Introduction
Ovarian cancer remains one of the most difficult
oncologic challenges, as it is often discovered at advanced stages.
It is diagnosed often with intraperitoneal dissemination of cancer
cells and the presence of ascitic fluid. The cancer cells present
in the intraperitoneal space often display intrinsic
chemoresistance. The ascitic fluid is a complex exudative liquid
known to contain various growth factors and cytokines (1). It has been shown that ascites could
contribute to intraperitoneal cancer cell implantation and tumor
development by promoting cell growth, invasion and drug resistance
(2,3). One of the characteristics of all
ascites including ovarian cancer ascites is its inability to clot.
This may be due to the presence of several fibrinolytic and
proteolytic enzymes such as plasminogen activators and matrix
metalloproteinases that are not fully neutralized by their
inhibitors (4,5). The absence of clotting of ascites in
the peritoneal site favors tumor cell dissemination and their
implantation on the peritoneal surface.
It was reported that endothelial protein C receptor
(EPCR) binds protein C (PC) secreted by liver cells (6) and thereby inhibits the coagulation
pathway by generation of activated protein C (aPC). Proteolytic
cleavage of thrombin bound to thrombomodulin acts on PC to generate
aPC. Endothelial protein C is an important regulator of
homeostasis, in addition to its involvement in the systemic
response to acute inflammation (7–11). On
endothelial cells, the aPC, together with its cofactor protein S,
degrades factors Va and VIIIa and thereby interferes with thrombin
generation and abolishes the coagulation cascade (12). Endogenous aPC is shown to limit
cancer cell extravasation through S1P-1-mediated vascular
endothelium (13) and also plays a
role in innate immune system (14,15).
Originally, EPCR was shown to be expressed on
endothelial cells of large blood vessels but not in liver
sinosuoidal and spleen endothelial cells (16). EPCR (CD201) expression is also
detected in some inflammatory cells such as monocytes and
neutrophils (17). Some authors
claim that EPCR could be considered as a cancer stem cell marker
(18). EPCR exists as membrane
bound as well as free soluble endothelial protein C receptor
(sEPCR) form (19). In fact, sEPCR,
can regulate the quantity of circulating aPC and influence the
clotting of plasma (20). Ligand
binding to EPCR promotes endocytosis of EPCR (21) via the Rab GTPase pathway (22). aPC/EPCR interaction can directly
modulate cell signaling and alter gene expression in inflammation
and apoptosis (23) and also
provide cytoprotection via PAR-1 activation (24).
We previously demonstrated the expression of EPCR in
a large number of cancer cell lines, in solid tumors (25) and malignant hemophaties. This led us
to suggest that plasmatic sEPCR can be considered as a biomarker of
cancer-associated hypercoagulability in human hematologic
malignancies (26).
In the present study, we investigated the role of
aPC, known to be a natural physiological anticoagulant, in the
activation of cancer cells and the loss of clotting properties of
ascitic fluid in patients with peritoneal ovarian
carcinomatosis.
Materials and methods
Cells
The human ovarian cancer cell line OVCAR-3NIH,
purchased from American Type Culture Collection (ATTC; Manassas,
VA, USA), was cultured in RPMI-1640 medium containing 10% fetal
calf serum (FCS), glutamine (03 mg/ml), penicillin (50 U/ml),
streptomycin (50 µg/ml) and incubated at 37°C with 5%
CO2.
Ascites
Peritoneal fluids were collected from 20 ovarian
cancer patients treated at the Hospital Hôtel-Dieu (Paris, France).
As evacuation of ascites is a part of the routine management of
patients, only oral consent was obtained. Cells from ascitic fluids
were pelleted by a short spin at 1,000 rpm, and the supernatant was
re-centrifuged for another 10 min and then collected. The pelleted
cells and the resulting supernatant were aliquoted and stored.
Cyto-ELISA
OVCAR-3 cells (6×104/well), in
FCS-enriched medium, were seeded in flat-bottomed 96-well plates
(Nunc). Confluent cell layers from 3-day cultures were washed with
phosphate-buffered saline (PBS). The cells were then exposed to
native protein C (Ceprotin®) or aPC (Xigris®)
both at concentrations of 10 ng/ml for 2, 5 and 10 min. In
parallel, culture medium with only FCS served as the control. The
cells adhering to the plate were fixed with 100 µl of 0.5
g/l glutaraldehyde and 0.4% Triton for 5 min at 4°C. Alternatively,
the cells were fixed with 100 µl of 100% ethanol for 5 min
at −20°C. The plates were washed with PBS containing 0.02% Tween-20
and 10 g/l BSA (PBS-T-Alb), and then (5-wells/test) were exposed
for 90 min to mouse anti-phosphorylated threonine or tyrosine
antibodies (1/100 diluted in PBS-T-ALb). The immune complexes were
detected by peroxidase-conjugated anti-mouse Ig (1:2,000). The
fixed peroxidase was revealed by o-phenylenediamine
benzidine (OPD) (0.2 g/l in 0.05 mol/l phosphate buffer, pH 5.0,
containing 0.5 ml/l H2O2). The absorbance was
measured at 492 nm. Control wells were constructed with isotype or
without cells. Between each step, the wells were washed three times
with PBS.
Wound healing assay
OVCAR-3 cells (6×104) were cultured in
the presence of RPMI-1640 containing 10% FCS in 24-well plates
coated with 0.2% gelatin. After 18 h, the semi-confluent cells were
dislodged by a cell scraper on a standardized surface as previously
described (27). The cells were
then incubated in RPMI-1640 containing only 2% FCS (to reduce cell
proliferation) in the presence or absence of PC or aPC (10 ng/ml).
The effect of protein C on cell migration was evaluated
(Microvision Instruments, Evry, France) by measuring the number of
cells migrating to the wound edge after 6, 18, 27 and 48 h.
Droplet test
To elucidate the mechanism of cell migration, we
developed a ‘droplet model’ of Matrigel for the OVCAR-3 cells. The
cell-Matrigel suspension (droplet) mimics an ex vivo cell
cluster implant on the peritoneal membrane surface.
