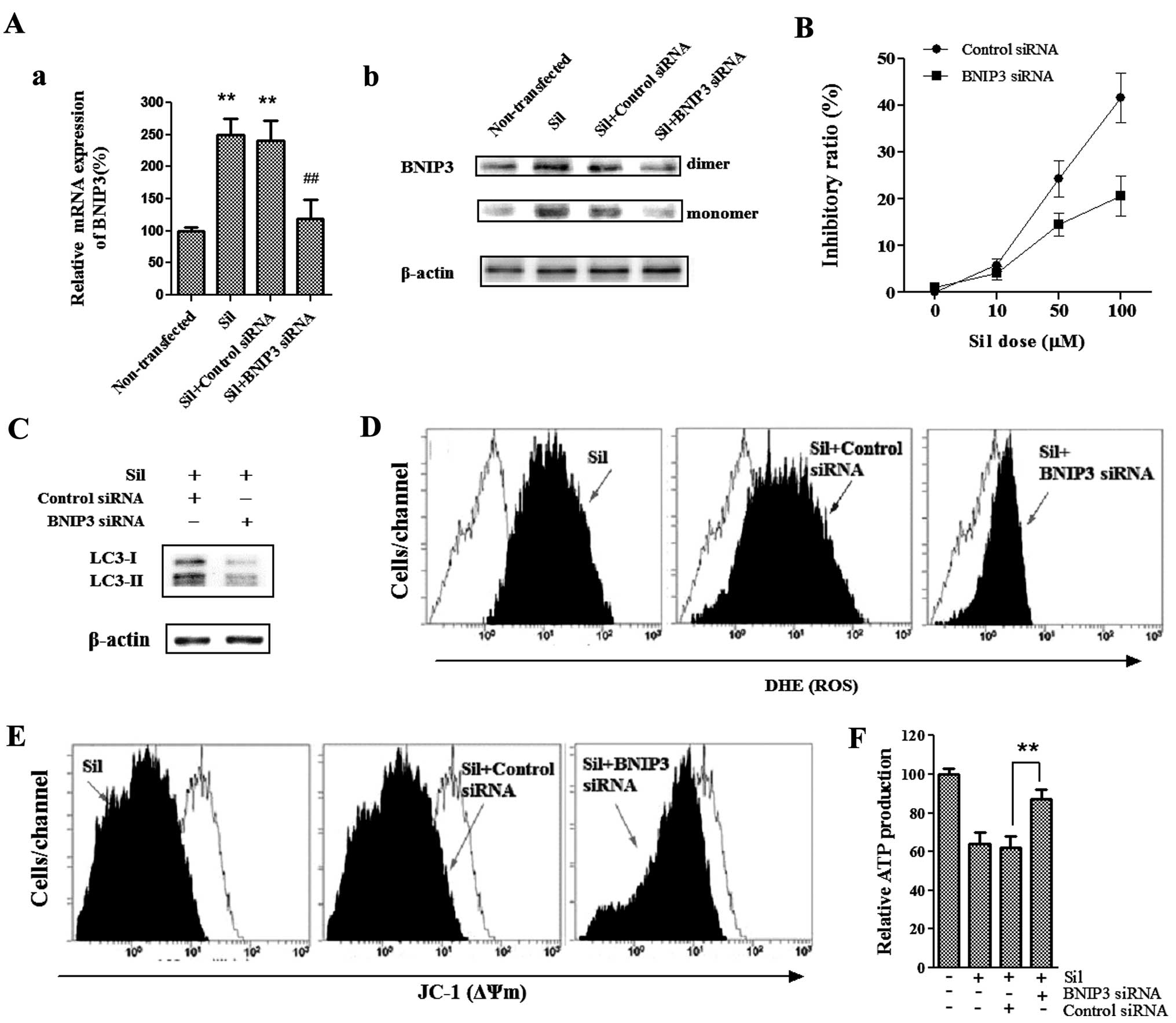
BNIP3 mediates silibinin-induced cell
death
To determine whether BNIP3, a pro-death protein and
a member of the BH3-only Bcl-2 family, is required for
silibinin-induced cell death, we used BNIP3-specific siRNA to knock
down BNIP3 and then analyzed the effects of loss of BNIP3 on
silibinin-enhanced autophagic cell death as well as ROS production
and mitochondrial dysfunction. As shown in Fig. 4A, silibinin upregulated BNIP3
protein and transcript levels, whereas BNIP3 siRNA blocked BNIP3
upregulation induced by silibinin. We found that the cells
pretreated with BNIP3 siRNA were more resistant to
silibinin-induced cell death as compared to those pretreated with
the control siRNA (Fig. 4B).
