Introduction
Cyclin-dependent kinases (CDKs), are members of the
serine-threonine protein kinase family and are responsible for
taking control of cell cycle regulation in eukaryotic cells. CDKs
show their action by interacting with cyclins and different
CDK-cyclin complexes regulate the cell cycle in the G1, S and G2/M
phases (1). New anticancer therapy
strategies refer to the inhibition of CDK-cyclin complexes as an
important target to prevent uncontrolled proliferation and induce
apoptosis in cancer cells (2).
Olomoucine, roscovitine and purvalanol are examples of CDK
inhibitors (CDKIs) designed and investigated for their apoptotic
potential on cancer cells (3).
Purvalanol is a purine-derived CDKI that binds with a high
selectivity and competitively to the ATP binding side of CDK1/2 and
leads to G2/M cell cycle arrest (4). Τheir impact on the apoptotic cell
death mechanism requires further elucidation. Natural polyamines,
putrescine, spermidine and spermine, play essential roles in the
regulation of cell growth and proliferation. Increased levels of
polyamines in cells are considered to be involved in cancer
progression. Intracellular polyamine levels are under the control
of several catabolic enzymes, such as spermidine/spermine-N-acetyl
transferase (SSAT). Although purvalanol-induced cell cycle arrest
and apoptotic cell death were demonstrated in prostate (5), breast (6) and colon cancer cells (7), the exact molecular mechanism of
purvanol-induced apoptosis has not been elucidated yet.
Endoplasmic reticulum (ER) is an essential organelle
responsible for protein synthesis, folding, post-translational
modification of proteins and protein trafficking in eukaryotes
(8). ER alerts a self-protective
mechanism that is called ER stress during nutrient deprivation,
pathogen infection, alterations in redox status, intraluminal
Ca2+ levels and folding defective protein conditions
(9). ER stress response is
initiated by activation of three types of ER membrane receptors;
inositol-requiring enzyme 1 (IRE1α), PRKR-like ER kinase (PERK) and
activating transcription factor-6 (ATF-6). Activated and released
IRE1α acts as an RNase to initiate transcription of XBP1 mRNA and
it becomes a transcriptional activator for unfolded protein
response (UPR) gene targets, such as BiP and calreticulin (10). Activation of ER membrane receptor
PERK, during prolonged UPR, phosphorylates eIF2α to attenuate mRNA
transcription within the cell (11). Recently, autophagy, a cytoprotective
intracellular degradation system, is assumed to be activated by
both IRE1α and PERK as an alternative way to degrade accumulated
misfolded proteins (12).
Concominantly, during UPR stress, ATF-6 is released from BiP and
translocates from the ER to the Golgi. Cleaved ATF-6 migrates to
the nucleus and transactivates various chaperones and major ER
stress markers such as the CAAT-enhancer binding protein (CHOP)
gene. CHOP is a transcription factor that regulates the expression
of the Bcl-2 family members. Overexpression of CHOP induces
apoptosis and its deficiency has been shown to protect cells from
apoptotic cell death. Prolonged severe ER stress could eventually
activate caspase-12 and caspase-dependent apoptosis (13). Thus, our aim in the present study
was to understand the molecular basis of purvalanol-induced ER
stress which leads to cell death in HCT 116 colon cancer cells.
Materials and methods
Drugs and antibodies
Purvalanol was purchased from Tocris Bioscience
(Bristol, UK), dissolved in DMSO to make a 10 mM stock solution and
stored at −20°C. Phospho-Rb, p53, p21, Bad, Bid, Bik, PUMA, Mcl-1,
Bcl-xl, pro-caspase-3, pro-caspase-7, PARP, Beclin-1, p62, LC3,
Atg-5, Atg-12, PERK, XBP1, caspase-12, JNK, ATF-6, CHOP, BiP,
IRE1α, calnexin, calreticulin, PDI, c-jun, GAPDH and β-actin (each
diluted 1:1,000) rabbit antibodies were purchased from Cell
Signaling Technology (CST, Danvers, MA, USA). HRP-conjugated
secondary anti-rabbit antibodies (diluted 1:3,000) were from
CST.
Cell culture
HCT 116 colon cancer cells (CCL 247) were purchased
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). Cells were maintained in McCoy’s 5A medium (Gibco,
Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Pan
Biotech, Aidenbach, Germany) and 100 U/100 mg/ml
penicilin/streptomycin and grown in the presence of 5%
CO2 in humidified air at 37°C (Heracell 150; Thermo
Electron Corporation, Waltham, MA, USA).
Cell viability assay
The effects of purvalanol on cell viability were
determined by colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT;
Roche, Indianapolis, IN, USA) assay. Cells were seeded at a density
of 1×104 cells/well in 96-well plates, allowed to attach
overnight and treated for 24 and 48 h with various concentrations
of purvalanol (0–30 μM). After purvalanol treatment, 10
μl of MTT reagent (5 mg/ml) was added to the cell culture
medium for 4 h. Following removal of media, 200 μl DMSO was
added to dissolve the formazan crystals, which are produced due to
activated mitochondria. The absorbance of the suspensions was
determined at 570 nm with a microplate reader (Bio-Rad, Hercules,
CA, USA).
Trypan blue dye exclusion assay
Cells were seeded on 6-well plates (5×104
cells/well) and treated with purvalanol (15 μM) for 96 h.
Every 24 h, the cells were trypsinized and stained with trypan blue
and viable and dead cells were counted under light microscopy. Data
were platted on a graph indicating the number of cells (y-axis) vs.
time (x-axis).
Fluorescence microscopy
PI staining
HCT 116 cells (1×105) were seeded into
12-well plates and treated with purvalanol (0–30 μM) for 24
h. Following a dose-dependent purvalanol treatment, the cells were
washed once with 1X PBS and stained with propidium iodide (PI) (50
mg/ml stock concentration in 1X PBS) fluorescent probe and were
incubated for 10 min in the dark. Purvalanol-induced cell death was
detected with fluorescence microscopy (Olympus, Tokyo, Japan).
DAPI staining
The cells were seeded in 12-well plates at a density
of 1×105 cells/well and treated with purvalanol (0–30
μM) for 24 h. After treatment, the cells were washed once
with 1X PBS. The cells were stained with 1 μl/ml
4′,6-diamidino-2-phenylindole (dAPI) (1 mg/ml stock concentration
in 1X PBS) fluorescent probe and were incubated for 10 min in the
dark. Dose-dependent purvalanol-induced nuclear DNA fragmentation
was visualized using fluorescence microscopy.
DiOC6 staining
HCT 116 (1×105) cells were seeded into
12-well plates. Following exposure of cells to purvalanol (0–30
μM), they were washed once with 1X PBS, and then stained
with 4 nM 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] (40 nM stock concentration in DMSO;
Calbiochem, La Jolla, CA, USA) fluorescent probe. Mitochondrial
membrane potential (MMP) disruption was visualized by fluorescence
microscopy.
