Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide (1). Non-small
cell lung cancer (NSCLC) accounts for ~85% of all lung malignancies
and adenocarcinoma is the most common histological subtype.
Although recent progress in diagnosis and treatment including
molecular-targeted therapy have provided a considerable survival
benefit, ~40% of NSCLC are diagnosed at an advanced stage, with an
overall 5-year survival of ~15% and recurrence rates remain high
(2). Therefore, further
identification of key molecular alterations in NSCLC is
required.
Tyrosine phosphorylation is an important signaling
mechanism in the regulation of various biological processes.
Activation of protein tyrosine kinase (PTK) is a common feature of
cancer and many drugs targeting PTKs, such as epidermal growth
factor receptor (EGFR)-tyrosine kinase inhibitors, have been
introduced. Protein tyrosine phosphatases (PTPs) also regulate
tyrosine phosphorylation and are involved in cancer. Recent
evidence has shown the relevance of PTPs either as tumor
suppressors or oncoproteins (3).
Epigenetic and genetic alterations in genes coding PTPs may be
associated with cancer phenotypes.
PTPRH, also known as stomach cancer-associated PTP-1
(SAP-1), was first identified as a transmembrane-type PTP abundant
in a subset of pancreatic and colorectal cancer cell lines
(4). This enzyme belongs to the R3
subtype receptor-type PTP, together with PTPRB (also known as
VE-PTP), PTPRJ (also known as DEP-1) and PTPRO and localizes to the
microvilli of the brush border in the gastrointestinal tract
(5). Ablation of PTPRH
inhibits tumorigenesis in mice heterozygous for an adenomatous
polyposis coli mutation (Apcmin/+) (6). Although it has been suggested that
PTPRH regulates intestinal tumorigenesis, the mechanism is
unclear. In contrast, PTPRH was found to be downregulated in
advanced human hepatocellular carcinoma (7) and inhibited the proliferation of
cultured cells (8,9). Thus, the role of PTPRH is
largely unknown and needs to be clarified in diseases including
lung cancer.
Epigenetic changes such as aberrant DNA methylation
in human cancers have been described (10). DNA hypermethylation in the 5′-UTR
CpG-rich regions can block the expression of tumor suppressor
genes. In NSCLC, silencing of specific genes such as
RASSF1A, CDKN2A, RARβ, MGMT,
APC, DAPK, FHIT, and CDH13 due to DNA
hypermethylation around their promoter regions has been frequently
observed (11). However, the role
of DNA hypomethylation in permitting overexpression of specific
genes is relatively poorly understood in NSCLC.
In the present study, we investigated the expression
status of PTPRH in NSCLC. Next, it was elucidated that
overexpression of PTPRH was caused by an underlying
mechanism involving DNA hypomethylation. Then, the regulatory
mechanism was confirmed in vitro using a DNA methylation
inhibitor. Furthermore, it was determined that PTPRH DNA
methylation was an independent prognostic factor for NSCLC
patients.
Materials and methods
Patients and tissue samples
Two independent cohorts of lung cancer patients were
investigated. The first cohort (LC-C1) comprised 89 paired samples
of tumorous lung and corresponding non-cancerous tissues from
patients with primary NSCLC who underwent lung resection at the
Department of Thoracic Surgery, Keio University Hospital, Japan,
between 2001 and 2006. Approval for institutional review of these
samples was obtained in accordance with the requirements of the
Keio University Institutional Review Board (IRB #16-90-1). The
second cohort (LC-C2) consisted of 145 paired samples of tumorous
lung and corresponding non-cancerous tissues from patients with
primary lung adenocarcinoma (LADC) who underwent lung resection at
the National Cancer Center Hospital, Japan, between 1997 and 2008.
These tissue specimens were provided by the National Cancer Center
Biobank, Japan and the present study was also approved by the
Ethics Committee of the National Cancer Center, Japan. All patients
in the present study provided written informed consent.