The ovarian cancer cells were incorporated in
ice-cold Matrigel® (50,000 cells/200 µl Matrigel)
to which was added either native or aPC at concentrations of 10 ng,
respectively. A droplet of 10 µl Matrigel-cell suspension
was then delivered into each well of a 96-well microplate
previously coated with 0.2% of gelatin and cooled down until the
gel had solidified. Then, 0.2 ml of culture medium containing 2%
FCS was gently added to the wells.
We studied the effect of 4 signaling inhibitors
including 3 inhibitors of Ras signaling [UO126 (a MEK-1/2 kinase
inhibitor), PD98059 (an ERK inhibitor) and FTI-277 (a farnesyl
transferase inhibitor)] and one inhibitor of Rho GTPase (GGTI-298,
ageranyl-geranyl transferase inhibitor). All inhibitors were from
Calbiochem (France) and were used at a final concentration of 10
µM added to the OVCAR cell suspension 15 min before adding
the Matrigel. The number of cells migrating out of the droplet were
observed and found to be time-dependent. They were counted after 18
h.
Cell cycle (sub-G1) evaluation
OVCAR-3 cells were seeded in culture flasks and
incubated with PC or aPC at 10 ng/ml for 24 h. Phase distribution
(G1, S and G2) was performed using DNA content using flow cytometry
and analyzed by Multi Cycle AV software as previously described
(28). The coefficient of variation
(CV) of the mean value of DNA-associated fluorescence of the G1
population (width of the peak) is a reflection of the accuracy of
the DNA content measurement.
Anticoagulant activity of living
cells
Activated partial thromboplastin time (aPTT)
evaluates intrinsic coagulation pathway efficiency; coagulation of
a plasma sample is measured after addition of phospholipid
(cephalin) and calcium. We previously described a method, based on
aPTT, to estimate EPCR expression by endothelial and cancer cells
(29), i.e. cells previously
incubated with APC were added to a plasma and activated partial
thromboplastin clotting time was measured. Cells expressing EPCR
bind aPC, inducing a prolongation of aPTT. OVCAR-3 cells were
seeded in 24-well macroplates, and after 24 h the cells were washed
with RPMI-1640 and incubated with the addition of 50 µg/ml
aPC for 15 min at 4°C. After a further washing, the effects of
OVCAR-3 cells (either remaining attached to the Petri dish or
detached by Accutase) were tested on the cephalin clotting time of
normal plasma.
OVCAR-3 (10×104) cells were added to 0.1
ml normal plasma. After an incubation period of 3 min at 37°C,
cephalin and CaCl2 were added to trigger coagulation.
The clotting time was then measured at 37°C by a spectrophotometer
at 595 nm. The results are presented both in real-time and as the
ratio of cells + aPC/cells + PC.
Determination of sEPCR, D-dimer and
soluble fibrin (SF) in the ascitic fluid
sEPCR antigen was measured by Asserachrom
sEPCR-ELISA immunoassay as recommended by the commercial supplier
