Introduction
Hepatocellular carcinoma (HCC) is one of the most
common cancers, and accounts for 600,000 deaths annually (1). Recently, the discovery of new targets
in the molecular pathways in HCC has achieved significant results
(2).
The growth of tumor cells is regulated by various
proteins called cyclin-dependent protein kinases (Cdks) that
consist of a family of heterodimeric serine/threonine kinases
(3). The cell cycle consists of DNA
synthesis (S) and mitotic (M) phases separated by gap phases in the
order G1-S-G2-M (4). DNA damage at
the G2/M checkpoint prevents cells from entering mitosis (M-phase).
The activity of the cyclin B-Cdc2 complex is pivotal in regulating
G2-phase transition (5).
The Forkhead box protein M1 (FoxM1) is a member of
the forkhead transcription factor family, which has been shown to
have an important role in controlling the cell cycle. In
particular, FoxM1 controls mitotic entry through the periodic
upregulation of a group of genes that are maximally expressed as
cells progress through late G2 and into M phase (6,7). The
FoxM1 transcript undergoes alternative splicing to produce three
alternative isoforms (8).
There are a number of target genes of FoxM1
implicated in mitosis, such as cyclin B, Aurora B kinase, CENPA and
PLK1 (9,10). Cyclin B is an important gene in G2/M
transition. Aurora B kinase is a protein that functions in the
attachment of the mitotic spindle to the centromere. Centromere
protein A (CENPA) is a protein that encodes centromere protein.
Polo-like kinase 1 (PLK1) belongs to a family of serine-threonine
kinases and plays a critical role in mitotic progression. Human
Cdc25 genes consist of three isoforms: Cdc25A, B and C (11,12).
In mammalian cells, all three Cdc25 isoforms are associated with
cell cycle regulation because of their functions in the
dephosphorylation of Cdk1 and Cdk2. Cdc25A regulates both G1/S and
S/G2 transition (13), while Cdc25B
and Cdc25C regulate late G2 and mitosis, respectively (14–16).
Moreover, Cdc25C is an upstream factor of Cdc2 and can be
phosphorylated by PLK1 (17).
Deubiquitinating enzymes (DUBs) are proteases that
regulate ubiquitin or ubiquitin-like gene products. The human
genome encodes nearly 100 DUBs with specificity for ubiquitin in 5
families (18). Ubiquitin specific
protease 39 (USP39) is one of the DUBs without ubiquitin protease
activity, as it lacks three important residues for protease
activity in DUBs (19). USP39
encodes a conserved protein of the U4/U6.U5 tri-snRNP, with 65%
overall homology to the yeast Sad1p splice factor, and it has been
implicated in the assembly of mature spliceosome. In vitro,
USP39 appears to be essential for pre-mRNA splicing, but not for
the stability of the spliceosome complex once it is formed
(20). USP39 is also essential in
the spindle assembly checkpoint and regulates the Aurora B mRNA
levels in U2OS cells (19). In
addition, zebrafish USP39 mutation can induce G1/S arrest by rb1
splicing defect, and the e2f4 is a target of USP39. USP39 mutation
contributes to adenohypophyseal sensitivity to rb1 and e2f4 that
causes pituitary tumorigenesis (20). USP39 may act as an oncogenic factor
in breast cancer, and downregulation of USP39 was found to induce
the apoptosis of MCF-7 breast cancer cells (21). Thus, USP39 may be a potential
molecular target for cancer therapy. However, the roles of USP39 in
HCC have been rarely studied. In the present study, we aimed to
investigate the potential roles of USP39 in hepatocellular cancer.
We found that the expression levels of USP39 were higher in tumor
tissues than in adjacent normal tissues, and the expression of
USP39 was strongly associated with the pathological grade. Thus, we
hypothesized that USP39 may act as a new target in liver cancer. To
verify our hypothesis, we stably ablated USP39 expression by shRNA
in hepatocellular carcinoma cell line HepG2 and investigated the
changes in cell proliferation in vitro and tumor growth
in vivo. We demonstrated that inhibition of USP39 suppressed
the tumorigenesis of HCC via the inhibition of FoxM1 splicing.
Materials and methods
Tissue array and evaluation of
immunostaining
HCC tissue arrays were purchased from the National
Engineering Center for Biochips (Shanghai, China). The expression
of USP39 in the tissues was evaluated by immunohistochemical
staining using the USP39-specific antibody. The staining was scored
according to the staining intensity and the percentage of positive
cells, and the final staining scores were calculated as the score
of the staining intensity multiplied by the score of the percentage
of positive cells.
Cell growth assay
Cell growth was measured by multiparametric
high-content screening (HCS). HepG2 cells were infected with either
the NC lentivirus or the USP39 siRNA lentivirus and were seeded at
2,000 cells/well in 96-well plates, and then incubated at 37°C with
5% CO2 for 5 days. The plates were processed with
ArrayScan™ HCS software (Cellomics Inc.) and kept at 4°C for up to
24 h before each day’s analysis. The system can identify stained
cells and report the intensity and distribution of fluorescence in
each individual cell. In each well, at least 800 cells were
analyzed. Images and data were stored in a Microsoft SQL database
for easy retrieval.
Cell culture and MTT assay
Human HCC cell lines, HepG2, SMMC-7721, BEL-7402 and
Huh-7, were purchased from the Shanghai Institutes for Biological
Sciences (China). All cell lines were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), 100 U/ml of
penicillin and 100 µg/ml of streptomycin (complete medium)
and maintained at 37°C with 5% CO2.
HCC cells (1×105 in 0.2 ml/well) were
seeded in a 96-well plate in three replicates, and cultured in
complete medium at 37°C for 1–8 days. One hundred microliters of
MTT solution (5 mg/ml) was added into each well and incubated at
37°C for 4 h. The supernatant was then removed and 150 µl of
DMSO was added per well. The plate was oscillated for 30 min at
room temperature. Absorbance at 490 nm was measured and the values
were determined after background subtraction. The experiment was
repeated at least three times.
Vector construction and transfection
The siRNA targeting the USP39 sequences (KD,
ACCAAGTTGCCTCCATATCTA and KD#, CCAGACAACTATGAGATCATCGATT) and the
non-silencing sequence (TTCTCCGAACGTGTCACGT) were transformed into
short hairpin RNA (shRNA) (stem-loop-stem structure) and cloned
into the pGCSIL-GFP lentiviral vector (GeneChem Co., Ltd.,
Shanghai, China) after AgeI/EcoRI digestion. The
recombinant plasmid and two virus packaging plasmids (GeneChem)
were transfected into the human 293T cell line using Lipofectamine
3000 (Invitrogen Life Technologies, Grand Island, NY, USA)
following the manufacturer’s instructions. After 3 days of
incubation, the lentivirus from the culture medium was collected.
For stable infection, the HCC cell lines were cultured in 6-well
plates, and infected with the USP39 shRNA-expressing lentivirus
(USP39-shRNA) or the non-silencing shRNA-expressing lentivirus
(control) with a multiplicity of infection (MOI) of 10. Five days
after infection, the cells were observed under fluorescence
microscopy (DMI4000B; Leica Microsystems, Germany). For USP39
overexpression, full length of USP39 cDNA was amplified by PCR and
cloned into pEGFP-N2 (BD, Franklin Lakes, NJ, USA). The recombinant
plasmids were transfected into cells using Lipofectamine 3000
(Invitrogen) following the manufacturer’s instructions.