Clinicopathological parameters in both cohorts are summarized in
Table I.
 | Table IClinicopathological parameters of
patients with NSCLCs in LC-C1 and LC-C2. |
Table I
Clinicopathological parameters of
patients with NSCLCs in LC-C1 and LC-C2.
| Clinicopathological
parameters | LC-C1 (n=89) | LC-C2 (n=145) |
|---|
| Age (years) |
| Median | 68 | 61 |
| Interquartile
range | 60–75 | 55–66 |
| Gender |
| Male | 56 | 81 |
| Female | 33 | 64 |
| Smoking status
(pack-year) |
| Median | 30 | 13 |
| Interquartile
range | 0–60 | 0–41 |
| Histological
type |
|
Adenocarcinoma | 54 | 145 |
| Squamous cell
carcinoma | 24 | 0 |
| Large cell
carcinoma | 6 | 0 |
| Others | 5 | 0 |
| Tumor size
(cm) |
| Median | 3 | 2.8 |
| Interquartile
range | 2.5–4.0 | 2.2–4.5 |
| Tumor stage |
| T1 | 41 | 64 |
| T2 | 30 | 63 |
| T3-4 | 18 | 18 |
| Nodal status |
| N0 | 60 | 94 |
| N1 | 14 | 24 |
| N2-3 | 15 | 27 |
| Metastatic
status |
| M0 | 87 | 145 |
| M1 | 2 | 0 |
| Pathological TNM
stage |
| I | 50 | 83 |
| II | 12 | 31 |
| III–IV | 27 | 31 |
| EGFR
mutation |
| Wild-type | 63 | 73 |
| Mutant | 26 | 48 |
| K-ras
mutation |
| Wild-type | 84 | 109 |
| Mutant | 5 | 12 |
Cell lines
Three human lung cancer cell lines were used: A549
(adenocarcinoma), VMRC-LCD (adenocarcinoma) and EBC-1 (squamous
cell carcinoma). A549 cells were purchased from the American Type
Culture Collection (ATCC; manassas, VA, USA). VMRC-LCD and EBC-1
cells were purchased from the Health Science Research Resources
Bank (Osaka, Japan). All cell lines were cultured according to the
supplier's instructions.
cDNA microarray analysis
GeneChip Human Genome 2.0 array (Affymetrix, Inc.,
Santa Clara, CA, USA) was used to monitor the expression profiles
of the LC-C1 samples. Total RNA was extracted from tumorous tissues
and paired non-cancerous lung tissues using TRIzol (Life
Technologies, Carlsbad, CA, USA). The labeled cRNA was prepared
using standard Affymetrix protocols. The signal intensities of the
probe sets were normalized using the Affymetrix Power Tools RMA
method implemented using Resolver software (Rosetta Biosoftware,
Seattle, WA, USA).
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR) analysis
Total RNA extracted from 62 tumorous and 17
non-cancerous tissues in LC-C1, 111 tumorous and 96 non-cancerous
tissues in LC-C2 and lung cancer cell lines, was
reverse-transcribed to cDNA using TaqMan reverse transcription
reagents or SuperScript III reverse transcriptase (both from Life
Technologies). For qRT-PCR analysis, TaqMan gene expression assays
were used for human PTPRH (Hs00936195_m1, #4331182) and
human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; #4310884E,
both from Life Technologies) to normalize input cDNA. Quantitative
analysis was performed using Applied Biosystems 7500 Fast-Real Time
PCR system (Life Technologies). All assays were performed in
triplicate.
Infinium assay
Genomic DNA was extracted from all LC-C2 tissue
samples and lung cancer cell lines using a QIAamp DNA mini kit
(Qiagen, Valencia, CA, USA). Bisulfite conversion using an EZ DNA
Methylation-Gold kit (Zymo Research, Irvine, CA, USA) was carried
out on 500 ng aliquots of DNA. Subsequently, DNA methylation status
at 27,578 CpG loci was examined at single-CpG resolution using the
Infinium HumanMethylation27 Bead array (Illumina, San Diego, CA,
USA). An Evo robot (Tecan, Männedorf, Switzerland) was used for
automated sample processing. After whole genome amplification and
hybridization, the specifically hybridized DNA was
fluorescence-labeled by a single-base extension reaction and
detected using a BeadScan reader (Illumina) in accordance with the
manufacturer's protocols. The data were then assembled using
GenomeStudio methylation software (Illumina). At each CpG site, the
ratio of the fluorescence signal was measured using a methylated
probe relative to the sum of the methylated and unmethylated
probes, such as the β-value, which ranges from 0 to 1 and reflects
the methylation level of an individual CpG site. The reliability of
DNA methylation levels (β-values) determined by the Infinium assay
was verified in our previous studies (12–14).