(Diagnostic Stago, Inc.). Fibrin degradation product, D-dimer
concentration, was determined by
STA®-Liatest® D-Di, and SF monomers were
quantified as previously reported (30).
Results
The results that follow focus on the role of aPC in
ovarian cancer cell activation as well as a loss of clotting
observed for ascitic fluids. The investigation concerned several
issues focused on the following:
Evaluation of sEPCR and fibrin
degradation products in the peritoneal fluid of ovarian cancer
patients
All the samples tested (n=20) contained sEPCR.
Eighty-five percent of the peritoneal fluid had an sEPCR
concentration of 71±23 ng/ml which was lower than that of normal
plasma (baseline values 100±28 ng/ml) (6). The remaining 15% samples revealed a
sEPCR concentration which was similar to that of normal plasma
(Table I). In parallel, D-dimer, a
fibrin degradation product and SF were quantified (Table I). The 20 patients studied showed
values for D-dimer (60%) and for SF (50%) which were superior to
baseline values (500 ng/ml for D-dimer and 250 ng/ml for SF). The
results presented in Table I
indicated that in the peritoneal fluids there were ongoing
processes of coagulation followed by fibrinolysis.
 | Table ISamples with an sEPCR concentration
similar to that of normal plasma. |
Table I
Samples with an sEPCR concentration
similar to that of normal plasma.
| Samples | D-Di (ng/ml) | SF (ng/ml) | EPCR (ng/ml) |
|---|
| N.1 | 710 | 770 | 44 |
| N.2 | 420 | 100 | 57 |
| N.3 | 220 | 10 | 22 |
| N.4 | 350 | 260 | 43 |
| N.5 | 980 | 710 | 57 |
| N.6 | 2,310 | 210 | 61 |
| N.7 | 290 | 590 | 247 |
| N.8 | 3,760 | 2,280 | 68 |
| N.9 | 510 | 190 | 96 |
| N.10 | 780 | 240 | 83 |
| N.11 | 470 | 1,210 | 93 |
| N.12 | 760 | 160 | 106 |
| N.13 | 7,440 | 2,240 | 92 |
| N.14 | 810 | 190 | 72 |
| N.15 | 1,250 | 420 | 250 |
| N.16 | 2,410 | 700 | 154 |
| N.17 | 440 | 720 | 80 |
| N.18 | 250 | 60 | 91 |
| N.19 | 760 | 70 | 56 |
| N.20 | 1,560 | 440 | 85 |
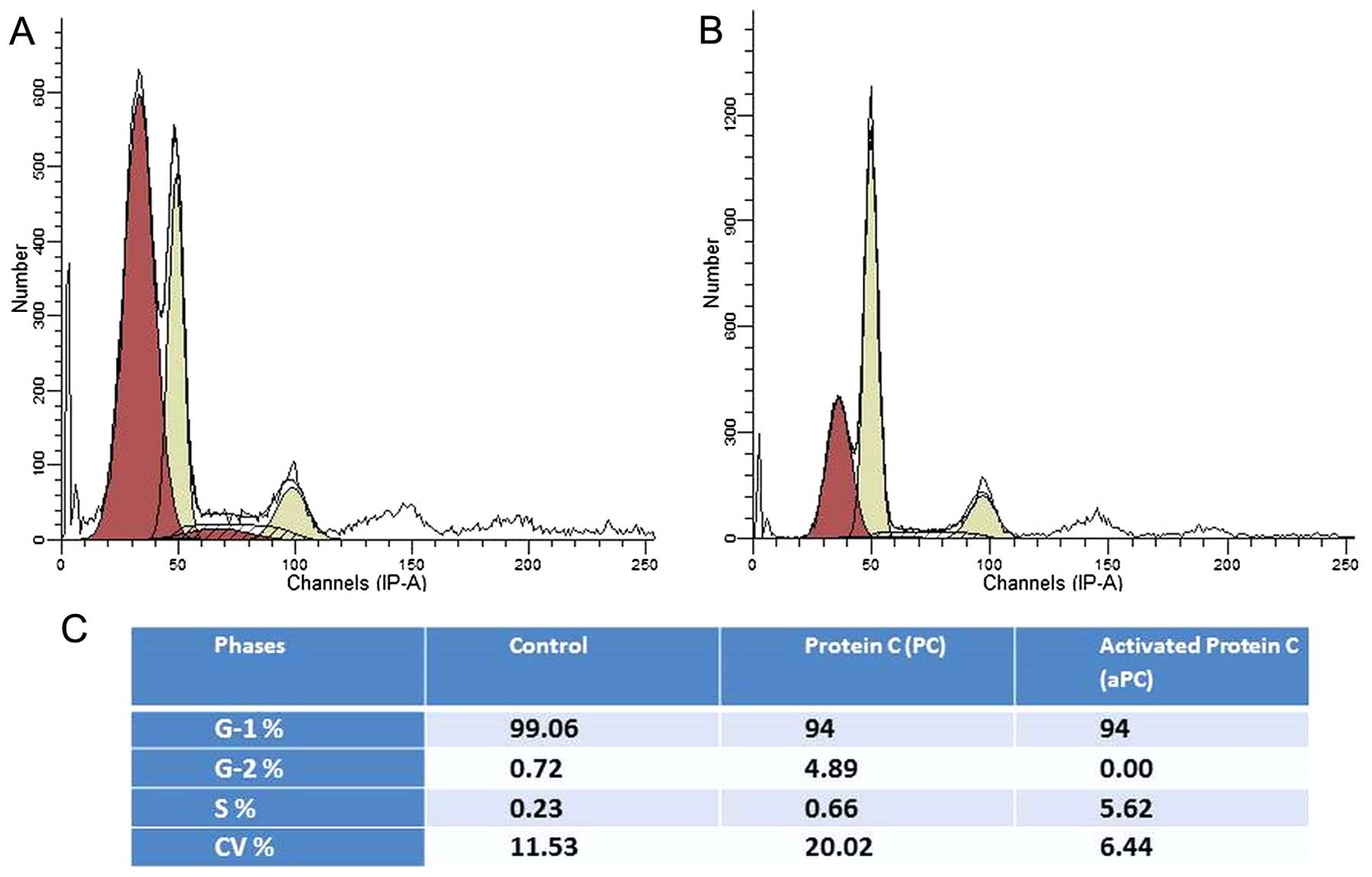
Protein C induces cell cycle
activation
When cancer cells were cultured in medium containing
1% of FCS for 24 h, all cells entered into the G1 phase. Upon
addition of culture medium containing protein C, 4.8% of the cells
in the G1 phase underwent transition to the G2 phase (Fig. 1A), whereas when aPC was added, 5.62%
of the cells in the G1 phase entered into the S phase (Fig. 1B). These results suggest that
protein C influences the ovarian cancer cell cycle.
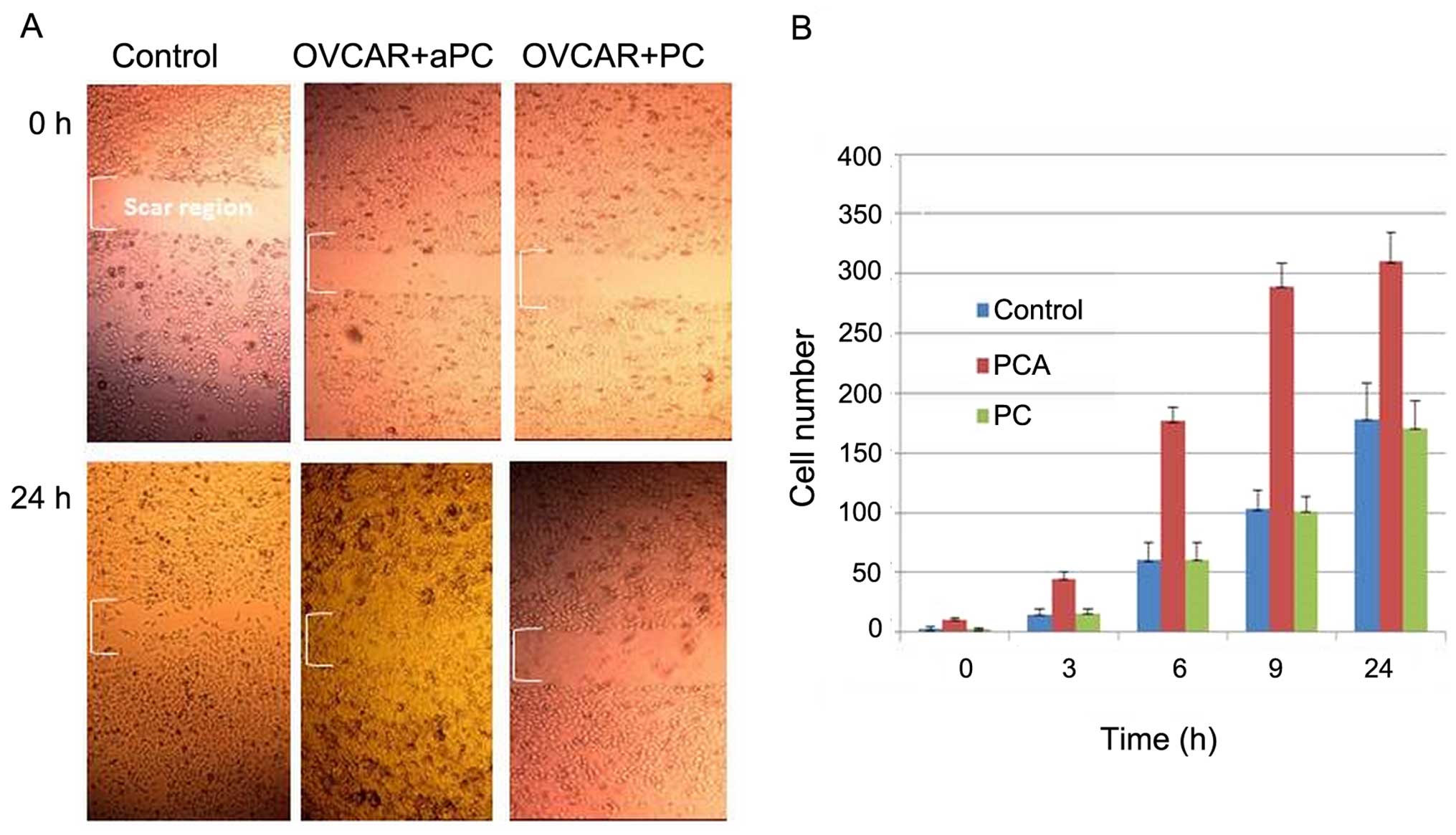
aPC induces OVCAR-3 cell migration
The influence of protein C on OVCAR-3 cell migration
at 3, 6, 9 and 24 h is presented in Fig. 2. It was observed that aPC enhanced
cell migration into the wound, whereas in the presence of PC, cell
migration was found to be similar to that of the control. The
histogram in Fig. 2B depicts the
effect of aPC and PC on ovarian cancer cells migrating into the
scar region as a function of time (0, 3, 6, 9 and 24 h).