Tumor xenografts
BALB/c nude mice aged 4–6 weeks were obtained from
the Laboratory Animal Centre of the Affiliated Drum Tower Hospital
of Nanjing University Medical School and maintained in a standard
pathogen-free condition. All experiments were approved by the
Institutional Animal Care and Use Committee of the Affiliated Drum
Tower Hospital of Nanjing University Medical School. Tumor cells
(2×106) in 0.2 ml serum-free RPMI-1640 medium were
subcutaneously injected into the flank of each mouse. The right
flank was injected with tumor cells containing USP39 shRNA and
control tumor cells were injected into the left flank of each
mouse. Each group consisted of 10 mice. Tumor growth was monitored
by measuring the length (L) and width (W) of each tumor using a
caliper, and tumor size was calculated by the formula L ×
W2 × (π/6). At the end of the experiment, the tumors
were isolated from the mice and fixed in 4% paraformaldehyde and
paraffin-embedded.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from the HCC cell lines
using TRIzol (Invitrogen) following the manufacturer’s
instructions. qRT-PCR was performed as reported previously
(22). Briefly, 1 µg of
total RNA was transcribed using random primers and Primescript
reverse transcriptase (Takara, Dalian, China). Quantitative PCR
reaction for the indicated genes was carried out using SYBR-Green
qPCR kit (Takara) on a fluorescent temperature cycler (Mx3000P
Real-Time PCR system; Stratagene, La Jolla, CA, USA). The following
primers were used to detect the expression of USP39 (forward,
5′-CCAGCGATGGCAAC TAC-3′ and reverse, 5′-ACCACAACGGAAACACG-3′); and
GAPDH (forward 5′-TGACTTCAACAGCGACACCCA-3′ and reverse,
5′-CACCCTGTTGCTGTAGCCAAA-3′).
PCR reaction was performed with the parameters of
denaturation: 95°C for 5 min, followed by 45 cycles of 95°C for 15
sec and 60°C for 1 min. Using GAPDH as an endogenous control,
relative gene expression was determined by the comparative Ct
method. The melting curve of a product is sequence-specific and can
be used to distinguish nonspecific from specific PCR products. Gene
expression was analyzed with the Stratagene analysis software. The
experiment was repeated at least three times.
Flow cytometric analysis
Cells transfected with the lentivirus were
harvested, washed twice with cold PBS, fixed with cold 70% ethanol
overnight, and resuspended with PBS. The suspension was filtrated
through a 400-mesh membrane. The cells were stained with propidium
iodide (PI) or Annexin V-APC (eBioscience, Inc.), and then analyzed
using a BD FACSCalibur flow cytometer (BD Biosciences, San Diego,
CA, USA). The experiment was repeated at least three times.
Western blotting
Total proteins were extracted from the tumor cell
lines in RIPA buffer containing fresh protease and phosphatase
inhibitors. The protein concentration was determined using the BCA
assay (Pierce, Rockford, IL, USA). Twenty micrograms of protein was
separated on 10% SDS-PAGE and transferred onto a PVDF membrane. The
membrane was blocked with 3% BSA in 10 mM Tris-HCl (pH 7.4)
containing 0.05% Tween-20 and incubated with a primary antibody at
4°C for 12 h. After washing with Tris-HCl buffer for 3 times, the
membrane was incubated with a corresponding peroxidase-conjugated
secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
and developed in SuperSignal West Pico Chemiluminescent Substrate
(Pierce). The protein was visualized by autoradiography and
quantified by densitometric analysis using a Versadoc Imaging
System Model 3000. The experiment was repeated at least three
times.
Statistical analyses
All statistical analyses were performed using SPSS
11.0 software (SPSS Inc., Chicago, IL, USA). Differences among
categorical variables were analyzed using one-way ANOVA/SNK test or
independent-sample Student’s t-test. The xenografted tumors were