Furthermore, silencing of BNIP3 significantly reduced the
silibinin-enhanced autophagy-related LC3-II/LC3-I ratio (Fig. 4C), suggesting that BNIP3 was
essential for the silibinin-induced autophagic cell death noted in
the MCF7 cells. Since silencing of BNIP3 effectively reversed the
cell death and autophagy induced by silibinin, we next investigated
the involvement of BNIP3 in silibinin-induced ROS production, as
well as loss of ΔΨm and ATP production. Notably, BNIP3 siRNA
greatly abrogated the ability of silibinin to induce an increase in
ROS production (Fig. 4D), as well
as loss of ΔΨm (Fig. 4E) and
decrease in ATP production (Fig.
4F). Together these data provide evidence that BNIP3 plays a
critical role in the silibinin-induced autophagic cell death, as
well as ROS generation, mitochondrial dysfunction and defective ATP
production.
Discussion
Silibinin, a popular dietary supplement isolated
from milk thistle seed extracts, has shown considerable efficacy in
treating various types of cancer. Recently, it has been reported
that silibinin effectively induced apoptosis, type Ι of PCD, in a
human breast carcinoma cell line (22), suggesting its potential as a
therapeutic agent against breast cancer. However, the mechanisms
underlying the antitumor effect of silibinin are poorly defined.
Notably, our present study demonstrated that silibinin induced MCF7
human breast cancer cells to undergo autophagic cell death (PCD
type II), as well as ROS-dependent disruption of ΔΨm and loss of
ATP production through a mechanism involving BNIP3.
It is well-known that excessive levels of autophagy
can lead to cytoplasmic vacuolation resulting in cell death
(23). In the present study,
silibinin-induced autophagic cell death was demonstrated, as 3-MA
and Baf-A1, two autophagy inhibitors acting at different stages of
autophagy, effectively abrogated silibinin-induced cell death. In
addition, we investigated a set of autophagy hallmarks, including
the conversion of LC3-I to LC3-II, Atg12-Atg5 formation, and
Beclin-1 to further confirm the induction of autophagy. The
Atg12-Atg5 conjugate is known as one of the key stages for the
autophago-somes and it acts as an E3-like enzyme for lipidation of
Atg8 family proteins and their association to vesicle membranes in
autophagy (24).
Microtubule-associated protein 1-light chain 3 (LC3) is the
mammalian homolog of the yeast protein Atg8. When autophagy is
activated, LC3-I is activated by Atg7 and modified into the active
form LC3-II (membrane-bound) which is the component of
autophagosomes (25). Consistent
with these data, we found that both the level of LC3-II and the
LC3-II/LC3-I ratio which closely correlate with the number of
autophagosomes, as well as Atg12-Atg5 formation were increased
after exposure to silibinin. Our finding that elevated expression
of Beclin-1 which was accompanied by a decreased level of Bcl-2 is
significant, since Beclin-1 participates in the regulation of the
early stages of autophagosome formation (26), and Bcl-2 is reported to inhibit
autophagy by interacting with Beclin-1 and can repress mTOR to
activate autophagy via preventing Ca2+ release from ER
(27). It has been reported that
the PCD pathways I and II may both be induced by the same stimuli
and/or in the same cell types (28). Therefore, it is possible that
silibinin-induced autophagy was accompanied by apoptosis, and the
interplay between apoptosis and autophagy occurred, which warrants
further investigation.