DNA methylation analysis with the
MassARRAY system
Bisulfite treatment using an EpiTect Bisulfite Kit
(Qiagen GmbH, Hilden, Germany) was carried out on 250 ng of genomic
DNA extracted from LC-C2 samples, in accordance with the
manufacturer's protocol. DNA methylation levels containing the CpG
site cg11261264 in the Infinium assay were evaluated quantitatively
using the MassARRAY platform (Sequenom, San Diego, CA, USA). This
method utilizes base-specific cleavage and matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry (MALDI-TOF
MS) (15). Specific PCR primers for
bisulfite-converted DNA were designed using the EpiDesigner
software package (http://www.epidesigner.com; Sequenom). The forward and
reverse primers used were 5′-TTGTGTGTTGTTTGAAGTTAGTGTTT-3′ and
5′-CTAAACCTAAAACTCCTAAATCCCC-3′, respectively. A T7-promoter tag
(5′-CAGTAATACGACTCACTATAGGGAGAAGGCT-3′) was added to the reverse
primer for in vitro transcription and a 10-mer tag was added
to each forward primer to balance the PCR. To overcome PCR bias in
DNA methylation analysis, we optimized the annealing temperature
and type of DNA polymerase: 0, 50 and 100% methylated control DNA
(EpiTect methylated human control DNA; Qiagen) was used as template
to test the linearity of the protocol. HotStar Taq DNA
polymerase (Qiagen) was used for the PCR. The PCR products were
used as a template for in vitro transcription and RNase
A-mediated cleavage reaction using an EpiTyPER reagent kit
(Sequenom). The fragmented samples were dispensed onto a
SpectroCHIP array and then detected on a MassARRAY analyzer compact
MALDI-TOF MS instrument. The data were visualized using EpiTYPER
Analyzer software v1.0 (Sequenom). The DNA methylation level (%) at
each CpG site was determined by comparing the signal intensities of
methylated and non-methylated templates. Experiments were performed
in triplicate for each sample-CpG site and the mean value for the
three experiments was used as the DNA methylation level.
5-Aza-2′-deoxycytidine (5-aza-dC)
treatment
A549, VMRC-LCD and EBC-1 cells were seeded at a
density of 9×105 cells/15 cm dish on day 0 and then
allowed to attach for a 24-h period. Then, 5-aza-dC (Sigma-Aldrich,
St. Louis, MO, USA) was added to a final concentration of 5
µM. Cells were passaged at a subculture ratio of 1:2 on day
3. 5-Aza-dC was added again to the same final concentration 24 h
after replating. Since toxicity was observed during preliminary
experiments, the final concentration of 5-aza-dC was reduced to 0.5
µM for EBC-1 cells. Genomic DNA and total RNA were extracted
from all cells on days 3 and 6.
Statistical analysis
Differences in mRNA expression or DNA methylation
levels between cancerous and non-cancerous tissues were
investigated using the Wilcoxon signed-rank or Mann-Whitney U test,
respectively. Correlation between PTPRH mRNA expression
levels obtained by microarray and qRT-PCR, as well as correlation
between DNA methylation and mRNA expression levels of PTPRH,
were examined using Spearman's correlation test. DNA methylation
levels obtained by the MassARRAY system were compared with those by
the Infinium assay using the Pearson's correlation test. The
correlation between mRNA expression or DNA methylation levels of
PTPRH and clinicopathological factors was examined using
Spearman's correlation, Mann-Whitney U and Kruskal-Wallis test.
Survival curves for patients with LADC were analyzed by the
Kaplan-Meier method and log-rank test. The PTPRH DNA
methylation cutoff level was determined in order to maximize the
sensitivity and specificity for recurrence by receiver operating
characteristic (ROC) curve analysis (16). Multivariate analysis of the
influence of variables on recurrence-free survival was performed
using the Cox proportional hazards model. Statistical analyses were
performed using SPSS 20.0 (SPSS, IBM, Chicago, IL, USA) and
GraphPad Prism 5.0 software (GraphPad Software, La Jolla, CA, USA).
All P-values were two-sided and P<0.05 was considered
statistically significant.