analyzed with the paired t-test. The immunoreactive scores for
USP39 for tissue array were analyzed using non-parametric
Mann-Whitney U, Kruskal-Wallis H and Wilcoxon tests. P<0.05 was
considered statistically significant.
Results
Expression of USP39 in the clinical HCC
tissues
Using a USP39-specific antibody, we analyzed the
USP39 protein level and distribution in the HCC tissue array by
immunohistochemical staining. The array included 116 HCC patient
samples including cancer and corresponding adjacent normal tissues,
6 cancer tissue samples and 2 adjacent normal tissue samples. The
HCC patients included 104 males and 18 females, with a median age
of 52 years (range, 14–73 years). Only 75 patients in the array had
TNM staging. As shown in Table I
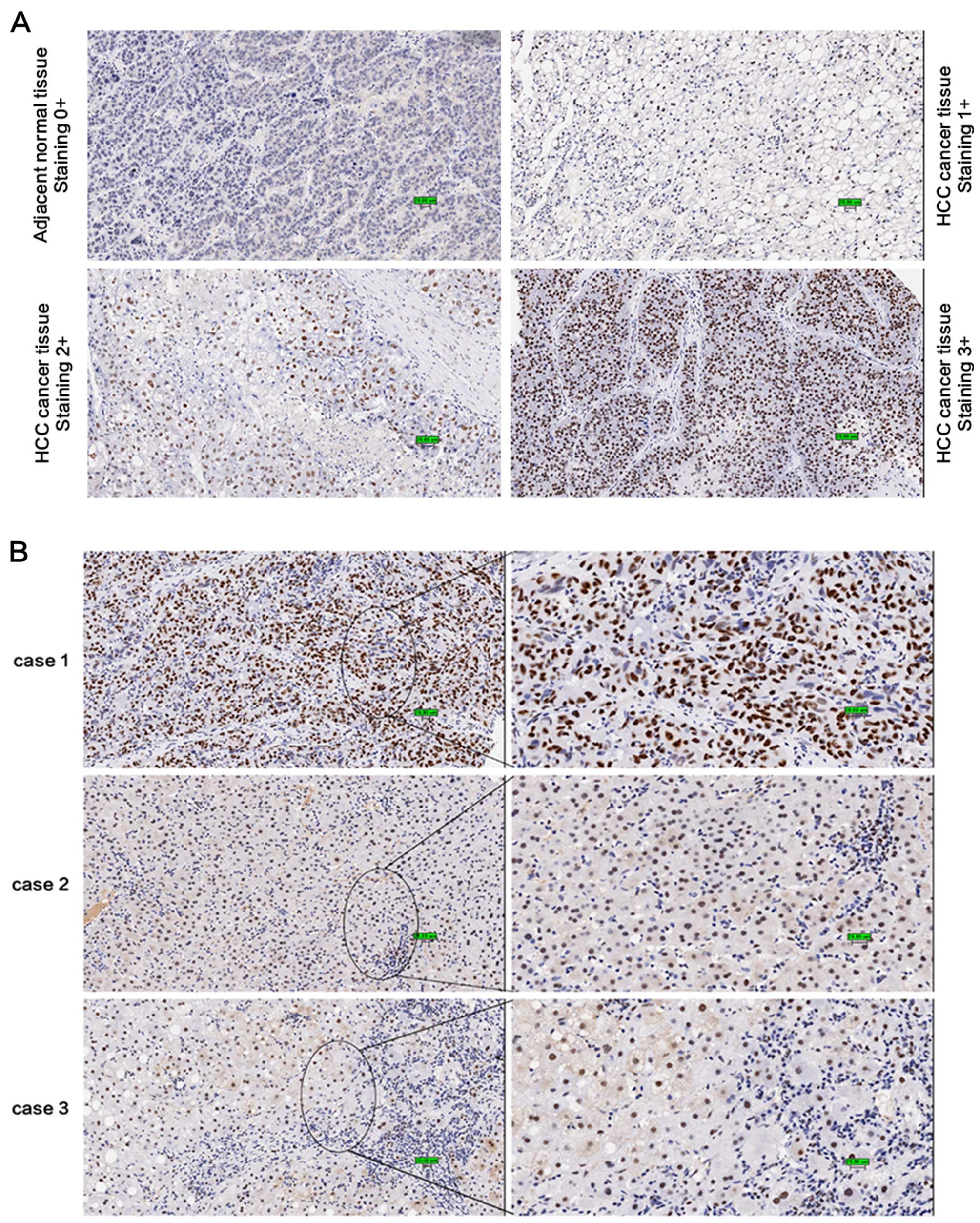
and Fig. 1A, USP39 was strongly
expressed in the HCC tumor tissues; 103 of the 122 cancer tissues
had a score of ≥2, while USP39 expression in the adjacent normal
tissues was markedly decreased compared with that in the cancer
tissues (Z=−7.499, P<0.001). As shown in Fig. 1B, USP39 was highly expressed in the
tumor cells but weakly expressed in the normal liver cells (case 1
and 2). Notably, the expression of USP39 was associated with the
pathological grade of HCC (Z=9.219, P<0.05; Table I); well-differentiated tumor cells
showed a high level of expression (case 1) while poorly
differentiated tumor cells exhibited a low level of expression
(case 3). Our results also indicated that the expression of USP39
was higher in older individuals (age ≥50) than that in younger
individuals (age <50). Yet, USP39 expression did not appear to
be associated with gender, tumor size and TNM stage. Thus, we can
draw a conclusion that the expression of USP39 may be associated
with the tumorigenesis of HCC (Fig.
1).
 | Table ICorrelation of the expression of
USP39 with clinicopathological features in the HCC tissue
array. |
Table I
Correlation of the expression of
USP39 with clinicopathological features in the HCC tissue
array.
|
Characteristics | N | USP39 expression
| Mean rank | Z | P-value |
|---|
| 0 | 1 | 2 | 3 | None |
|---|
| Gender |
| Male | 104 | 2 | 14 | 5 | 81 | 2 | 61.14 | −1.204 | 0.229 |
| Female | 18 | 1 | 2 | 3 | 11 | 1 | 53.18 | | |
| Age (years) |
| <50 | 49 | 2 | 9 | 6 | 31 | 1 | 52.65 | −2.614 | 0.009 |
| ≥50 | 73 | 1 | 7 | 2 | 61 | 2 | 64.97 | | |
| Tumor size
(cm) |
| <5 | 67 | 1 | 8 | 4 | 52 | 2 | 59.97 | −0.729 | 0.466 |
| ≥5 | 52 | 2 | 7 | 4 | 38 | 1 | 56.63 | | |
|
Differentiation |
| 1 | 6 | 0 | 3 | 0 | 3 | 0 | 42.50 | 9.219 | 0.027 |
| 2 | 75 | 3 | 11 | 7 | 53 | 1 | 56.66 | | |
| 3 | 40 | 0 | 2 | 1 | 35 | 2 | 68.92 | | |
| 4 | 1 | 0 | 0 | 0 | 1 | 0 | 73.5 | | |
| TNM stage |
| I | 23 | 0 | 2 | 0 | 20 | 1 | 38.64 | 1.703 | 0.636 |
| II | 26 | 1 | 3 | 1 | 21 | 0 | 34.90 | | |
| III | 22 | 0 | 2 | 1 | 18 | 1 | 36.93 | | |
| IV | 4 | 0 | 0 | 0 | 4 | 0 | 42.00 | | |
| Location |
| Tumor tissue | 122 | 3 | 16 | 8 | 92 | 3 | | −7.499 | 0.000 |
| Adjacent
tissue | 118 | 43 | 37 | 6 | 31 | 1 | | | |
Expression of USP39 in HCC cell
lines
We examined the level of USP39 expression in 5 HCC
cell lines including HepG2, SMMC-7721, BEL-7402, QGY-7701 and Huh7
using qRT-PCR. High expression of USP39 mRNA was observed in all 5
HCC cell lines (Fig. 2A).
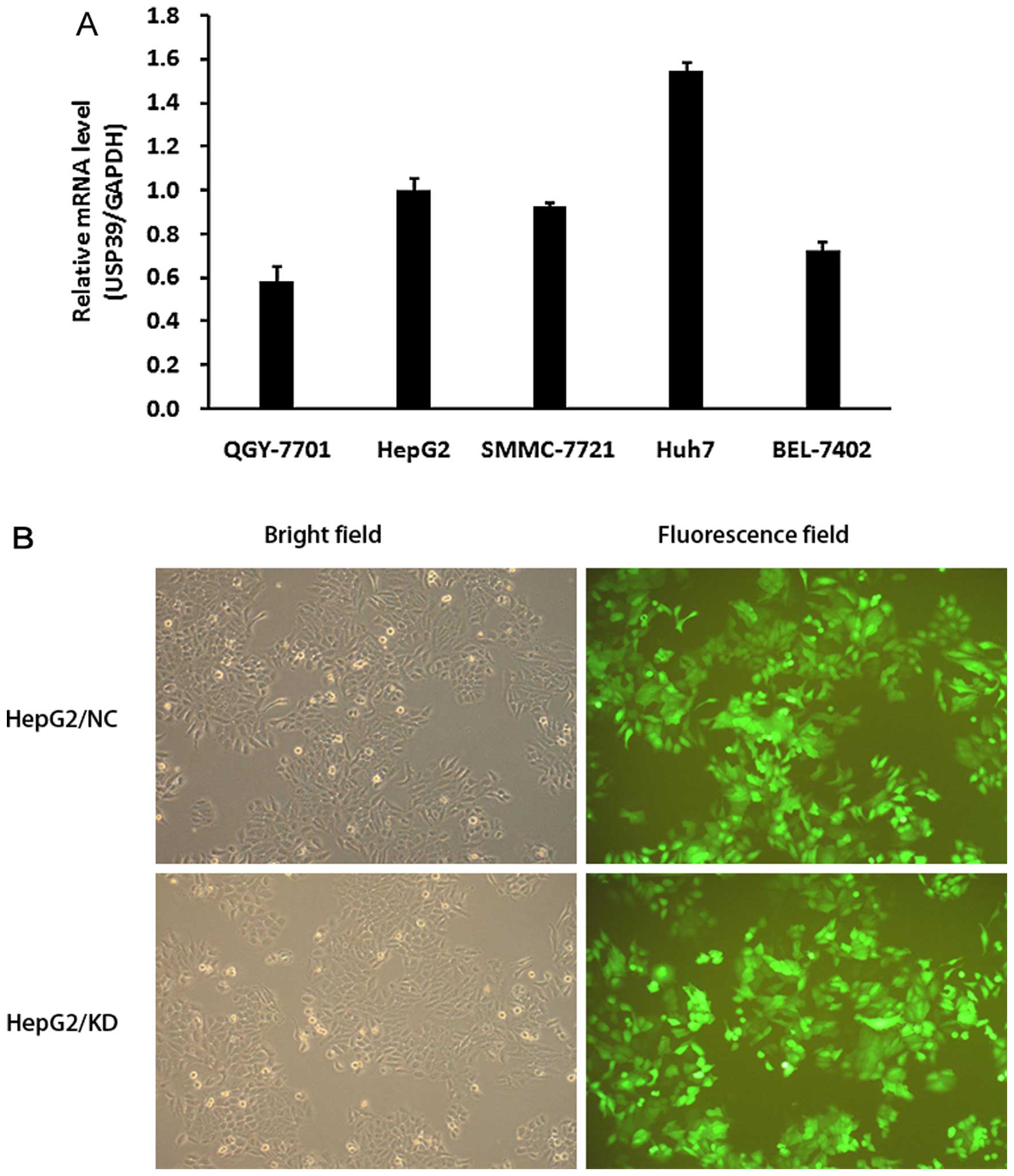 | Figure 2Detection of USP39 in HCC cell lines
and transfection of USP39-lentiviral knockdown. (A) The mRNA levels
of USP39 in HCC cell lines: QGY-7701, SMMC-7721, HepG2, Huh7 and
BEL-7402. USP39 expression was extremely high in Huh7, high in
HepG2, SMMC-7721 and BEL-7402, and moderate in QGY-7701. (B)
Lentiviral infection. Representative GFP expression in HepG2/NC
cells (upper panels) and HepG2/KD cells (lower panel). Left panels,
bright field; right panel, fluorescence field. Magnification, ×100.