Besides its role in removing mitochondria damaged by
oxidative stress, autophagy also plays a role in the catabolism of
oxidized proteins (29). Previous
data indicate that ROS plays an essential role in the activation of
autophagy (30). It was
demonstrated that ROS can induce autophagy through several distinct
mechanisms involving Atg4-Atg8/LC3, Beclin-1, PI3K-Akt-mTOR,
catalase, and the mitochondrial electron transport chain, which
leads to both cell-survival and cell-death responses and could be
selective toward cancer cells (31,32).
In the present study, silibinin treatment caused a
concentration-dependent increase in intracellular ROS. Furthermore,
the antioxidants NAC and AA prevented not only ROS production but
also silibinin-induced autophagy, indicating that ROS production
seems to be a cause of silibinin-induced autophagic cell death.
Similar to what we observed, several pieces of evidence indicate
that silibinin induces autophagic death through the ROS pathway in
other cancer cell types, such as human fibrosarcoma HT1080
(33) and HeLa cells (34).
Notably, our findings demonstrated that the ATP
levels were significantly reduced by silibinin in the MCF7 cells.
ATP is a switch to cell death and mitochondrial dysfunction induces
changes in cellular energy metabolism in cancer cells (35). It has been reported that
high-polarized (high ΔΨm) mitochondria help the maintenance of
sufficient ATP levels (36).
Similar to previous findings, we noted that the loss of ΔΨm was
accompanied by a marked decline in the cytoplasmic ATP content,
indicating destruction of mitochondrial function by silibinin. In
addition to their critical role in ATP synthesis, mitochondria are
also the major source of ROS in most cell types (35). Mitochondrial dysfunction is usually
characterized by the opening of mPTP, a non-specific pore in the
inner mitochondrial membrane. The opening of mPTP results in
massive swelling of the inner membrane and subsequent rupture of
the outer mitochondrial membrane, causing release of mitochondrial
components into the cytoplasm including ROS (37). In this regard, we initially
speculated that the dissipation of ΔΨm caused by silibinin might
contribute to the generation of ROS. Surprisingly, we found that
inhibition of ROS production by antioxidants effectively
regenerated ΔΨm and maintained ATP levels, implying that ROS are
responsible for silibinin-stimulated mitochondrial dysfunction and
subsequent ATP depletion. This finding is supported by the facts
that excessive ROS trigger the opening of mitochondrial channels,
and in turn, leads to the simultaneous collapse of the ΔΨm and a
transient increase in ROS generation. This
mitochondrion-to-mitochondrion ROS signaling constitutes a positive
feedback mechanism involving the recently described process named
ROS-induced ROS-release (RIRR) which may release an ROS burst
finally leading to cell death (38).
BNIP3 is a mitochondrial BH3-only protein that
contributes to cell death through activation of the mitochondrial
pathway of apoptosis (11).
Although it belongs to the Bcl-2 family, its pro-cell death
activity is distinct from other family members. BNIP3 is also known
to induce mitochondrial autophagy (9). In the present study, our results
showed that silencing of BNIP3 significantly reduced the
silibinin-enhanced autophagy-related LC3-II/LC3-I ratio, implying
an essential role of BNIP3 in silibinin-induced autophagic cell
death in MCF7 cells. Notably, BNIP3 siRNA sustained ΔΨm and ATP
levels during autophagy, and prevented ROS production in the
silibinin-treated MCF7 cells. Similar to what we observed, Ghavami
et al showed that overexpression of ∆TM-BNIP3, a
dominant-negative BNIP3 mutant which prevents wild-type (wt) BNIP3
from targeting the mitochondria and antagonizes wt BNIP3-induced
effects, not only reversed cell death and autophagy, but also
reduced ROS production and mitochondrial damage in
S100A8/A9-treated cells including MCF7 (39).