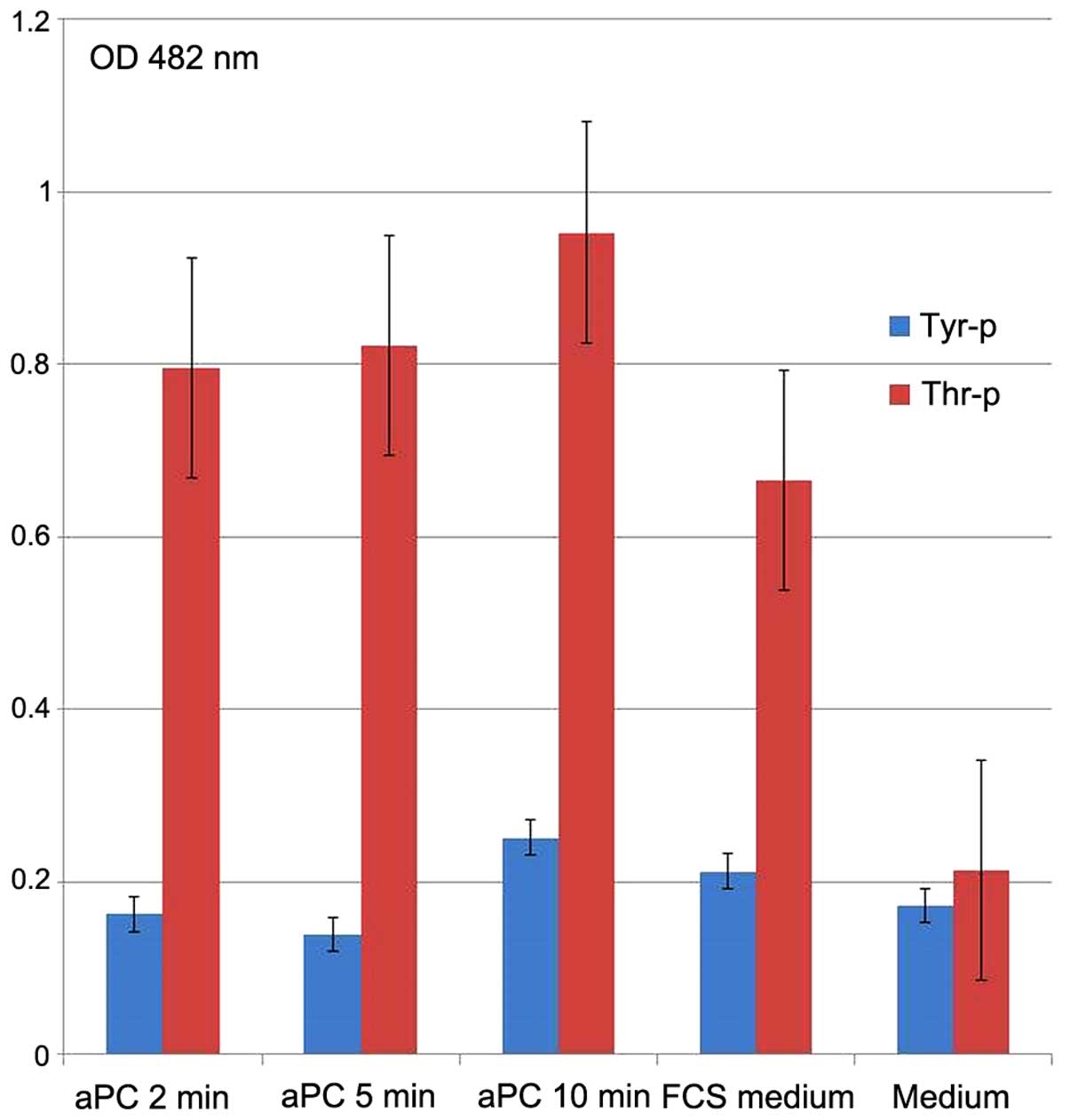
aPC induces threonine and tyrosine
phosphorylation of the OVCAR-3 cells
The effect of aPC on threonine and tyrosine
phosphorylation in the OVCAR-3 cancer cells was tested by
cyto-ELISA (Fig. 3). Compared to
tyrosine, threonine was strongly and significantly phosphorylated
when the cancer cells were incubated (10 µg/ml) with aPC.
Threonine phosphorylation started 2 min after aPC incubation, while
tyrosine phosphorylation occurred later, that is only after 10 min
of incubation. The medium with 10% bovine calf serum served as the
control and was found to induce threonine and tyrosine
phosphorylation.