HCC, hepatocellular carcinoma. |
Effects of USP39 on the proliferation and
colony formation of HCC cells
To verify the roles of USP39 in the tumorigenesis of
HCC in vitro, we inhibited USP39 expression in the HCC cell
lines by shRNA targeting USP39 (Fig.
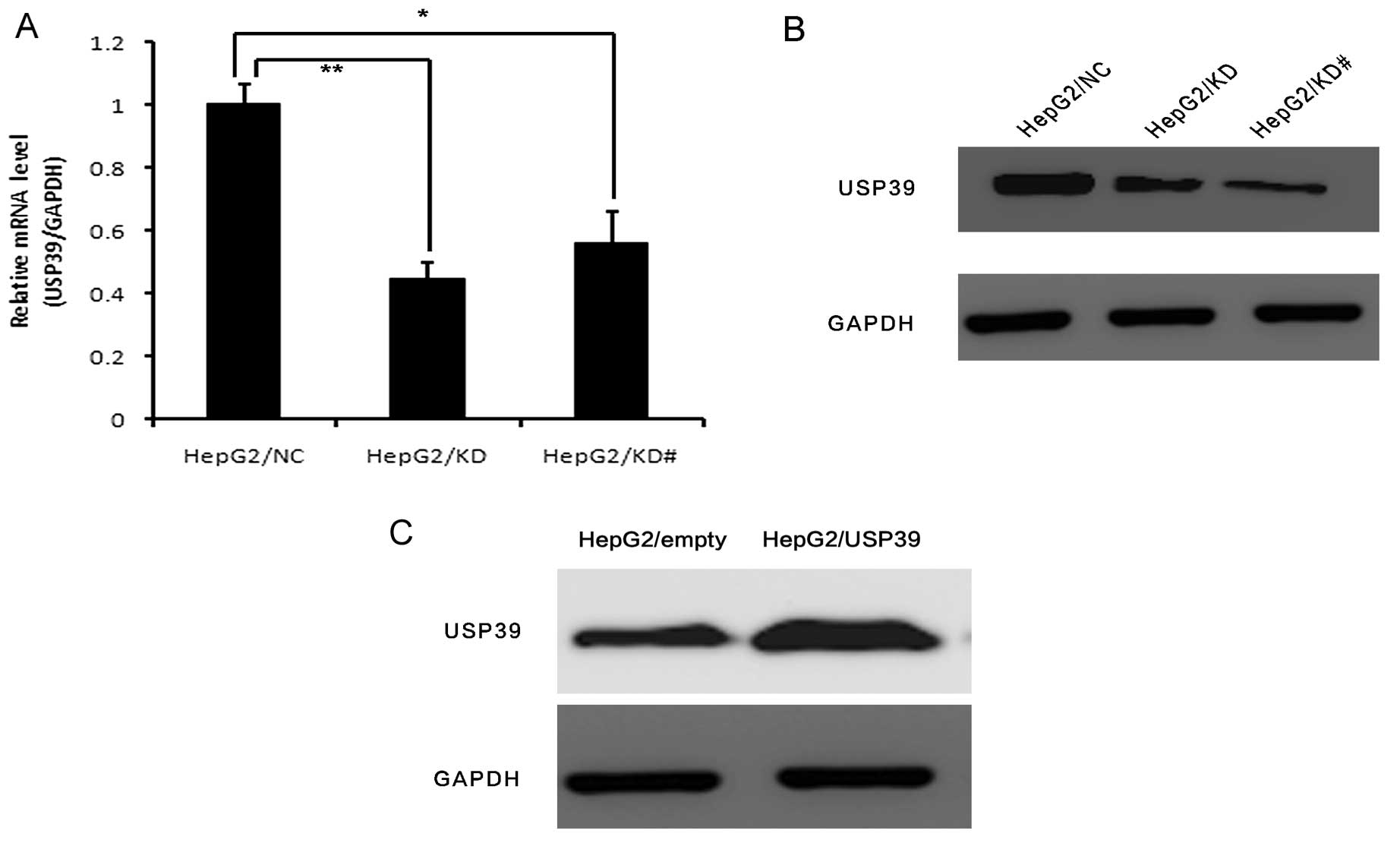
2B). USP39 expression was significantly decreased in the
HepG2/KD and HepG2/KD# cells compared to that in the HepG2/NC cells
at both the mRNA and protein levels (Fig. 3A and B). We also overexpressed USP39
in the HepG2 cells. USP39 expression was significantly increased in
the HepG2/USP39 cells compared to that in the HepG2/vector cells at
the protein level (Fig. 3C).