Cell cycle analysis by PI
staining
HCT 116 colon carcinoma cells at a density of
2×105 cells/well were seeded in 6-well plates, and then
subsequently treated with purvalanol (15 μM) for 12, 24 and
48 h. Both floating and adherent cells were collected and fixed
with 70% ethanol. Following incubation on ice for 30 min, the cells
were diluted with 1X PBS. Samples were then centrifuged at 1,200
rpm for 5 min. Pellets were resuspended in 1X PBS, RNase (100
μg/ml) and PI solution (40 μg/ml). Samples were kept
for 30 min at 37°C in the dark. Cell cycle distribution was
analyzed by Accuri C6 (BD Biosciences, Oxford, UK). 10,000
events/sample were acquired and evaluated using BD Accuri C6
software (BD Biosciences).
CHOP activation by FACS flow
analysis
HCT 116 colon carcinoma cells were seeded in 6-well
plates and transfected with CHOP promoter (−649/+136) pmCherry-1
plasmid (0.5 mg/ml) using the Fugene6 (Promega, Sunnyvale, CA,
USA). After a 24 h transfection, the cells were exposed to
purvalanol (15 μM) for 12, 24 and 48 h. Following purvalanol
treatment, mCherry-CHOP plasmid-transfected 10,000 cells/sample
were determined by flow cytometry using FL-3 (red fluorescence)
(FACScan, Attune; Applied Biosciences, Foster City, CA, USA).
GFP-LC3 localization by FACS flow
analysis
HCT 116 colon carcinoma cells were seeded in 6-well
plates and transfected with a green fluorescent protein
(GFP)-tagged LC3-expressing vector (0.5 μ/ml) using Fugene6.
After a 24 h transfection, the cells were exposed to purvalanol (15
μM) for 12, 24 and 48 h. Following purvalanol treatment,
GFP-LC3 plasmid-transfected 10,000 cells/sample were determined by
flow cytometry using FL-1 (green fluorescence) (FACScan, Attune;
Applied Biosciences).
Cell death ELISA assay
HCT 116 cells (1×104) were seeded in
96-well plates and treated with 15 μM purvalanol for 12, 24
and 48 h. Cytoplasmic histone-associated-DNA fragments (mono- and
oligonucleosomes) were determined using Cell Death detection ELISA
Plus assay, according to the manufacturer’s instructions (Roche).
Briefly, cell lysates were placed in a streptavidin-coated
microplate. A mixture of anti-histone-biotin and anti-DNA-POD was
added and incubated for 2 h at 15–25°C. Following the removal of
unbound antibodies by a washing procedure, POD was determined
photometrically at 405 nm with 2,2′-azino-di-[3-ethylbenzthiazoline
sulfonate (6)] diammonium salt
(ABTS) as a substrate. In order to determine the DNA fragments
following drug treatment in colon carcinoma cells, total DNA
content was isolated.
Protein extraction and
immunoblotting
HCT 116 colon carcinoma cells were treated with the
appropriate concentrations of purvalanol in a time-dependent
manner. First, all the samples were washed with ice-cold 1X PBs and
lysed on ice in a solution containing 20 mM Tris-HCl (pH 7.5), 150
mM NaCl, Nonidet P-40 0.5%, (v/v), 1 mM EDTA, 0.5 mM PMSF, 1 mM DTT
and protease inhibitor cocktail (Complete, Roche). After cell
lysis, the cell debris were removed by centrifugation for 15 min at
13,200 rpm, and protein concentrations were determined by the
Bradford protein assay (BioRad). Total protein lysates (30
μg) were separated on a 12% SDS-PAGE and transferred onto
PVDF membranes (Roche). The membranes were then blocked with 5%
milk blocking solution in Tris buffer saline (TBS)-Tween 20 (Sigma)
and incubated with appropriate primary and horseradish peroxidase
(HRP)-conjugated secondary antibodies (CST) in antibody buffer
containing 5% (v/v) milk blocking solution. Following a gentle
washing step with 1X TBS-Tween 20, the proteins were analyzed using
an enhanced chemiluminescence detection system (ECL). Bands were
exposed to Lumi-Film Chemiluminescent Detection (Roche).
Statistical analysis
All the experiments were statistically analyzed by
two-way ANOVA using GraphPad Prism 6 (GraphPad Software, La Jolla,
CA, USA). Statistically significant results by ANOVA were further
analyzed by Bonferroni post-hoc analysis (where indicated).
A p<0.05 was considered to indicate a statistically significant
result. Error bars in the graphs were generated using ± standard
deviation (SD) values. Band intensities were quantified using
imageJ software and normalized to β-actin or GAPDH.
Results
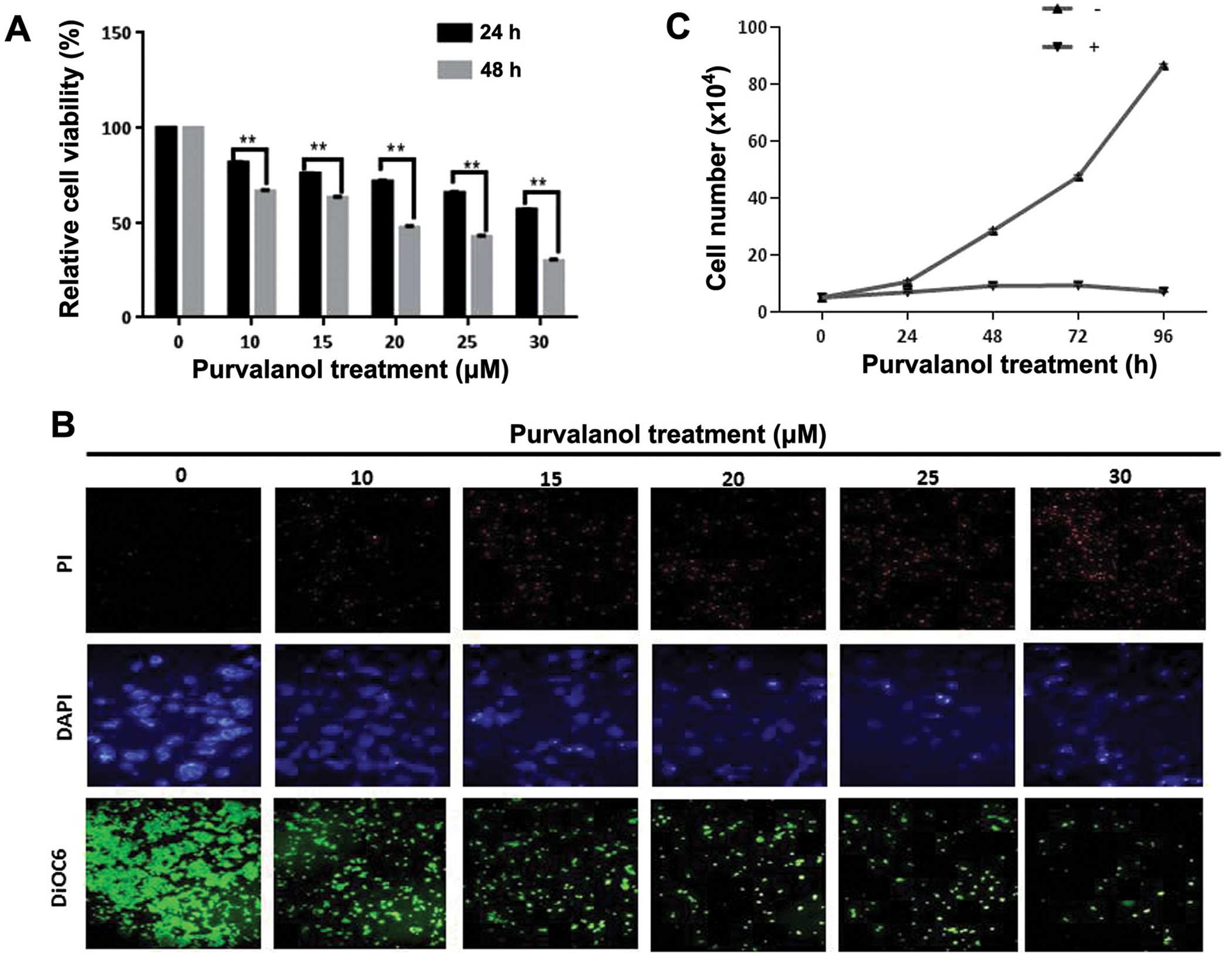
Purvalanol inhibits cell viability and
proliferation in a dose-dependent manner
In order to understand the time- and dose-dependent
effects of purvalanol treatment on HCT 116 cell viability, we
performed an MTT assay. According to the MTT assay, 15 μM of
purvalanol decreased the cell viability by 25% in HCT 116 cells
within 24 h. Exposure to purvalanol for 48 h decreased cell
viability by 40% (Fig. 1A).