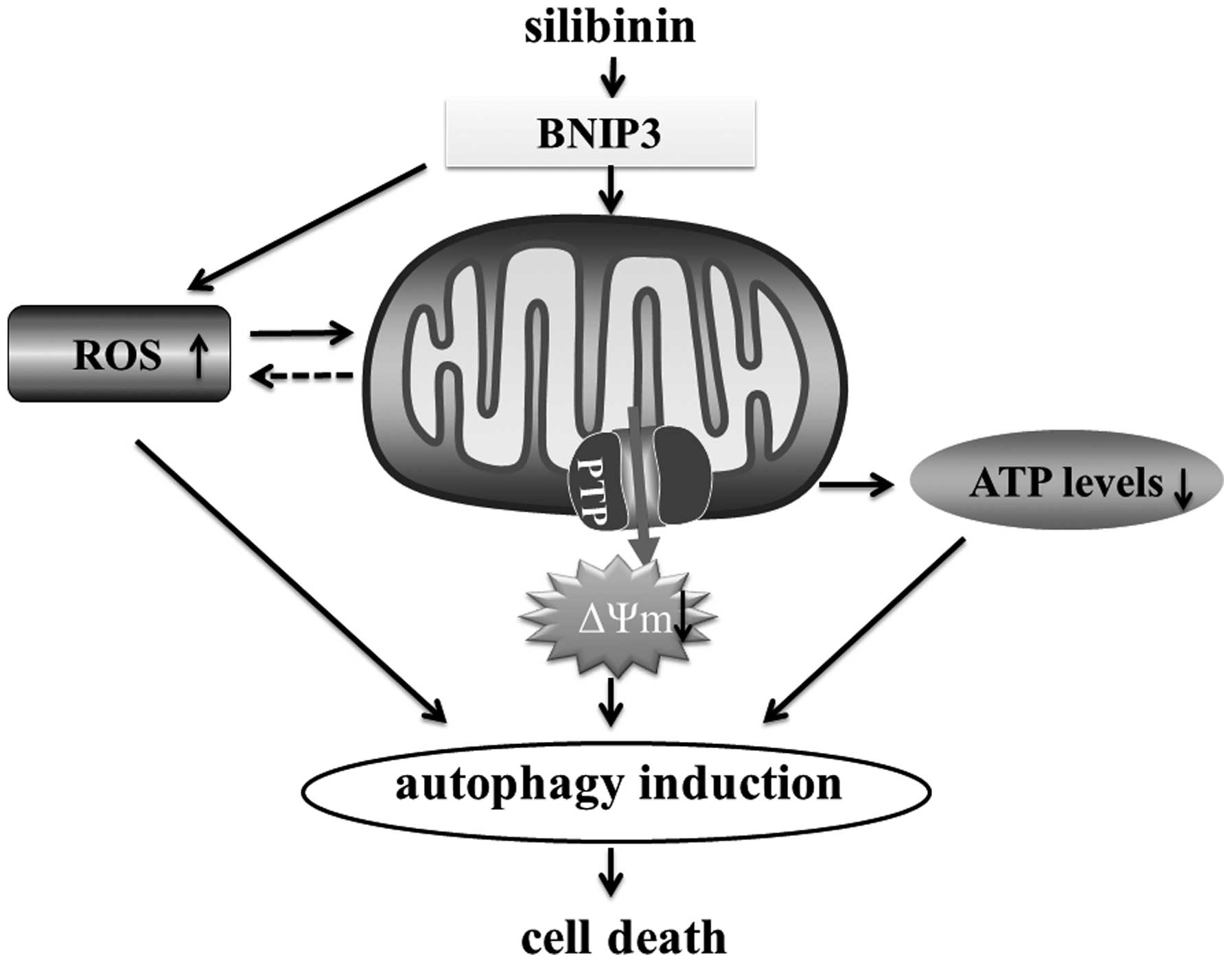
In conclusion, the present study shed new light on
the mechanisms involved in silibinin-triggered cell death. The
present study suggests that silibinin-induced autophagic cell death
involves an increase in ROS production, subsequently followed by
mitochondrial dysfunction and ATP depletion. Finally, we suggest
that BNIP3 is the critical factor that mediates the ROS-dependent
mitochondrial death pathway (Fig.