USP39 knockdown inhibits the growth of
HepG2 cells and USP upregulation induces the opposite effects
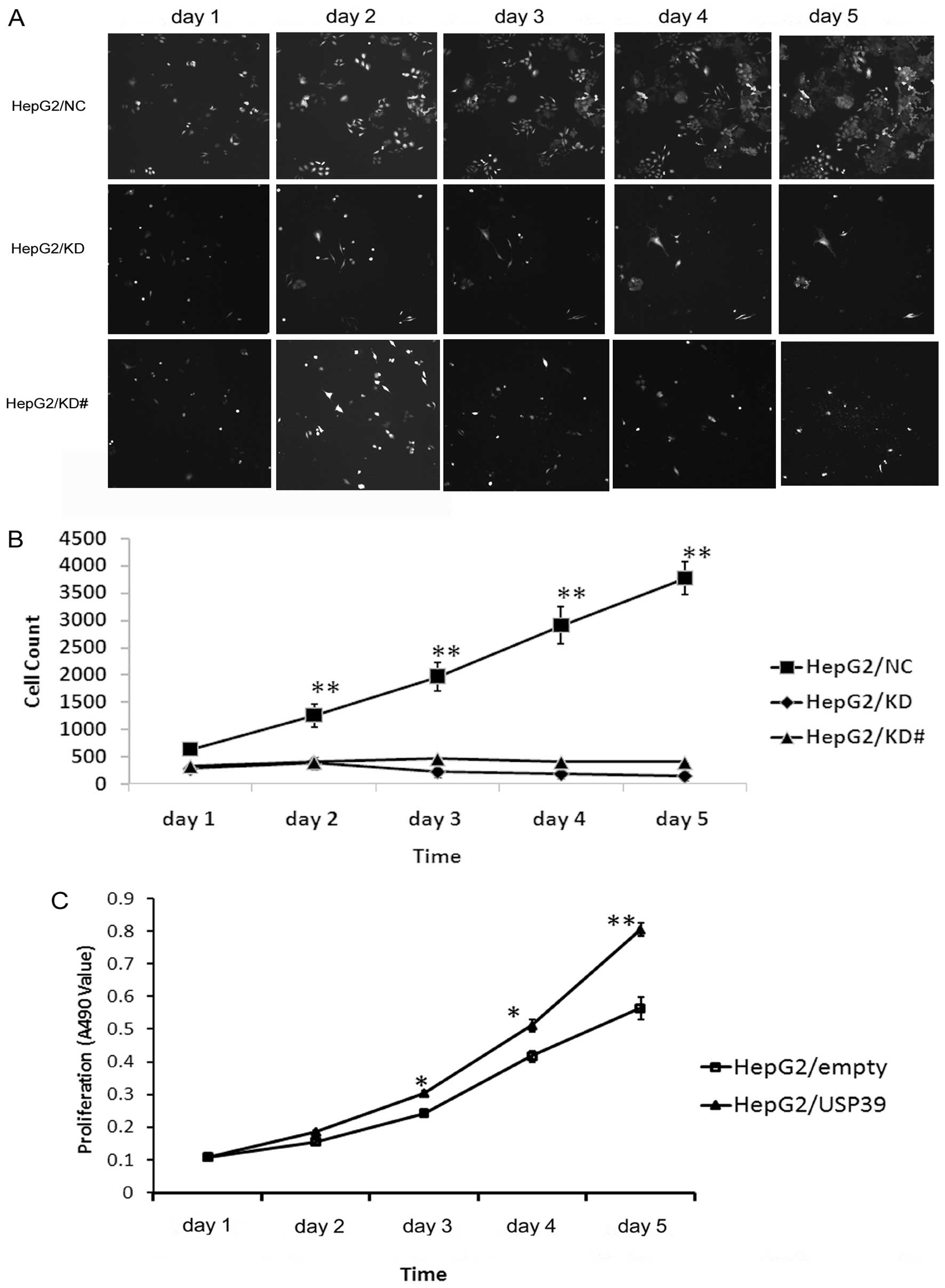
To explore the function of USP39 on cell growth,
HepG2 cells transfected with either the USP39-siRNA lentivirus or
the NC lentivirus were monitored by Cellomics assay. HepG2 cells
containing USP39-siRNA and NC were seeded in 96-well plates, and
cell growth was assayed every day for 5 days (Fig. 4A). The results showed that the cell
growth was inhibited after downregulation of USP39 (Fig. 4B). MTT assay indicated that the
growth of the HepG2 cells was significantly increased after USP39
was upregulated (Fig. 4C). Thus,
these results indicated that USP39 knockdown suppressed the growth
of HCC cells and USP39 overexpression had opposite effects.
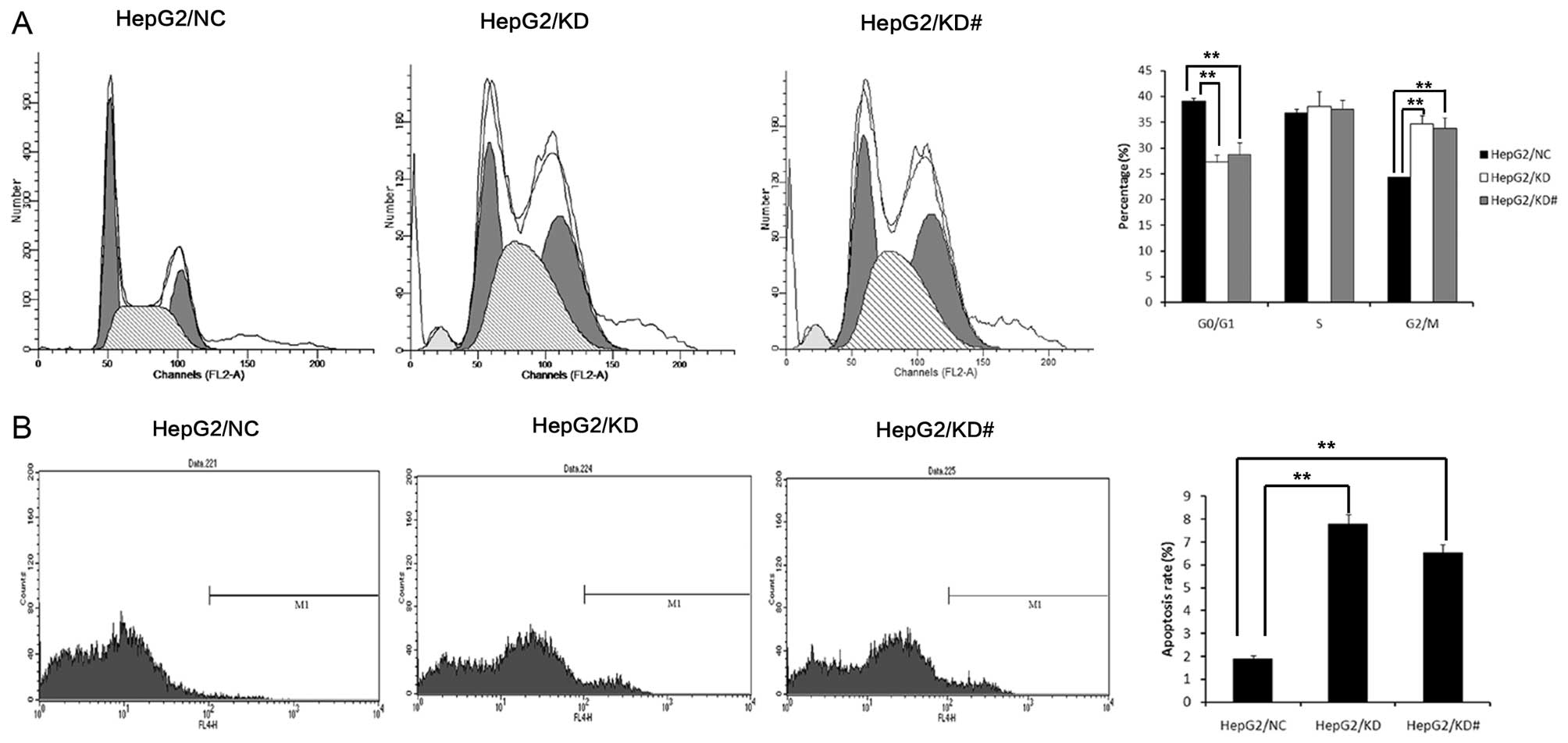
USP39 induces G2/M arrest
USP39 was initially reported to regulate the cell
cycle checkpoint (19–21). Moreover, we examined cell cycle
change by FCM. We found that USP39 knockdown in the HepG2 cells
showed a lower cell population in the G0/G1 stage and a higher cell
population in the G2/M stage (Fig.