Moreover, PI and DAPI staining of cells after dose-dependent
purvalanol treatment for 24 h showed that cell death and nuclear
condensation were increased in the HCT 116 colon cancer cells. In
addition, we also determined that MMP disruption was increased in
dose-dependent manner in the HCT 116 colon cancer cells (Fig. 1B). Cell survival analysis with
trypan blue dye exclusion assay showed that 15 μM purvalanol
inhibited cell proliferation after 24 h and this cytostatic effect
was maintained until 72 h (Fig.
1C).
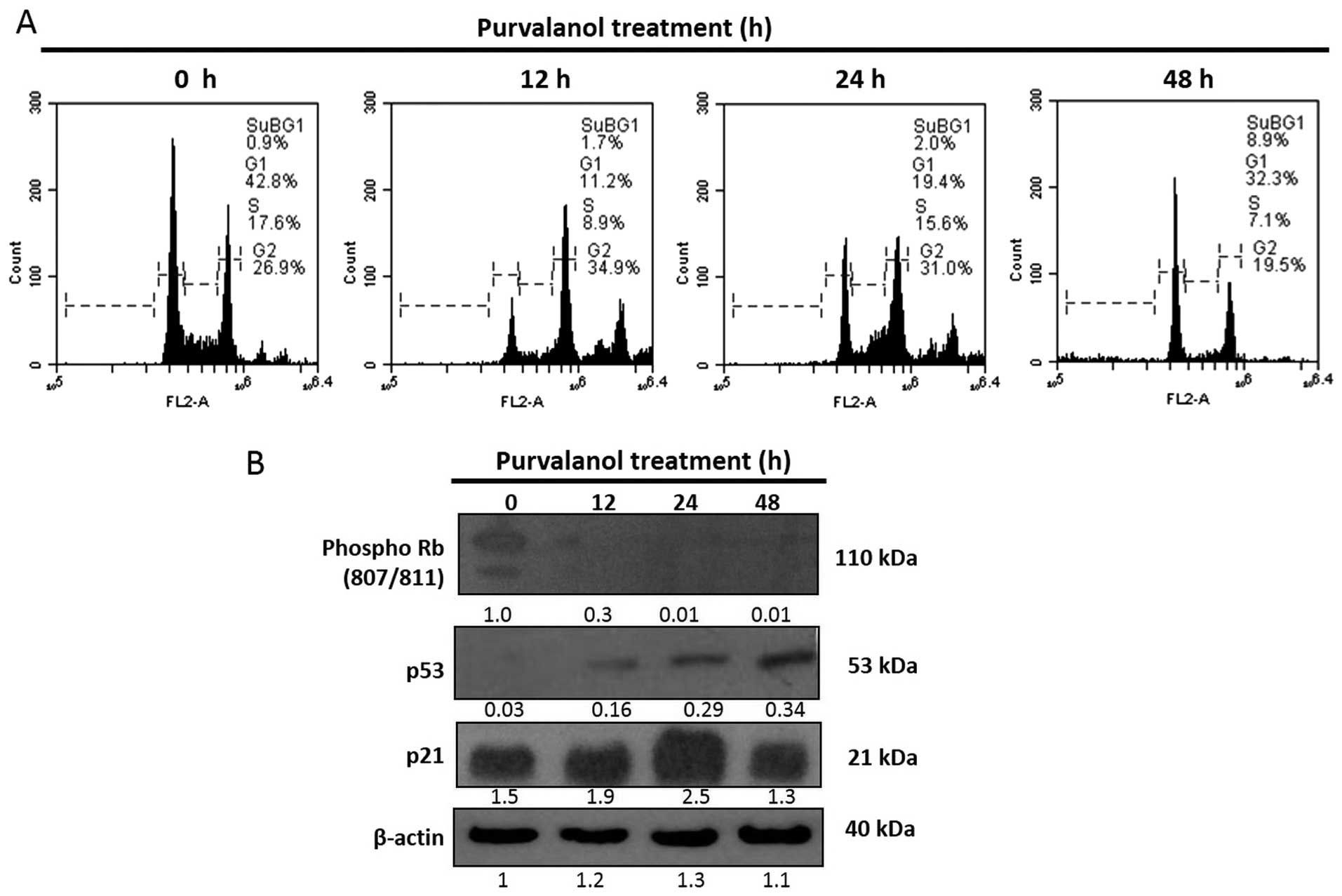
Purvalanol induces cell cycle arrest in a
time-dependent manner
The effect of purvalanol on the cell cycle profile
of HCT 116 cells was examined following PI staining by FACS flow
analysis. 15 μM of purvalanol increased the subG1 population
in a time-dependent manner (12, 24 and 48 h), to 1.7, 2 and 8.9%,
respectively (Fig. 2A). In order to
confirm FACS flow analysis, we analyzed major cell cycle
coordinating proteins such as phosphorylated Rb, p53 and p21 by
immunob-lotting. According to the immunoblotting results,
purvalanol treatment increased the expression of p53 in a
time-dependent manner, but significant upregulation was determined
after a 48 h purvalanol treatment in the HCT 116 colon cancer
cells. However, expression of p21 was increased only after a 24 h
purvalanol treatment. Moreover, protein expression of the
phosphorylated form of Rb (807/811) was sharply downregulated only
after 12 h in HCT 116 colon cancer cells (Fig. 2B).