5).
Acknowledgments
The present study was supported by the Zhejiang
Provincial Natural Science Foundation of China (grant nos.
LQ14H290002 and LY12H29010) and the Zhejiang Provincial
Administration of Traditional Chinese Medicine (grant no.
2014ZB012).
References
|
1
|
Tinoco G, Warsch S, Glück S, Avancha K and
Montero AJ: Treating breast cancer in the 21st century: emerging
biological therapies. J Cancer. 4:117–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al: Nomenclature Committee on Cell Death
2009: Classification of cell death: recommendations of the
Nomenclature Committee on Cell Death 2009. Cell Death Differ.
16:3–11. 2009. View Article : Google Scholar :
|
|
3
|
Reggiori F and Klionsky DJ:
Autophagosomes: biogenesis from scratch? Curr Opin Cell Biol.
17:415–422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuan J, Lipinski M and Degterev A:
Diversity in the mechanisms of neuronal cell death. Neuron.
40:401–413. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burton TR and Gibson SB: The role of Bcl-2
family member BNIP3 in cell death and disease: NIPping at the heels
of cell death. Cell Death Differ. 16:515–523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gustafsson AB: Bnip3 as a dual regulator
of mitochondrial turnover and cell death in the myocardium. Pediatr
Cardiol. 32:267–274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vande Velde C, Cizeau J, Dubik D, Alimonti
J, Brown T, Israels S, Hakem R and Greenberg AH: BNIP3 and genetic
control of necrosis-like cell death through the mitochondrial
permeability transition pore. Mol Cell Biol. 20:5454–5468. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kubli DA, Ycaza JE and Gustafsson AB:
Bnip3 mediates mitochondrial dysfunction and cell death through Bax
and Bak. Biochem J. 405:407–415. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Milić N, Milosević N, Suvajdzić L, Zarkov
M and Abenavoli L: New therapeutic potentials of milk thistle
(Silybum marianum). Nat Prod Commun. 8:1801–1810. 2013.
|
|
13
|
Jeong JC, Shin WY, Kim TH, Kwon CH, Kim
JH, Kim YK and Kim KH: Silibinin induces apoptosis via
calpain-dependent AIF nuclear translocation in U87MG human glioma
cell death. J Exp Clin Cancer Res. 30:442011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim S, Choi MG, Lee HS, Lee SK, Kim SH,
Kim WW, Hur SM, Kim JH, Choe JH, Nam SJ, et al: Silibinin
suppresses TNF-alpha-induced MMP-9 expression in gastric cancer
cells through inhibition of the MAPK pathway. Molecules.
14:4300–4311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
16
|
Meng G, Sun Y, Fu W, Guo Z and Xu L:
Microcystin-LR induces cytoskeleton system reorganization through
hyperphosphorylation of tau and HSP27 via PP2A inhibition and
subsequent activation of the p38 MAPK signaling pathway in
neuroendocrine (PC12) cells. Toxicology. 290:218–229. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Waterhouse NJ, Goldstein JC, von Ahsen O,
Schuler M, Newmeyer DD and Green DR: Cytochrome cmaintains
mitochondrial transmembrane potential and ATP generation after
outer mitochondrial membrane permeabilization during the apoptotic
process. J Cell Biol. 153:319–328. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gong X, Ivanov VN, Davidson MM and Hei TK:
Tetramethylpyrazine (TMP) protects against sodium arsenite-induced
nephrotoxicity by suppressing ROS production, mitochondrial
dysfunction, pro-inflammatory signaling pathways and programed cell
death. Arch Toxicol. June 25–2014.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duan W, Jin X, Li Q, Tashiro S, Onodera S
and Ikejima T: Silibinin induced autophagic and apoptotic cell
death in HT1080 cells through a reactive oxygen species pathway. J
Pharmacol Sci. 113:48–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scherz-Shouval R and Elazar Z: ROS,
mitochondria and the regulation of autophagy. Trends Cell Biol.