5A). HepG2/KD cells showed an increased apoptosis rate compared
with the HepG2/NC cells (Fig.
5B).
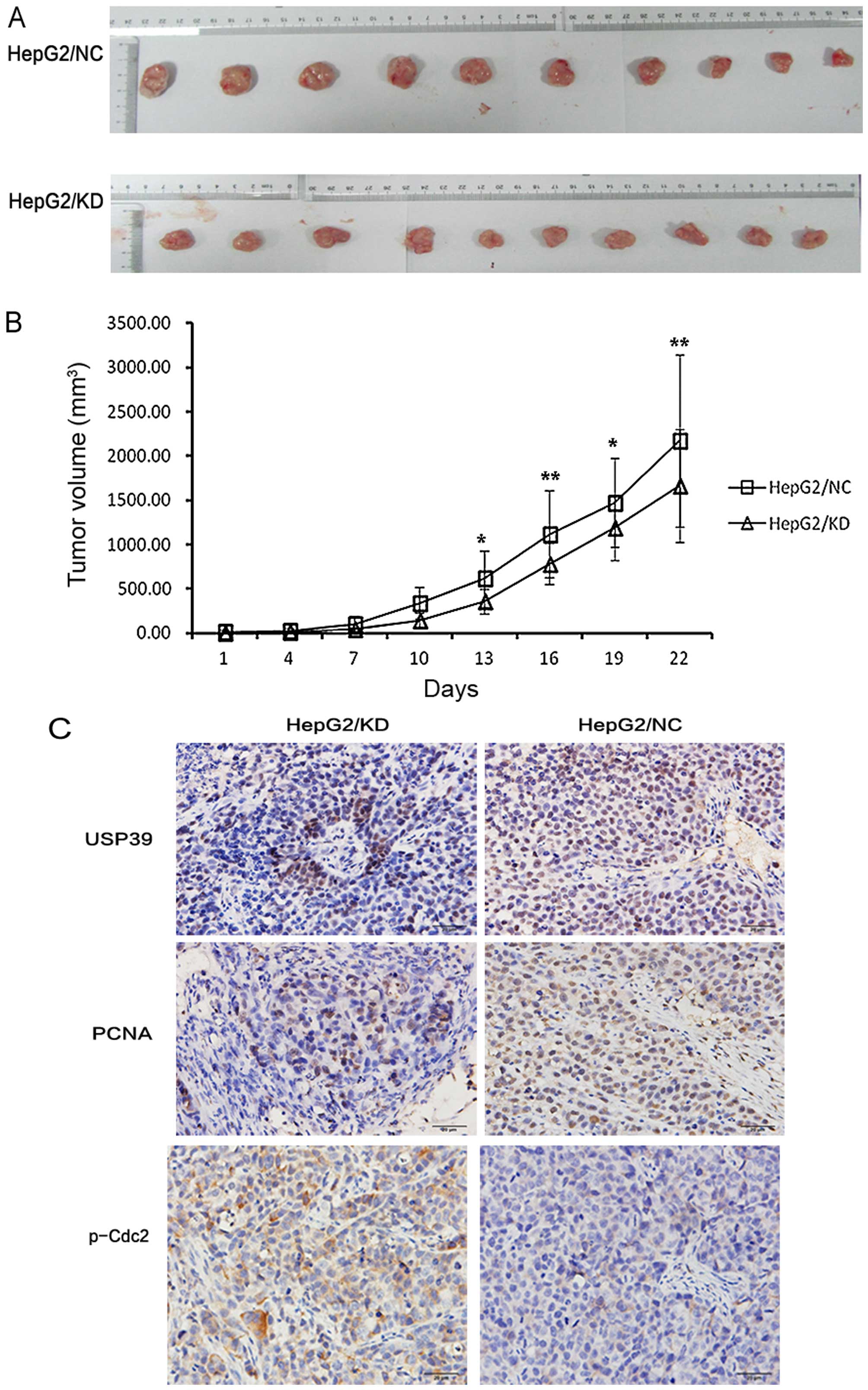
Effect of USP39 on xenograft tumor
growth
To investigate the effect of USP39 on xenograft
tumor growth, we injected HepG2 cells containing the USP39 shRNA or
the non-silencing target RNA into nude mice. Immunohistochemical
result showed that USP39 expression in the HepG2/KD-xenografted
tumors was significantly reduced compared to that in the
xenografted tumors derived from HepG2/NC cells (Fig. 6C). Moreover, cell proliferation in
the HepG2/KD-xenografted tumors was significantly decreased as
demonstrated by anti-PCNA staining (Fig. 6C). Furthermore, USP39 knockdown
significantly decreased xenografted tumor growth in the HepG2 cells
(Fig. 6A and B). These results
indicate that USP39 contributes to HCC tumor growth in
vivo.