Purvalanol activates ER stress
In order to evalute the time-dependent effect of
purvalanol on ER stress via activated Ca2+ metabolism,
we performed immunoblotting. According to the immunblotting
results, purvalanol upregulated both PDI and calnexin expression
levels within 1 h of purvalanol exposure in the HCT 116 colon
cancer cells. However, calre-ticulin, an ER resident protein with
Ca2+ buffering effect, was downregulated in a
time-dependent manner following 15 μM of purvalanol
treatment (Fig. 3A). We next
examined the expression profiles of ER membrane proteins and their
downstream target key molecules by immunoblotting. BiP expression
was upregulated after a 3 h purvalanol treatment and this effect
was increased in a time-dependent manner. Moreover, a 1 h
purvalanol treatment induced PERK expression and phosphorylation of
eIF-2α within a 3 h drug exposure in the HCT 116 cells. Another ER
membrane receptor, IRE1α, was also upregulated following
time-dependent purvalanol treatment. Late response of cells to
purvalanol treatment related with ER stress was determined by the
cleavage of ATF-6 after 12 h. CHOP, a transcriptional target of
both cleaved ATF-6 and ATF-4, was found to be significantly
upregulated following a 12 h purvalanol treatment (Fig. 3B). In order to examine the
purvalanol-induced CHOP activation, we performed FACS flow analysis
after transfection of the CHOP promoter (−649/+136) pmCherry-1
plasmid (Fig. 3C). According to
FACS flow analysis, purvalanol-induced CHOP activation via ATF-4
transcriptional activity was determined after 48 h.
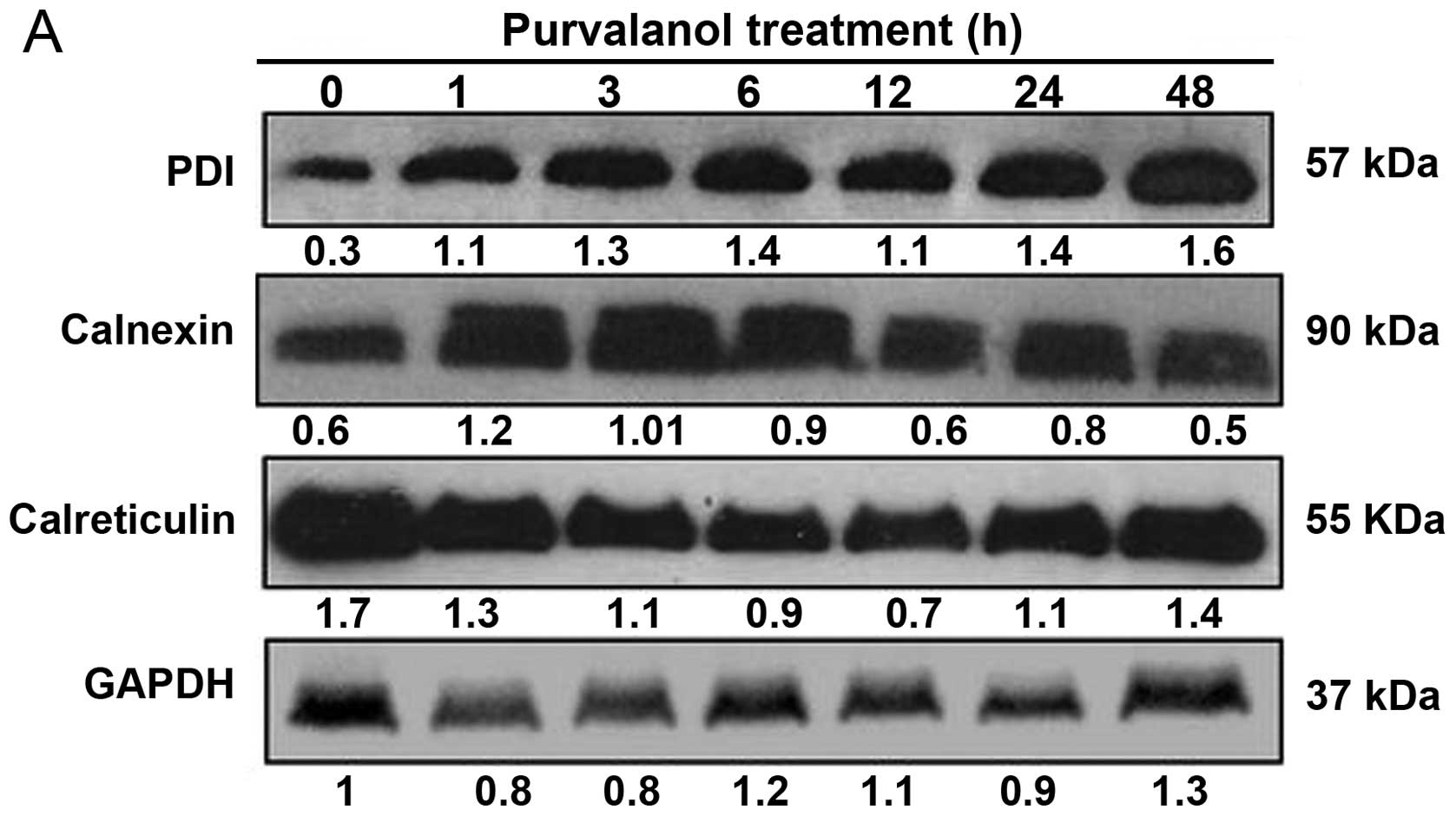 | Figure 3Purvalanol triggers ER stress in a
time-dependent manner. Following time-dependent purvalanol
treatment, total proteins were isolated and separated on 12% SDS
gel, transferred onto PVDF membranes and blotted with (A) PDI,
calnexin, calreticulin, (B) ATF-6, BiP, IRE1a, PERK, XBP1, JNK,
eIF-2a, ATF-4, caspase-12 and CHOP antibodies. GAPDH and β-actin
were used as a loading control. (C) Cells (3×105) were
transfected with the mCherry CHOP plasmid and following 24 h, the