17:422–427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yousefi M, Ghaffari SH, Zekri A, Hassani
S, Alimoghaddam K and Ghavamzadeh A: Silibinin induces apoptosis
and inhibits proliferation of estrogen receptor (ER)-negative
breast carcinoma cells through suppression of nuclear factor kappa
B activation. Arch Iran Med. 17:366–371. 2014.PubMed/NCBI
|
|
23
|
Kondo Y and Kondo S: Autophagy and cancer
therapy. Autophagy. 2:85–90. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuma A, Mizushima N, Ishihara N and Ohsumi
Y: Formation of the approximately 350-kDa Apg12-Apg5.Apg16
multimeric complex, mediated by Apg16 oligomerization, is essential
for autophagy in yeast. J Biol Chem. 277:18619–18625. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kabeya Y, Mizushima N, Yamamoto A,
Oshitani-Okamoto S, Ohsumi Y and Yoshimori T: LC3, GABARAP and
GATE16 localize to autophagosomal membrane depending on form-II
formation. J Cell Sci. 117:2805–2812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tassa A, Roux MP, Attaix D and Bechet DM:
Class III phosphoinositide 3-kinase - Beclin1 complex mediates the
amino acid-dependent regulation of autophagy in C2C12 myotubes.
Biochem J. 376:577–586. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thorburn A: Apoptosis and autophagy:
regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar :
|
|
28
|
Ferraro E and Cecconi F: Autophagic and
apoptotic response to stress signals in mammalian cells. Arch
Biochem Biophys. 462:210–219. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kiffin R, Bandyopadhyay U and Cuervo AM:
Oxidative stress and autophagy. Antioxid Redox Signal. 8:152–162.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar
|
|
31
|
Li ZY, Yang Y, Ming M and Liu B:
Mitochondrial ROS generation for regulation of autophagic pathways
in cancer. Biochem Biophys Res Commun. 414:5–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Azad MB, Chen Y and Gibson SB: Regulation
of autophagy by reactive oxygen species (ROS): implications for
cancer progression and treatment. Antioxid Redox Signal.
11:777–790. 2009. View Article : Google Scholar
|
|
33
|
Duan WJ, Li QS, Xia MY, Tashiro S, Onodera
S and Ikejima T: Silibinin activated p53 and induced autophagic
death in human fibrosarcoma HT1080 cells via reactive oxygen
species-p38 and c-Jun N-terminal kinase pathways. Biol Pharm Bull.
34:47–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan S, Li L, Chen S, Yu Y, Qi M, Tashiro
S, Onodera S and Ikejima T: Silibinin induced-autophagic and
apoptotic death is associated with an increase in reactive oxygen
and nitrogen species in HeLa cells. Free Radic Res. 45:1307–1324.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wen S, Zhu D and Huang P: Targeting cancer
cell mitochondria as a therapeutic approach. Future Med Chem.
5:53–67. 2013. View Article : Google Scholar :
|
|
36
|
Van Blerkom J, Davis P and Alexander S:
Inner mitochondrial membrane potential (DeltaPsim), cytoplasmic ATP
content and free Ca2+ levels in metaphase II mouse
oocytes. Hum Reprod. 18:2429–2440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Halestrap AP, Clarke SJ and Javadov SA:
Mitochondrial permeability transition pore opening during
myocardial reperfusion - a target for cardioprotection. Cardiovasc
Res. 61:372–385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ghavami S, Eshragi M, Ande SR, Chazin WJ,
Klonisch T, Halayko AJ, McNeill KD, Hashemi M, Kerkhoff C and Los
M: S100A8/A9 induces autophagy and apoptosis via ROS-mediated
cross-talk between mitochondria and lysosomes that involves BNIP3.
Cell Res. 20:314–331. 2010. View Article : Google Scholar
|