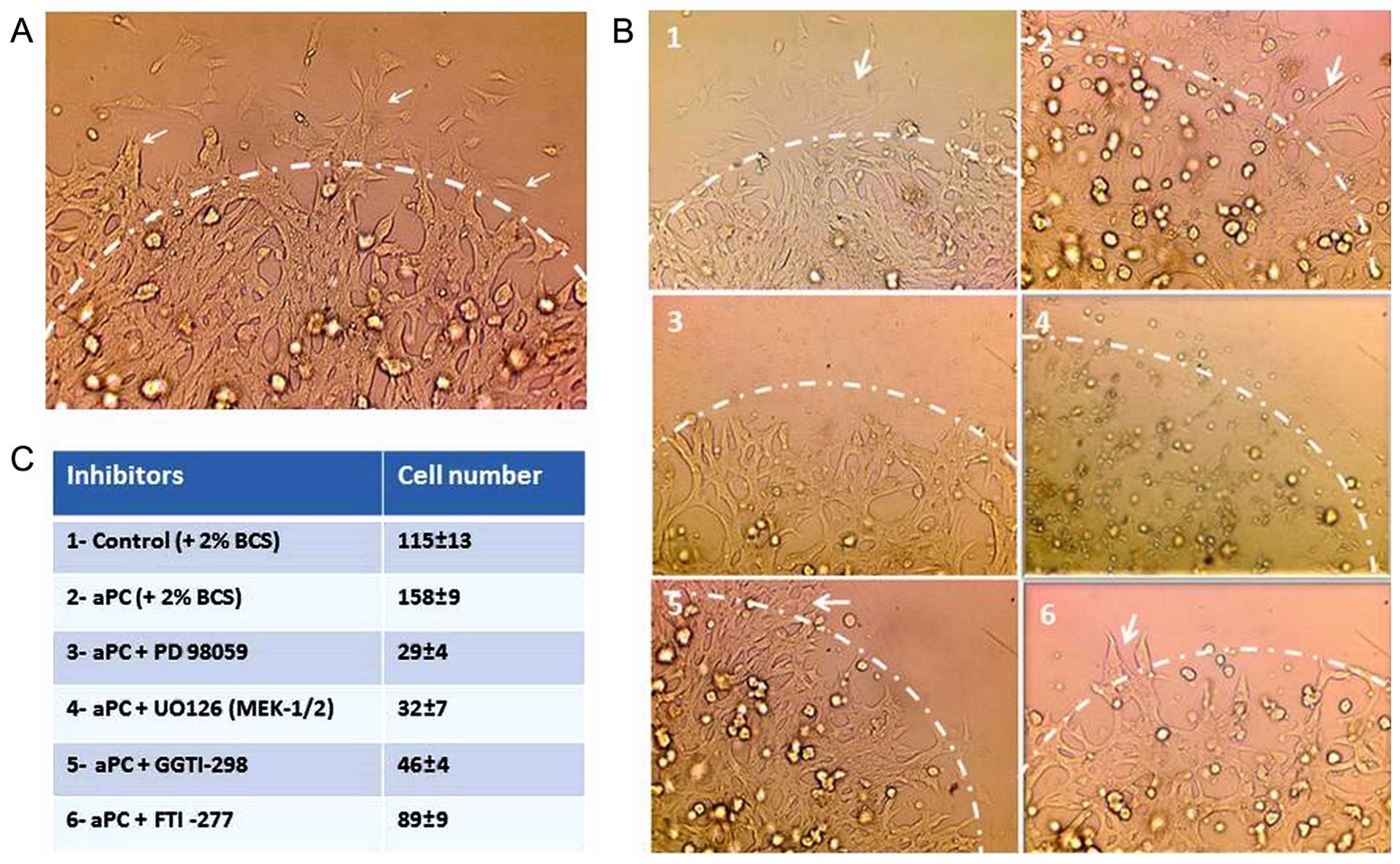
aPC upregulates cell migration via
MEK-ERK and Rho-GTPase pathways in OVCAR-3 cells
The effect of inhibitors of several signaling
pathways on cell migration was tested by droplet test (Fig. 4A and B). As presented in Fig. 4A, the cells were incorporated in the
droplet. The cell migration from the droplet started after 2–3 h
towards the outer periphery of the droplets (arrows). The outer of
the periphery is indicated for clarity by a discontinuous line
drawing. Fig. 4B indicates the
control OVCAR-3 cells (Fig. 4B1)
and the OVCAR-3 cells treated with aPC (Fig. 4B2). The cancer cell migration was
inhibited by PD 98059 an ERK inhibitor (Fig. 4B3), UO126 an MEK-1/2 kinase
inhibitor (Fig. 4B4) and GGTI-298
an inhibitor of Rho GTPases (Fig.
4B5), whereas the migration was unaffected by FTI-277 an
inhibitor of farnesyl transferase (Fig.
4B6) and rapamycin a raptor-mTor complex inhibitor (data not
shown). The number of cells migrating outside the droplets, in five
experiments, is presented in Fig.
4C.