cells were treated with 15 μM purvalanol for 12, 24 and 48
h. Purvalanol-induced CHOP activation was examined by FACS flow
analysis at FL-3 channel. |
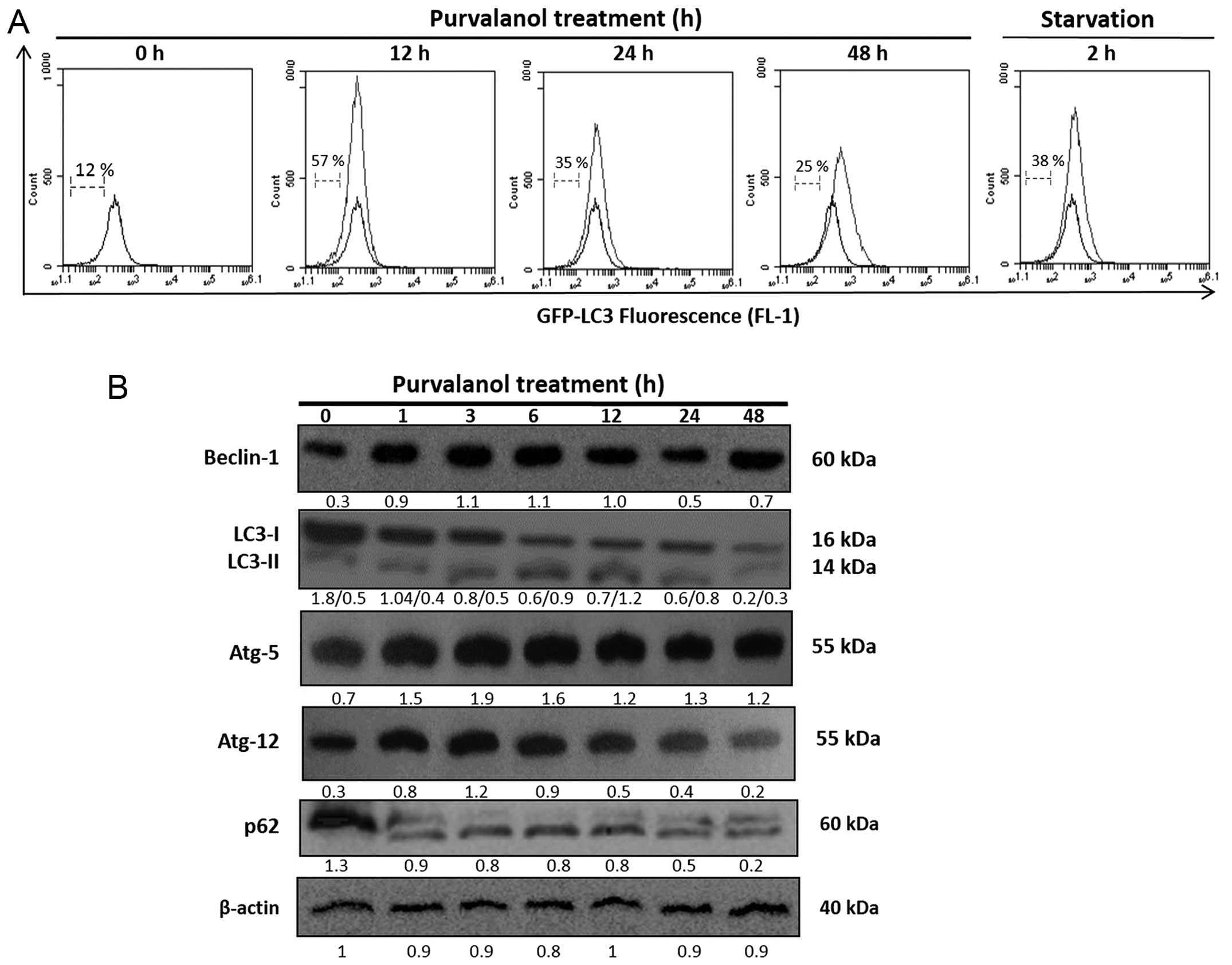
Purvalanol-induced ER stress activates
autophagy in HCT 116 colon cancer cells
To evaluate the effect of purvalanol on autophagic
regulation related with its influence on ER membrane receptor
activation, we examined the involvement of LC3 via GFP-LC3 plasmid
transfection in the HCt 116 cells. According to FACS flow analysis,
purvalanol induced GFP-LC3 intensity within a 12 h drug exposure
and this effect was maintained for 24 h (Fig. 4A). Moreover, Beclin-1, Atg-5 and
Atg-12 expression levels were upregulated; p62 degradation and LC3
cleavage were observed at early time-points following purvalanol
treatment in the HCT 116 colon cancer cells (Fig. 4B).
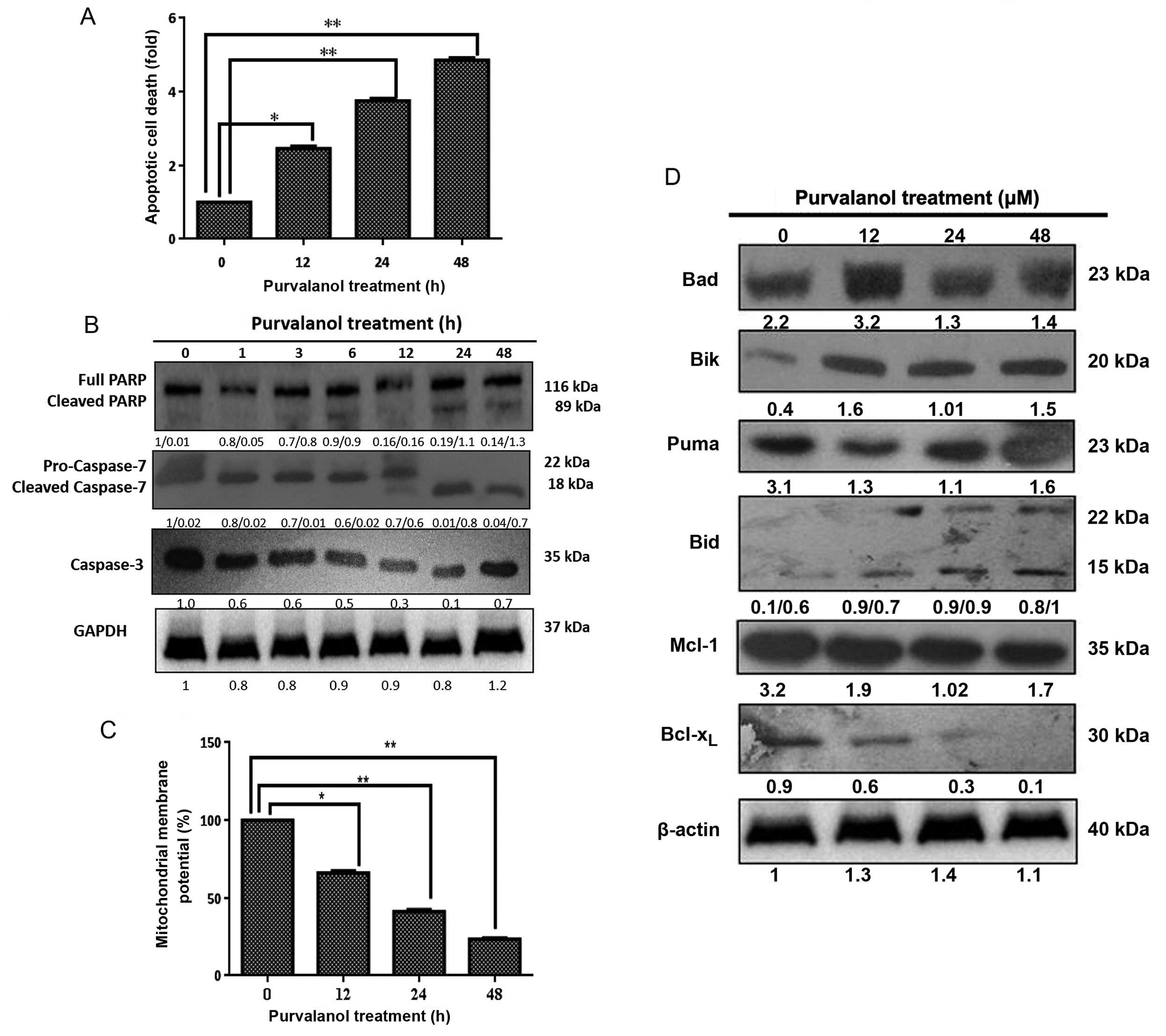
Prolonged purvalanol treatment causes
apoptotic cell death by modulating the Bcl-2 family members in HCT
116 colon cancer cells
The apoptotic induction due to purvalanol was
determined by Cell Death ELISA assay. As shown in Fig. 5A, exposure of cells to 15 μM
purvalanol for 12, 24 and 48 h induced apoptosis by 2.5-, 3.5- and
5.5-fold compared to the untreated control cells (p<0.05 and
p<0.001), respectively. The apoptotic induction of purvalanol
was found to be caspase-dependent. As shown in Fig. 5B, caspase-7 and caspase-3 were
activated by causing PARP cleavage. As shown in Fig. 5C, 15 μM purvalanol for 12, 24
and 48 h induced MMP loss by 75, 55 and 40%, respectively
(p<0.05 and p<0.001). To analyze the functional role of Bcl-2
family members in mitochondria-mediated apoptosis due to purvalanol
treatment, the modulation of the expression levels of anti- and
pro-apoptotic proteins was examined in the HCT 116 cells by western
blot analysis. Purvalanol down-regulated Bcl-xl and Mcl-1 and
upregulated Bik, Bad, Bid, Bad and PUMA protein expression levels
(Fig. D). Therefore, we conclude
that purvalanol induced mitochondria-mediated apoptosis by
activating caspases and modulating Bcl-2 family members.