USP39-induced G2/M arrest depends on
FoxM1
To further investigate the molecular mechanisms of
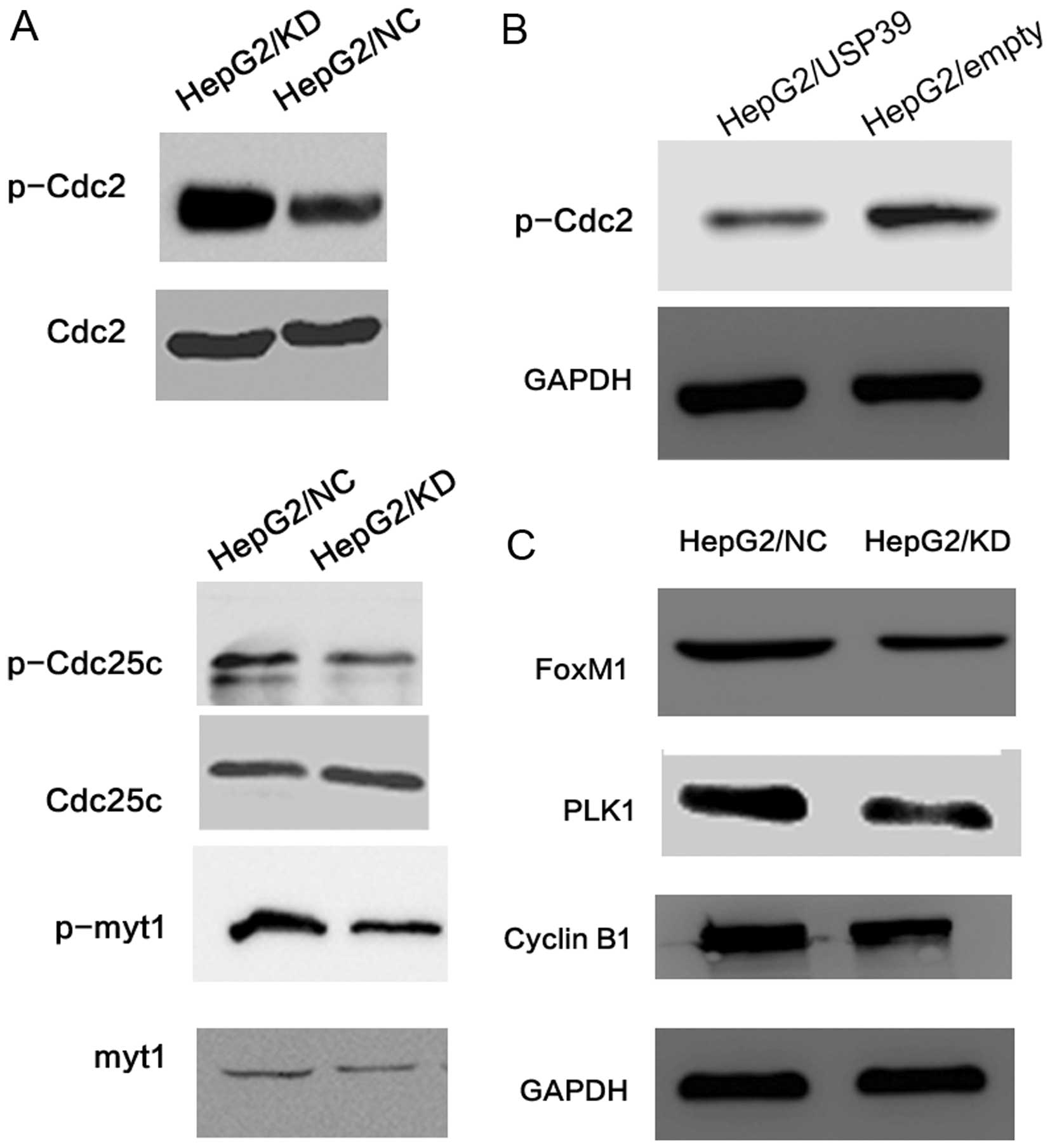
USP39 on cell proliferation, we examined the expression of various
important cell cycle proteins. We found that the level of p-Cdc2 in
the HepG2/KD cells was upregulated while the levels of p-myt1 and
p-Cdc25c were downregulated when compared to the levels in the
HepG2/NC cells. Yet, the protein levels of total Cdc2, myt1 and
Cdc25c were not changed (Fig. 7A).
When USP39 was upregulated in the HepG2 cells, the p-Cdc2 level was
decreased compared with the level in the control cells (Fig. 7B). Additionally, p-Cdc2 was
upregulated in the HepG2/KD-xenografted tumors (Fig. 6C). PLK1, the upstream factor of
Cdc2, was decreased in the HepG2/KD cells compared with the
HepG2/NC cells (Fig. 7C).
Furthermore, FoxM1 which controls the level of PLK1, was decreased
in the HepG2/KD cells. Cyclin B1, another target gene of FoxM1, was
also decreased in the HepG2/KD cells (Fig. 7C).
Discussion
Since USP39 is an important factor for SR-related
proteins targeting a set of key regulatory genes by modulating RNA
splicing (23), this research aimed
to discover the roles of USP39 in HCC. In the present study, we
demonstrated for the first time that USP39 knockdown led to a
defect in mRNA splicing and further caused cell cycle arrest in the
G2/M phase in HCC. Firstly, we compared the expression of USP39 in
tumor tissues and corresponding adjacent normal liver tissues from
more than 100 HCC patients. Notably, the expression of USP39 was
associated with the pathological grade, suggesting that USP39 may
play an important role in HCC.
To confirm the role of USP39 in HCC, we selected one
effective shRNA sequence to study in the subsequent research, and
we successfully established stable USP39-knockdown and
USP39-overexpressing HepG2 cells. We found that USP39 knockdown
significantly inhibited the growth and colony formation of the
HepG2 cells. Conversely, USP39 overexpression significantly
enhanced the growth and colony formation of the HepG2 cells.
Moreover, USP39 knockdown led to G2/M arrest and induced cell
apoptosis in the HepG2 cells. These data indicate that USP39
knockdown inhibited HCC growth more likely through inducing G2/M
phase arrest. Furthermore, the tumor growth was decreased in the
USP39/KD-engrafted mice compared to the growth in the control mice.
Thus, USP39 ablation inhibited the growth of HCC cells in
vitro and in vivo.
Since sustained proliferative signaling is one of
the hallmarks of cancer, inhibition of signaling is an effective
approach in cancer therapy (24).
Checkpoints of the cell cycle control the proper timing of cell
cycle events by enforcing the dependency of late events on the
completion of early events (25).
Consequently, a checkpoint block can result in cell cycle arrest
and significantly alter the activity of cell proliferation. A
previous study revealed that downregulation of USP39 inactivated
the G0/G1 arrest in the zebrafish in vivo by splicing the
rb1 that plays an important role in the transition from G0 to G1
phase (20). A similar effect was
observed in human breast cancer in vitro (21). Nevertheless, our research showed
that USP39 knockdown induced significant G2/M arrest in the HCC
cell lines, along with the suppression of Cdc2 activity (the level
of p-Cdc2 at Tyr15 site upregulation). We also verified a similar
result of p-Cdc2 expression in the xenograft tumors obtained from
the nude mice as that in vitro. It is well-known that the
G2/M transition is regulated by a complex of Cdc2 and cyclin B1,
and a decrease in Cdc2 activity ultimately leads to G2/M arrest
(26,27).
There are three major phosphorylation sites (Thr161,
Tyr15 and Thr14) by which Cdc2 kinase activity is regulated
(28). Phosphorylation at Thr14 and
Tyr15 is carried out by Wee1 and Myt1 protein kinases and results
in the inhibition of Cdc2 (29–31).
PLK1 starts the mitotic cascade by phosphorylating and activating
Cdc25C phosphatase and inhibits myt1 activity, which in turn
establishes a feedback amplification loop that influences the cell
cycle (32). A previous study
demonstrated that Cdc2 is activated by the dephosphorylation at
Tyr15. In the present study, we found that USP39 knockdown induced
G2/M arrest in HepG2 cells, and increased the expression of p-Cdc2
(Thr15). The expression of PLK1 was decreased after USP39
knockdown. FoxM1 was reported to regulate expression of PLK1
(33) and to undergo alternative
splicing (8). Moreover, expression
of FoxM1 was also decreased after USP39 knockdown in the HepG2
cells. Cyclin B1, another target gene of FoxM1, was also decreased
following USP39 knockdown. van Leuken et al found that USP39
could control the mRNA level of Aurora B, and Aurora B is also a
target gene of FoxM1 (19). Thus,
we can tentatively conclude that USP39 may contribute to the mRNA
splicing of FoxM1.
In summary, by using in vitro and in
vivo approaches, we provide evidence that USP39 knockdown
inhibited the tumor growth in human HCC by inducing G2/M arrest, at
least partly, due to FoxM1 splicing. USP39 expression was
associated with the pathological tumor stage of HCC, suggesting
that USP39 may be considered as a promising molecular target for
HCC.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (no. 31300103), the Clinical
Medical Center for Hepatobiliary Disease of Jiangsu Province (no.