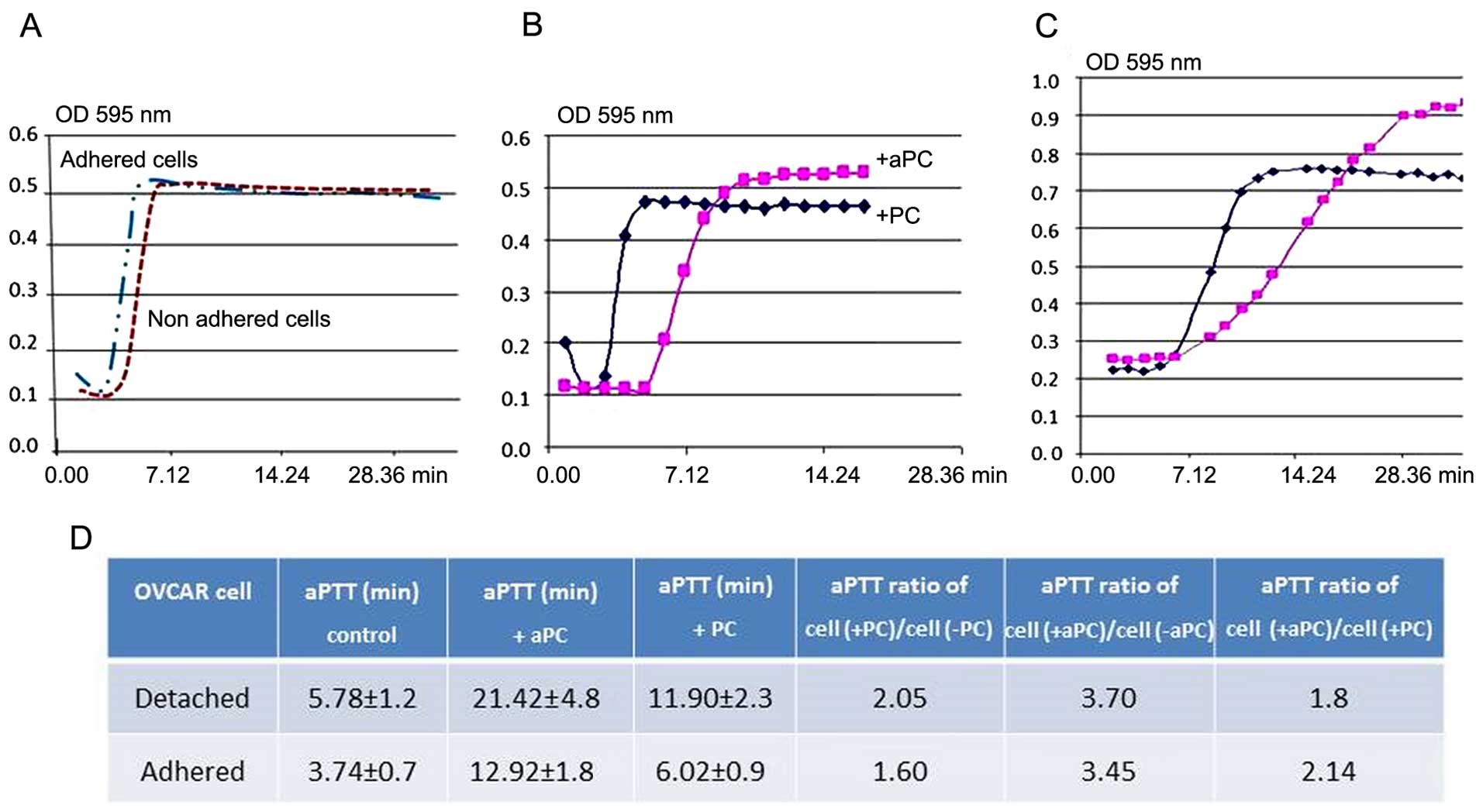
Anticoagulant activity induced by OVCAR-3
cells
The anticoagulant property of OVCAR-3 cells was
assessed by measuring the prolongation of aPTT of normal plasma
induced by aPC. As shown in Fig.
5A, the effect of OVCAR-3 cells, detached and incubated either
with PC or APC, in fibrin polymerization curve of normal plasma is
shown. The same experiment was also performed using OVCAR-3 cells
in adherent conditions. When both detached (Fig. 5B) and adherent (Fig. 5C) cells, were incubated with aPC,
aPTT was prolonged as compared to cells incubated with protein C.
As documented in Fig. 5D, PC added
to the adherent or detached ovarian cancer cells induced a 2-fold
increase in cephalin clotting time, whereas aPC under the same
conditions induced a 3.7-fold increase (5.78–11.90 min for PC and
21.42 min for aPC). These results indicate the anti-coagulant
action of EPCR-aPC on the OVCAR-3 cells.
Discussion
The peritoneal cavity is a space between the
visceral peritoneum and the parietal peritoneum. Ten percent of all
cases of ascites are of cancer origin (31). It is an exudate commonly observed at
the late stage of cancer. Ascites may originate in the peritoneum
(carcinomatosis) or may come from cancer that has spread from
another site of the body. In ovarian carcinomatosis, the cancer
cells adhere to mesenteric cells and form micronodules on the
peritoneum (32).
Peritoneal fluids contain, besides cells, several
proteolytic enzymes including metalloproteases and serine proteases
(33). These fibrinolytic enzymes
provoke increased extracellular matrix degradation and facilitate
tumor cell invasion and metastasis (34) and as a consequence detach malignant
cells which leave a maternal nodule for forming a secondary nodule
on the peritoneal surface. Hence, considerable diminution of
clotting of the peritoneal fluid plays a major role in the
pathology and poor prognosis of ovarian carcinomatosis. We
previously demonstrated that EPCR on endothelial cells has a
physiological anticoagulant activity as ascertained in vitro
by aPTT test (29). In contrast
sEPCR, by its ability to trap aPC from plasma, can be considered as
a cancer-associated hypercoagulability factor (26).
Cell migration was found to be inhibited when a
neutralizing antibody against the EPCR antibody was added to the
culture medium. In addition, we also showed that cell migration,
induced by the binding of aPC to EPCR, was blocked by anti-ERK,
MEK-1/2, and Rho-GTPase inhibitors when they were added while
performing the droplet test. Our results indicate that the
ERK-MEK-1/2 and Rho-GTPase signaling pathways significantly
participate in the aPC/EPCR-PAR-1 induced cell migration. We found
that the droplet test was a useful and informative model for
studying cell migration. In addition, in another set of
experiments, we also found that aPC-EPCR interaction increased
cancer cell adhesion on the bottom of gelatin-coated culture flasks
(data not shown). Here, we showed that the interaction of aPC-EPCR
in ovarian cancer cells resulted in accelerated cell migration as
evaluated by the kinetics of the wound closure.
When cells were synchronized and arrested in the G1
phase, their incubation with protein C or aPC induced cell cycle
activation and passage from G1 to S or G2 phases after 18 h. These
results on the activation of the cell cycle are in good concordance
with our previous observation showing that aPC induces OVCAR cell
proliferation (6). In a similar
approach, but using human keratinocytes, Xue et al (34) showed that aPC stimulated the
proliferation, migration and wound closure (35) again confirming that protein C
induces enhanced cell migration.
The protein C system participates in the degradation
of factors Va and VIIIa (9) thereby
inhibiting fibrin formation. It also induces inhibition of
plasminogen activator inhibitor-1 (PAI-1) (36). In order to estimate the ability of
EPCR to bind aPC on the surface of living endothelial cells in
vitro (29) we used a method
that we had previously developed, based on prolongation of cephalin
clotting time of plasma when aPC bound cells are added. This
aPTT-based method was optimized to assess EPCR presence and
functionality on the OVCAR-3 cell membrane. Our results showed that
aPC bound on living ovarian cancer cells induced a prolongation of
plasma clotting time suggesting that ovarian cancer cells use
physiological anticoagulants such as aPC for their homeostasis.