Discussion
Dysregulation of molecular machinery of the cell
cycle process plays an important role in tumor progression and
malignancy (1). Thus, control of
the cell cycle by pharmacological CDKIs, is believed to be an
important therapeutic target for cancer therapy. CDKIs have been
shown to be promising chemotherapeutic agents in various cancer
types such as colon (7), prostate
(5) and breast cancer (6). Purvalanol, a new generation CDKI, has
been shown to cause cell cycle arrest at the G2/M checkpoint of the
cell cycle by inhibiting the ATP binding site of CDK1 and CDK2
(14) and to induce apoptosis
through activating caspases and cytochrome-c release in
gastric cancer cells (15). In our
previous study, we determined that 30 μM purvalanol induced
apoptotic cell death by modulating the expression of the Bcl-2
family members via activating polyamine catabolic metabolism in HCT
116 colon cancer cells (7). Another
previous study from our laboratory showed that 25 μM
purvalanol induced apoptosis via activation of caspases, which led
to dysfunction of MMP in MCF-7 breast cancer cells (6). A lower dose of purvalanol (15
μM) used in this study also inhibited cell proliferation via
cell cycle arrest and induced cell death through MMP loss (Figs. 1 and 2). Although a number of reports have
demonstrated the molecular action of roscovitine, there are few
research studies concerning purvalanol-induced apoptotic cell
death. Thus, clarification of molecular targets of purvalanol
treatment on apoptosis and autophagic mechanisms might increase the
therapeutic efficiency of the drug.
During homeostatic conditions in eukaryotic cells,
ER is essential in biosynthesis and signaling functions, and
several resident chaperones, Ca2+ binding proteins and
folding enzymes such as calnexin, calreticulin and protein
disulfide isomerase (PDI) are known to monitor ER biological
functions (16). It was shown that
a lack of calnexin within the cell induced a resistant profile
against ER stress-mediated apoptosis as it acts with caspase-8 for
Bap31 protein cleavage (17). In
addition, although overexpression of calreticulin in HeLa cells was
shown to enhance the sensitivity against ceramide-induced apoptosis
(18), knockout of calreticulin
increased the resistance phenotype (19). Moreover, silencing of calreticulin
in bladder cancer cells suppressed cell proliferation, migration
and attachment, whereas overexpression of calreticulin enhanced
cell migration and attachment (20). Thus, in the present study, we
determined that purvalanol induced UPR as a first response. Both
PDI and calnexin expression profiles were upregulated following a 1
h purvalanol treatment and this effect was observed until 12 h. In
addition, calreticulin expression was downregulated at early
time-points following purvalanol treatment which modulated
Ca2+ levels and cell adhesion (Fig. 3A). These results suggest that
purvalanol might first trigger ER stress via ER chaperone
activation in HCT 116 colon cancer cells.
Accumulation of unfolded or misfolded proteins in
the ER lumen during hypoxia, oxidative injury, a high-fat diet or
viral infections was found to induce various intracellular
signaling pathways via activation of receptors localized on the ER
membrane (9). During ER stress,
disassociation of ER membrane receptors from their ligand BiP
caused receptor-mediated nuclear activation of various
transcription factors such as ATF-6, ATF-4 and CHOP (12). ER stress- activated ER membrane
receptor, ATF-6 p90, is activated following BiP disassociation and
cleavage to form ATF-6 p50, a regulatory protein of X-box binding
protein 1 (XBP-1) mRNA (21).
Cleaved and activated ATF-6 p50 protein translocates to the nucleus
and triggers UPR target genes such as BiP and XBP1 (22). Similar to this finding,
N-butylidenephthalide was shown to induce ER-stress by triggering
ATF-6 cleavage and BiP upregulation within 6 h after drug exposure
in LNCaP and PC3 prostate cancer cell lines (23). Both ATF-6 and IRE1α trigger the
transcription of ER chaperones during ER stress conditions via
acting on XBP1 mRNA (24).
Moreover, IRE1α interaction with TRAF-2 triggers apoptotic cell
death via JNK activation (25).
Although 6-shogaol induced apoptosis without any significant effect
on IRE1α in SMMC-7721 hepatocellular carcinoma cells (26),
20-O-(β-D-glucopyranosyl)-20(S)-protopanaxadiol activated the IRE1α
phosphorylation and splicing of XBP1 in Ht29 human colon cancer
cells (27). In addition,
upregulation of IRE-1α was determined after a 6-h
N-butylidenephthalide treatment in prostate cancer cell lines
(23). In the present study,
although we demonstrated upregulation of IRE-1α expression and an
increase in XBP1 splicing, a significant upregulation of JNK
expression was observed after a 24-h purvalanol treatment (Fig. 3B). Concominantly, purvalanol induced
apoptotic cell death via activation of caspase-3 and caspase-7 and
PARP cleavage (Fig. 5B). Thus, we
conclude that short-term purvalanol treatment induces
ATF-6-mediated ER stress and long-term purvalanol induces apoptotic
cell death through activation of JNK in HCT 116 colon cancer
cells.
The third ER membrane receptor, PERK, is dimerized,
and auto-phosphorylation of itself activates the phosphorylation of
eIF-2α that attenuates protein translation (11). PERK activation and eIF-2α
phosphorylation were determined after a 6-h
20-O-(β-D-glucopyranosyl)-20(S)-protopanaxadiol treatment in HT29
human colon cancer cells (27).
6-shogaol induced apoptosis via activation of PERK and its
downstream target eIF-2α in SMMC-7721 hepatocellular carcinoma
cells (26). In addition,
luteolin-induced eIF-2α phosphorylation within a 2 h drug treatment
triggered the ER stress-mediated apoptosis in NCI-H460 lung
carcinoma cells (28). We
determined that PERK expression was upregulated only after a 1 h
purvalanol treatment and phosphorylation of eIF-2α was also
observed during 1–6 h of purvalanol treatment (Fig. 3B). Purvalanol might induce UPR and
attenuate protein translation at early time-points in HCT 116 cells
by activating the PERK/eIF-2α/ATF-4 signaling axis.