ZX201105) and the Clinical Medical Center for Digestive Disease of
Jiangsu Province (no. BL2012001). We thank Drs Huiping Yu and Jun
Yang from the Department of Pathology, The Affiliated Drum Tower
Hospital, School of Medicine, Nanjing University, for their
assistance in the analysis for IHC staining.
References
|
1
|
Villanueva A, Minguez B, Forner A, Reig M
and Llovet JM: Hepatocellular carcinoma: Novel molecular approaches
for diagnosis, prognosis, and therapy. Annu Rev Med. 61:317–328.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huynh H: Molecularly targeted therapy in
hepatocellular carcinoma. Biochem Pharmacol. 80:550–560. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fisher D, Krasinska L, Coudreuse D and
Novák B: Phosphorylation network dynamics in the control of cell
cycle transitions. J Cell Sci. 125:4703–4711. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nurse P: A long twentieth century of the
cell cycle and beyond. Cell. 100:71–78. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martin SJ, McGahon AJ, Nishioka WK, LaFace
D, Guo X, Th’ng J, Bradbury EM and Green DR: p34cdc2 and apoptosis.
Science. 269:106–107. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Laoukili J, Stahl M and Medema RH: FoxM1:
At the crossroads of ageing and cancer. Biochim Biophys Acta.
1775:92–102. 2007.
|
|
7
|
Koo CY, Muir KW and Lam EW: FOXM1: From
cancer initiation to progression and treatment. Biochim Biophys
Acta. 1819:28–37. 2012. View Article : Google Scholar
|
|
8
|
Ye H, Kelly TF, Samadani U, Lim L, Rubio
S, Overdier DG, Roebuck KA and Costa RH: Hepatocyte nuclear factor
3/fork head homolog 11 is expressed in proliferating epithelial and
mesenchymal cells of embryonic and adult tissues. Mol Cell Biol.
17:1626–1641. 1997.PubMed/NCBI
|
|
9
|
Wang IC, Chen YJ, Hughes D, Petrovic V,
Major ML, Park HJ, Tan Y, Ackerson T and Costa RH: Forkhead box M1
regulates the transcriptional network of genes essential for
mitotic progression and genes encoding the SCF (Skp2-Cks1)
ubiquitin ligase. Mol Cell Biol. 25:10875–10894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Costa RH: FoxM1 dances with mitosis. Nat
Cell Biol. 7:108–110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nagata A, Igarashi M, Jinno S, Suto K and
Okayama H: An additional homolog of the fission yeast
cdc25+ gene occurs in humans and is highly expressed in
some cancer cells. New Biol. 3:959–968. 1991.PubMed/NCBI
|
|
12
|
Galaktionov K and Beach D: Specific
activation of cdc25 tyrosine phosphatases by B-type cyclins:
Evidence for multiple roles of mitotic cyclins. Cell. 67:1181–1194.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Molinari M, Mercurio C, Dominguez J,
Goubin F and Draetta GF: Human Cdc25 A inactivation in response to
S phase inhibition and its role in preventing premature mitosis.
EMBO Rep. 1:71–79. 2000. View Article : Google Scholar
|
|
14
|
Karlsson C, Katich S, Hagting A, Hoffmann
I and Pines J: Cdc25B and Cdc25C differ markedly in their
properties as initiators of mitosis. J Cell Biol. 146:573–584.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Souza CP, Ellem KA and Gabrielli BG:
Centrosomal and cytoplasmic Cdc2/cyclin B1 activation precedes
nuclear mitotic events. Exp Cell Res. 257:11–21. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoffmann I, Clarke PR, Marcote MJ,
Karsenti E and Draetta G: Phosphorylation and activation of human
cdc25-C by cdc2-cyclin B and its involvement in the
self-amplification of MPF at mitosis. EMBO J. 12:53–63.
1993.PubMed/NCBI
|
|
17
|
Perdiguero E and Nebreda AR: Regulation of
Cdc25C activity during the meiotic G2/M transition. Cell Cycle.
3:733–737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reyes-Turcu FE, Ventii KH and Wilkinson
KD: Regulation and cellular roles of ubiquitin-specific
deubiquitinating enzymes. Annu Rev Biochem. 78:363–397. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
van Leuken RJ, Luna-Vargas MP, Sixma TK,
Wolthuis RM and Medema RH: Usp39 is essential for mitotic spindle
checkpoint integrity and controls mRNA-levels of aurora B. Cell
Cycle. 7:2710–2719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ríos Y, Melmed S, Lin S and Liu NA:
Zebrafish usp39 mutation leads to rb1 mRNA splicing defect and
pituitary lineage expansion. PLoS Genet. 7:e10012712011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang H, Ji X, Liu X, Yao R, Chi J, Liu S,
Wang Y, Cao W and Zhou Q: Lentivirus-mediated inhibition of USP39
suppresses the growth of breast cancer cells in vitro. Oncol Rep.
30:2871–2877. 2013.PubMed/NCBI
|
|
22
|
Sun XT, Yuan XW, Zhu HT, Deng ZM, Yu DC,
Zhou X and Ding YT: Endothelial precursor cells promote
angiogenesis in hepatocellular carcinoma. World J Gastroenterol.
18:4925–4933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Makarova OV, Makarov EM and Lührmann R:
The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP
are essential for the assembly of mature spliceosomes. EMBO J.
20:2553–2563. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smits VA, Klompmaker R, Vallenius T,
Rijksen G, Mäkela TP and Medema RH: p21 inhibits Thr161
phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J
Biol Chem. 275:30638–30643. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guadagno TM and Newport JW: Cdk2 kinase is
required for entry into mitosis as a positive regulator of
Cdc2-cyclin B kinase activity. Cell. 84:73–82. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Atherton-Fessler S, Liu F, Gabrielli B,
Lee MS, Peng CY and Piwnica-Worms H: Cell cycle regulation of the
p34cdc2 inhibitory kinases. Mol Biol Cell. 5:989–1001. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Norbury C, Blow J and Nurse P: Regulatory
phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO
J. 10:3321–3329. 1991.PubMed/NCBI
|
|
30
|
McGowan CH and Russell P: Human Wee1
kinase inhibits cell division by phosphorylating p34cdc2
exclusively on Tyr15. EMBO J. 12:75–85. 1993.PubMed/NCBI
|
|
31
|
Wells NJ, Watanabe N, Tokusumi T, Jiang W,
Verdecia MA and Hunter T: The C-terminal domain of the Cdc2
inhibitory kinase Myt1 interacts with Cdc2 complexes and is
required for inhibition of G(2)/M progression. J Cell Sci.
112:3361–3371. 1999.PubMed/NCBI
|
|
32
|
Cogswell JP, Brown CE, Bisi JE and Neill
SD: Dominant-negative polo-like kinase 1 induces mitotic
catastrophe independent of cdc25C function. Cell Growth Differ.
11:615–623. 2000.
|
|
33
|
Dibb M, Han N, Choudhury J, Hayes S,
Valentine H, West C, Ang YS and Sharrocks AD: The FOXM1-PLK1 axis
is commonly upregulated in oesophageal adenocarcinoma. Br J Cancer.
107:1766–1775. 2012. View Article : Google Scholar : PubMed/NCBI
|