EPCR exists as a membrane-bound form as well as a
free sEPCR form. In fact, sEPCR can regulate the quantity of
circulating aPC (20). Curiously,
the sEPCR level in the peritoneal fluid of 85% patients was less
than that in the plasma of healthy individuals. Only 3 patients
(15%) had elevated levels of sEPCR (247, 250 and 154 ng/ml) which
was below the level observed in plasma of patients with ovarian
cancer (25). This indicates that
sEPCR availability for trapping aPC is considerably reduced.
Therefore, in ascitic fluids from ovarian cancer, free protein C
binds to membrane EPCR of ovarian cancer cells, inducing cell
migration ensuring the unclottability of peritoneal fluid by
inhibition of the fibrin formation pathway.
Evaluation of D-dimer and SF in the peritoneal
fluids of the ovarian cancer patients indicated that as soon as
fibrin was formed, it was degraded. Moreover, the presence of
EPCR-containing cancer cells in the peritoneal fluid limited the
formation of fibrin on the cell surface as deduced from our
observation indicating a marked increase in the cephalin clotting
time of plasma.
In the peritoneal cavity, under other circumstances,
aPC/EPCR interaction and cell activation can occur independent of
the presence of cancer cells. There are a number of reports
indicating that aPC/EPCR interaction via PAR-1 activation induces
anti-inflammatory activity and anti-apoptotic activity (37,38).
Peritoneal carcinomatosis is an inflammatory process and involves
numerous non-tumor cells such as inflammatory cells. Inflammatory
monocytes and neutrophils express EPCR on their membranes (17). The interaction of aPC/EPCR can
downregulate the pro-coagulant activity of these cells. It can also
induce cancer cytoprotection and enhance the malignant phenotype of
cancer cells. The secretion of hyaloronan by mesenteric cells
contributes to lubrication of the luminal peritoneal cavity.
Whether EPCR is present or not on these cells has not been reported
to date.
In conclusion, in ovarian carcinomatosis, aPC-EPCR
interaction renders cancer cells highly aggressive, and as a result
of inhibition of fibrin formation, the development of secondary
nodules is facilitated. We are at present engaged in studies to
further elucidate the role of EPCR in cancer homeostasis.
References
|
1
|
Matte I, Lane D, Laplante C, Rancourt C
and Piché A: Profiling of cytokines in human epithelial ovarian
cancer ascites. Am J Cancer Res. 2:566–580. 2012.PubMed/NCBI
|
|
2
|
Desjardins M, Xie J, Gurler H, Muralidhar
GG, Sacks JD, Burdette JE and Barbolina MV: Versican regulates
metastasis of epithelial ovarian carcinoma cells and spheroids. J
Ovarian Res. 7:702014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goncharenko-Khaider N, Matte I, Lane D,
Rancourt C and Piché A: Ovarian cancer ascites increase Mcl-1
expression in tumor cells through ERK1/2-Elk-1 signaling to
attenuate TRAIL-induced apoptosis. Mol Cancer. 11:842012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Puiffe ML, Le Page C, Filali-Mouhim A,
Zietarska M, Ouellet V, Tonin PN, Chevrette M, Provencher DM and
Mes-Masson AM: Characterization of ovarian cancer ascites on cell
invasion, proliferation, spheroid formation, and gene expression in
an in vitro model of epithelial ovarian cancer. Neoplasia.
9:820–829. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang L, Madigan MC, Chen H, Liu F,
Patterson KI, Beretov J, O'Brien PM and Li Y: Expression of
urokinase plasminogen activator and its receptor in advanced
epithelial ovarian cancer patients. Gynecol Oncol. 114:265–272.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fukudome K, Ye X, Tsuneyoshi N, Tokunaga
O, Sugawara K, Mizokami H and Kimoto M: Activation mechanism of
anticoagulant protein C in large blood vessels involving the
endothelial cell protein C receptor. J Exp Med. 187:1029–1035.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dahlbäck B and Villoutreix BO: Molecular
recognition in the protein C anticoagulant pathway. J Thromb
Haemost. 1:1525–1534. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fukudome K and Esmon CT: Identification,
cloning, and regulation of a novel endothelial cell protein
C/activated protein C receptor. J Biol Chem. 269:26486–26491.
1994.PubMed/NCBI
|
|
9
|
Taylor FB Jr, Peer GT, Lockhart MS,
Ferrell G and Esmon CT: Endothelial cell protein C receptor plays
an important role in protein C activation in vivo. Blood.
97:1685–1688. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li W, Zheng X, Gu J, Hunter J, Ferrell GL,
Lupu F, Esmon NL and Esmon CT: Overexpressing endothelial cell
protein C receptor alters the hemostatic balance and protects mice
from endotoxin. J Thromb Haemost. 3:1351–1359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng X, Li W, Gu JM, Qu D, Ferrell GL,
Esmon NL and Esmon CT: Effects of membrane and soluble EPCR on the
hemostatic balance and endotoxemia in mice. Blood. 109:1003–1009.
2007. View Article : Google Scholar
|
|
12
|
Griffin JH, Zlokovic BV and Mosnier LO:
Protein C anticoagulant and cytoprotective pathways. Int J Hematol.
95:333–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van Sluis GL, Niers TM, Esmon CT,
Tigchelaar W, Richel DJ, Buller HR, Van Noorden CJ and Spek CA:
Endogenous activated protein C limits cancer cell extravasation
through sphin-gosine-1-phosphate receptor 1-mediated vascular
endothelial barrier enhancement. Blood. 114:1968–1973. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Azzazene D, Al Thawadi H, Al Farsi H,
Besbes S, Geyl C, Mirshahi S, Pardo J, Faussat AM, Jeannette S,
Therwath A, Pujade-Lauraine E and Mirshahi M: Plasma endothelial
protein C receptor influences innate immune response in ovarian
cancer by decreasing the population of natural killer and TH17
helper cells. Int J Oncol. 43:1011–1018. 2013.PubMed/NCBI
|
|
15
|
Van Sluis GL, Brüggemann LW, Esmon CT,
Kamphuisen PW, Richel DJ, Büller HR, Van Noorden CJ and Spek CA:
Endogenous activated protein C is essential for immune-mediated
cancer cell elimination from the circulation. Cancer Lett.