CHOP, is a key ER stress-induced transcription
factor and transcription activation is through ATF-6 and ATF-4
(29). Deficiency of CHOP protected
cells from ER-induced apoptotic cell death (30) and overexpression of CHOP stimulated
cell cycle arrest and apoptosis (31). Activated CHOP expression has been
implicated to repress anti-apoptotic Bcl-2 protein (32) and induce apoptotic cell death by
activating Bim (33). We showed
that CHOP expression was significantly upregulated after a 6 h
purvalanol treatment and this effect was increased in a
time-dependent manner (Fig. 3B).
Moreover, when we analyzed the activation of CHOP via the CHOP
promoter (−649/+136) inserted mCherry expression plasmid, mCherry
expression was found to be elevated following a 48 h purvalanol
treatment (Fig. 3C). CHOP-mediated
ER stress-induced apoptotic cell death occurred after a 48 h
purvalanol treatment in the HCT 116 colon cancer cells.
ER stress-induced autophagy is assumed to be
activated as an alternative way to degradate accumulated
unfolded/misfolded protein aggregates instead of the proteasome
degradation system (12). Both
IRE1α and PERK pathways were demonstrated to be associated with ER
stress-induced autophagy regulation in cancer cells (34). In contrary, other studies reported
that only PERK-eIF-2α signaling was involved in autophagy induction
by ER stress (35). Autophagy, an
evolutionarily conserved process, characterized by massive
degradation of cytosolic contents initiates the fusion of
autophagosomes to endosomes and lysosomes, which engulf cytoplasmic
contents within a double-membrane vacuole (36). This physiological process is carried
out through interaction of various molecules such as Bcl-2,
Beclin-1, Atg-5 and Atg-12 (37).
During autophagic induction, LC3-I (16 kDa) is cleaved to form
LC3-II (14 kDa) that is involved in autophagic vacuole membrane
formation by interacting with cytoplasmic protein p62 (38). As autophagy is assumed to be a cell
survival mechanism against chemotherapeutic drugs in cancer cells,
inhibition of autophagy by specific inhibitors induced
drug-mediated apoptotic cell death in prostate (39), breast (40) and colon (41) cancer cells. Moreover, in NCI-H460
lung carcinoma cells, although luteolin induced ER stress activated
apoptosis, autophagy was shown to be activated and inhibition of
autophagy induced apoptotic cell death (28). Thus, in our present study we
determined that another CDKI, purvalanol, induced autophagy
induction by upregulation of Beclin-1, Atg-5, Atg-12 protein
expression and LC3-II formation during a 6–12 h purvalanol
treatment (Fig. 4B) only after
Perk/eIF-2α induced ER stress.
The inactive state of ER membrane receptors can be
stimulated by slight ER stress conditions leading to translation
inhibition. Under this situation, autophagy induction could occur
as a cell protection mechanism. However, prolonged ER stress can
also result in apoptotic cell death through various molecular key
players such as caspase-12, JNK and CHOP (42). Our previous studies found that
long-term exposure of purvalanol triggered mitochondrial-mediated
and caspase-dependent apoptotic cell death in MCF-7 cells following
treatment with 25 μM purvalanol (6) and in HCT 116 colon cancer cells after
of 30 μM purvalanol (7). In
the present study, a lower dose of purvalanol for a long-term
exposure triggered caspase-dependent apoptotic cell death via
modulation of Bcl-2 family members and this effect was significant
after a 24 h purvalanol treatment in the HCT 116 colon cancer cells
(Fig. 5A–D). Finally, we conclude
that purvalanol induced ER stress-mediated autophagy as a first
response in HCT 116 colon cancer cells against purvalanol.
Prolonged ER stress activated both CHOP and JNK- mediated apoptotic
cell death.
Acknowledgments
The present study was supported by the Istanbul
Kultur University Scientific Projects Support Center and the
TUBITAK Scientific Projects Support Center (2209 program).
Abbreviations:
|
ABTS
|
2,2′-azino-di-[3-ethylbenzthiazoline
sulfonate (6)] diammonium salt
|
|
ATF-6
|
activating transcription factor-6
|
|
CDK
|
cyclin-dependent kinase
|
|
CDKI
|
cyclin-dependent kinase inhibitor
|
|
CHOP
|
CAAT/enhancer-binding protein
|
|
DiOC6
|
3,3′-dihexyloxacarbocyanine iodide
|
|
DMSO
|
dimethyl sulfoxide
|
|
ER
|
endoplasmic reticulum
|
|
GFP
|
green fluorescent protein
|
|
HRP
|
horseradish peroxidase
|
|
IRE1
|
inositol-requiring enzyme 1
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LC3
|
microtubule-associated protein
1A/1B-light chain 3
|
|
MMP
|
mitochondrial membrane potential
|
|
MTT
|
3-4,5-dimethyl-2-thiazolyl-2,5-diphenyl-2H-tetrazolium bromide
|
|
UPR
|
unfolded protein response
|
|
PERK
|
PRKR-like ER kinase
|
|
PBS
|
phosphate- buffered saline
|
|
PI
|
propidium iodide
|
|
POD
|
peroxidase
|
|
PVDF
|
polyvinyldifluoride
|
|
Rb
|
retinoblastoma
|
|
SDS-PAGE
|
sodium dodecyl sulphate polyacrylamide
gel electrophoresis
|
|
TBS
|
Tris-buffered saline
|
|
XBP-1
|
X-box binding protein 1
|
References
|
1
|
Harper JW and Adams PD: Cyclin-dependent
kinases. Chem Rev. 101:2511–2526. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Knockaert M, Greengard P and Meijer L:
Pharmacological inhibitors of cyclin-dependent kinases. Trends
Pharmacol Sci. 23:417–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gray N, Détivaud L, Doerig C and Meijer L:
ATP-site directed inhibitors of cyclin-dependent kinases. Curr Med
Chem. 6:859–875. 1999.PubMed/NCBI
|
|
4
|
Gray NS, Wodicka L, Thunnissen AM, Norman
TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, Le Clerc S, Meijer L,
et al: Exploiting chemical libraries, structure, and genomics in
the search for kinase inhibitors. Science. 281:533–538. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arisan ED, Obakan P, Coker-Gurkan A,
Calcabrini A, Agostinelli E and Unsal NP: CDK inhibitors induce
mitochondria-mediated apoptosis through the activation of polyamine
catabolic pathway in LNCaP, DU145 and PC3 prostate cancer cells.
Curr Pharm Des. 20:180–188. 2014. View Article : Google Scholar
|
|
6
|
Obakan P, Arisan ED, Özfiliz P,
Çoker-Gürkan A and Palavan-Ünsal N: Purvalanol A is a strong
apoptotic inducer via activating polyamine catabolic pathway in
MCF-7 estrogen receptor positive breast cancer cells. Mol Biol Rep.
41:145–154. 2014. View Article : Google Scholar
|
|
7
|
Gürkan AC, Arisan ED, Obakan P and
Palavan-Ünsal N: Inhibition of polyamine oxidase prevented
cyclin-dependent kinase inhibitor-induced apoptosis in HCT 116
colon carcinoma cells. Apoptosis. 18:1536–1547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schroder M and Kaufman RJ: ER stress and
the unfolded protein response. Mutat Res. 569:29–63. 2005.
View Article : Google Scholar
|
|
10
|
Yoshida H: Molecular biology of the ER
stress response. Seikagaku. 76:617–630. 2004.In Japanese.
PubMed/NCBI
|
|
11
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An endoplasmic reticulum stress-specific
caspase cascade in apoptosis. Cytochrome c-independent activation
of caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iizuka D, Inanami O, Kashiwakura I and
Kuwabara M: Purvalanol A enhances cell killing by inhibiting
up-regulation of CDC2 kinase activity in tumor cells irradiated
with high doses of X rays. Radiat Res. 167:563–571. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Villerbu N, Gaben AM, Redeuilh G and
Mester J: Cellular effects of purvalanol A: a specific inhibitor of
cyclin-dependent kinase activities. Int J Cancer. 97:761–769. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Molinari M, Eriksson KK, Calanca V, Galli
C, Cresswell P, Michalak M and Helenius A: Contrasting functions of
calreticulin and calnexin in glycoprotein folding and ER quality
control. Mol Cell. 13:125–135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zuppini A, Groenendyk J, Cormack LA, Shore
G, Opas M, Bleackley RC and Michalak M: Calnexin deficiency and
endoplasmic reticulum stress-induced apoptosis. Biochemistry.
41:2850–2858. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pinton P, Ferrari D, Rapizzi E, Di
Virgilio F, Pozzan T and Rizzuto R: The Ca2+
concentration of the endoplasmic reticulum is a key determinant of
ceramide-induced apoptosis: significance for the molecular
mechanism of Bcl-2 action. EMBO J. 20:2690–2701. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakamura K, Bossy-Wetzel E, Burns K, Fadel
MP, Lozyk M, Goping IS, Opas M, Bleackley RC, Green DR and Michalak
M: Changes in endoplasmic reticulum luminal environment affect cell
sensitivity to apoptosis. J Cell Biol. 150:731–740. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu YC, Chen CN, Wang B, Hsu WM, Chen ST,
Chang KJ, Chang CC and Lee H: Changes in tumor growth and
metastatic capacities of J82 human bladder cancer cells suppressed
by down-regulation of calreticulin expression. Am J Pathol.
179:1425–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoshida H, Haze K, Yanagi H, Yura T and
Mori K: Identification of the cis-acting endoplasmic reticulum
stress response element responsible for transcriptional induction
of mammalian glucose-regulated proteins. Involvement of basic
leucine zipper transcription factors. J Biol Chem. 273:33741–33749.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haze K, Yoshida H, Yanagi H, Yura T and
Mori K: Mammalian transcription factor ATF6 is synthesized as a
transmembrane protein and activated by proteolysis in response to
endoplasmic reticulum stress. Mol Biol Cell. 10:3787–3799. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiu SC, Chen SP, Huang SY, Wang MJ, Lin
SZ, Harn HJ and Pang CY: Induction of apoptosis coupled to
endoplasmic reticulum stress in human prostate cancer cells by
n-butylidenephthalide. PLoS One. 7:e337422012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoshida H, Okada T, Haze K, Yanagi H, Yura
T, Negishi M and Mori K: Endoplasmic reticulum stress-induced
formation of transcription factor complex ERSF including NF-Y (CBF)
and activating transcription factors 6alpha and 6beta that
activates the mammalian unfolded protein response. Mol Cell Biol.
21:1239–1248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Urano F, Bertolotti A and Ron D: IRE1 and
efferent signaling from the endoplasmic reticulum. J Cell Sci.
113:3697–3702. 2000.PubMed/NCBI
|
|
26
|
Hu R, Zhou P, Peng YB, Xu X, Ma J, Liu Q,
Zhang L, Wen XD, Qi LW, Gao N, et al: 6-Shogaol induces apoptosis
in human hepatocellular carcinoma cells and exhibits anti-tumor
activity in vivo through endoplasmic reticulum stress. PLoS One.
7:e396642012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang R, Chung Y, Kim HS, Kim DH, Kim HS,
Chang WY and Hyun JW:
20-O-(beta-D-glucopyranosyl)-20(S)-protopanaxadiol induces
apoptosis via induction of endoplasmic reticulum stress in human
colon cancer cells. Oncol Rep. 29:1365–1370. 2013.PubMed/NCBI
|
|
28
|
Park SH, Park HS, Lee JH, Chi GY, Kim GY,
Moon SK, Chang YC, Hyun JW, Kim WJ and Choi YH: Induction of
endoplasmic reticulum stress-mediated apoptosis and non-canonical
autophagy by luteolin in NCI-H460 lung carcinoma cells. Food Chem
Toxicol. 56:100–109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma Y, Brewer JW, Diehl JA and Hendershot
LM: Two distinct stress signaling pathways converge upon the CHOP
promoter during the mammalian unfolded protein response. J Mol
Biol. 318:1351–1365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barone MV, Crozat A, Tabaee A, Philipson L
and Ron D: CHOP (GADD153) and its oncogenic variant, TLS-CHOP, have
opposing effects on the induction of G1/S arrest. Genes Dev.
8:453–464. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Puthalakath H, O’Reilly LA, Gunn P, Lee L,
Kelly PN, Huntington ND, Hughes PD, Michalak EM, Mckimm-Breschkin
J, Motoyama N, et al: ER stress triggers apoptosis by activating
BH3-only protein Bim. Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vidal RL and Hetz C: Crosstalk between the
UPR and autophagy pathway contributes to handling cellular stress
in neurodegenerative disease. Autophagy. 8:970–972. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carra S, Brunsting JF, Lambert H, Landry J
and Kampinga HH: HspB8 participates in protein quality control by a
non-chaperone-like mechanism that requires eIF2{alpha}
phosphorylation. J Biol Chem. 284:5523–5532. 2009. View Article : Google Scholar
|
|
36
|
Martinet W, Agostinis P, Vanhoecke B,
Dewaele M and De Meyer GR: Autophagy in disease: a double-edged
sword with therapeutic potential. Clin Sci (Lond). 116:697–712.
2009. View Article : Google Scholar
|
|
37
|
Ku B, Woo JS, Liang C, Lee KH, Hong HS, E
X, Kim KS, Jung JU and Oh BH: Structural and biochemical bases for
the inhibition of autophagy and apoptosis by viral BCL-2 of murine
gamma-herpesvirus 68. PLoS Pathog. 4:e252008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kumar D, Shankar S and Srivastava RK:
Rottlerin induces autophagy and apoptosis in prostate cancer stem
cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett.
343:179–189. 2014. View Article : Google Scholar
|
|
40
|
Cui Q, Tashiro S, Onodera S, Minami M and
Ikejima T: Autophagy preceded apoptosis in oridonin-treated human
breast cancer MCF-7 cells. Biol Pharm Bull. 30:859–864. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU- induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schönthal AH: Endoplasmic reticulum stress
and autophagy as targets for cancer therapy. Cancer Lett.
275:163–169. 2009. View Article : Google Scholar
|