306:106–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laszik Z, Mitro A, Taylor FB Jr, Ferrell G
and Esmon CT: Human protein C receptor is present primarily on
endothelium of large blood vessels: implications for the control of
the protein C pathway. Circulation. 96:3633–2640. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Balazs AB, Fabian AJ, Esmon CT and
Mulligan RC: Endothelial protein C receptor (CD201) explicitly
identifies hematopoietic stem cells in murine bone marrow. Blood.
107:2317–2321. 2006. View Article : Google Scholar
|
|
18
|
Schaffner F, Yokota N, Carneiro-Lobo T,
Kitano M, Schaffer M, Anderson GM, Mueller BM, Esmon CT and Ruf W:
Endothelial protein C receptor function in murine and human breast
cancer development. PLoS One. 8:e610712013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fukudome K, Kurosawa S, Stearns-Kurosawa
DJ, He X, Rezaie AR and Esmon CT: The endothelial cell protein C
receptor. Cell surface expression and direct ligand binding by the
soluble receptor. J Biol Chem. 271:17491–17498. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saposnik B, Lesteven E, Lokajczyk A, Esmon
CT, Aiach M and Gandrille S: Alternative mRNA is favored by the A3
haplotype of the EPCR gene PROCRand generates a novel soluble form
of EPCR in plasma. Blood. 111:3442–3451. 2008. View Article : Google Scholar :
|
|
21
|
Nayak RC, Sen P, Ghosh S, Gopalakrishnan
R, Esmon CT, Pendurthi UR and Rao LV: Endothelial cell protein C
receptor cellular localization and trafficking: potential
functional implications. Blood. 114:1974–1986. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schuepbach RA and Riewald M: Coagulation
factor Xa cleaves protease-activated receptor-1 and mediates
signaling dependent on binding to the endothelial protein C
receptor. J Thromb Haemost. 8:379–388. 2010. View Article : Google Scholar
|
|
23
|
Joyce DE, Gelbert L, Ciaccia A, DeHoff B
and Grinnell BW: Gene expression profile of antithrombotic protein
C defines new mechanisms modulating inflammation and apoptosis. J
Biol Chem. 276:11199–11203. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Riewald M, Petrovan RJ, Donner A, Mueller
BM and Ruf W: Activation of endothelial cell protease activated
receptor 1 by the protein C pathway. Science. 296:1880–1882. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ducros E, Mirshahi S, Azzazene D,
Camilleri-Broët S, Mery E, Al Farsi H, Althawadi H, Besbess S,
Chidiac J, Pujade-Lauraine E, et al: Endothelial protein C receptor
expressed by ovarian cancer cells as a possible biomarker of cancer
onset. Int J Oncol. 41:433–440. 2012.PubMed/NCBI
|
|
26
|
Ducros E, Mirshahi SS, Faussat AM,
Mirshahi P, Dimicoli S, Tang R, Pardo J, Ibrahim J, Marie JP,
Therwath A, et al: Soluble endothelial protein C receptor (sEPCR)
is likely a biomarker of cancer-associated hypercoagulability in
human hematologic malignancies. Cancer Med. 1:261–267. 2012.
View Article : Google Scholar
|
|
27
|
Berthaut A, Mirshahi P, Benabbou N,
Azzazene D, Bordu C, Therwath A, Legeais JM and Mirshahi M:
Vascular endothelial growth factor receptor-1 (VEGFR-1) expression
in human corneal fibroblast decreased with age. Mol Vis.
15:1997–2007. 2009.PubMed/NCBI
|
|
28
|
Queille S, Drougard C, Sarasin A and
Daya-Grosjean L: Effects of XPD mutations on ultraviolet-induced
apoptosis in relation to skin cancer-proneness in repair-deficient
syndromes. J Invest Dermatol. 117:1162–1170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ducros E, Berthaut A, Mirshahi SS, Faussat
AM, Soria J, Agarwal MK and Mirshahi M: Aldosterone modifies
hemostasis via upregulation of the protein-C receptor in human
vascular endothelium. Biochem Biophys Res Commun. 373:192–196.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mirshahi S, Soria C, Kouchakji B, Kierzek
G, Borg JY, Varin R, Chidiac J, Drouet L, Mirshahi M and Soria J:
New combinational assay using soluble fibrin and D-dimer
determinations: a promising strategy for identifying patients with
suspected venous thromboembolism. PLoS One. 9:e923792014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ayantunde AA and Parsons SL: Pattern and
prognostic factors in patients with malignant ascites: a
retrospective study. Ann Oncol. 18:945–949. 2006. View Article : Google Scholar
|
|
32
|
Smolle E, Taucher V and Haybaeck J:
Malignant ascites in ovarian cancer and the role of targeted
therapeutics. Anticancer Res. 34:1553–1561. 2014.PubMed/NCBI
|
|
33
|
Casslén B, Bossmar T, Lecander I and
Astedt B: Plasminogen activators and plasminogen activator
inhibitors in blood and tumour fluids of patients with ovarian
cancer. Eur J Cancer. 30A:1302–1309. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Graves LE, Ariztia EV, Navari JR, Matzel
HJ, Stack MS and Fishman DA: Proinvasive properties of ovarian
cancer ascites-derived membrane vesicles. Cancer Res. 64:7045–7049.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xue M, Thompson P, Kelso I and Jackson C:
Activated protein C stimulates proliferation, migration and wound
closure, inhibits apoptosis and upregulates MMP-2 activity in
cultured human keratinocytes. Exp Cell Res. 299:119–127. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Van Hinsbergh VW, Bertina RM, Van
wijngaarden A, Van Tilburg NH, Emeis JJ and Haverkate F: Activated
protein C decreases plasminogen activator-inhibitor activity in
endothelial cell-conditioned medium. Blood. 65:444–451.
1985.PubMed/NCBI
|
|
37
|
Mosnier LO, Zlokovic BV and Griffin JH:
The cytoprotective protein C pathway. Blood. 109:3161–3172. 2007.
View Article : Google Scholar
|
|
38
|
Mohan Rao LV, Esmon CT and Pendurthi UR:
Endothelial cell protein C receptor: a multiliganded and
multifunctional receptor. Blood. 124:1553–1562. 2014. View Article : Google Scholar : PubMed/NCBI
|