Results
PTPRH mRNA expression is increased in
NSCLC
PTPRH gene expression in LC-C1 was
investigated by cDNA micro-array. The PTPRH mRNA expression
levels were significantly higher in tumorous tissues compared with
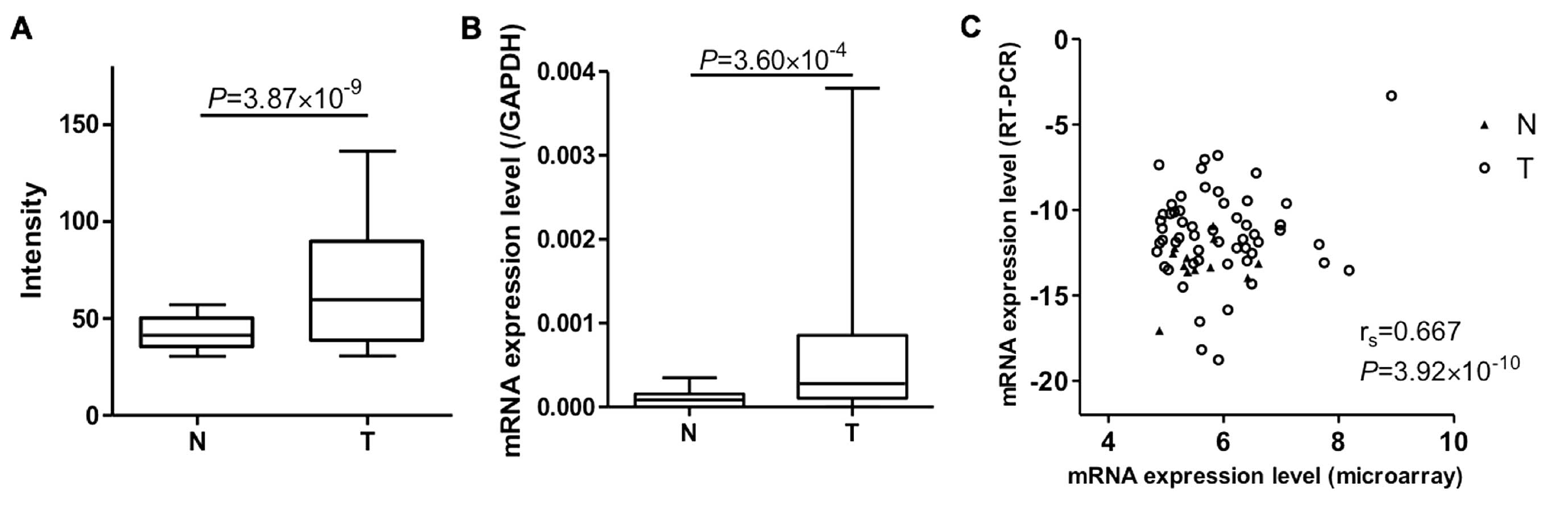
the corresponding non-cancerous tissues (Fig. 1A). We performed qRT-PCR on
PTPRH to confirm the microarray data for 62 cancerous and 17
non-cancerous tissues whose samples were still available. Again,
the PTPRH mRNA expression levels were significantly higher
in tumorous tissues compared with non-cancerous tissues (Fig. 1B). The PTPRH mRNA expression
data correlated well with that determined by qRT-PCR and microarray
analysis (rs=0.667; P=3.92×10−10; Fig. 1C).
The correlation between PTPRH gene expression
and clinicopathological parameters are summarized in Table II. Tumor-node-metastasis (TNM)
classification was performed according to the Union Internationale
Contre le Cancer (UICC)-6 staging system for NSCLC. Distribution of
age, gender, histological type, pathological TNM stage, EGFR
and K-ras mutation status except for smoking status did not
significantly correlate with PTPRH mRNA expression.
| Table IICorrelation between mRNA expression
levels (microarray) of PTPRH and clinicopathological
parameters of patients with non-small cell lung cancers. |
Table II
Correlation between mRNA expression
levels (microarray) of PTPRH and clinicopathological
parameters of patients with non-small cell lung cancers.
|
Clinicopatho-logical parameters | No. | Median intensity
(interquartile range) | Pa-value |
|---|
| Age (years) | | |
5.94×10−1b (rs=−0.057) |
| Gender |
| Male | 56 | 63.9
(41.4–95.1) |
9.56×10−2c |
| Female | 33 | 47.3
(37.7–84.2) | |
| Smoking status
(pack-year) | | |
6.73×10−3b (rs=0.287) |
| Histological
type |
|
Adenocarcinoma | 54 | 49.9
(35.4–84.9) |
1.51×10−1d |
| Squamous cell
carcinoma | 24 | 62.0
(46.1–91.2) | |
| Large cell
carcinoma | 6 | 84.9
(59.6–114.3) | |
| Others | 5 | 74.9
(64.0–97.3) | |
| Tumor stage |
| T1 | 41 | 49.5
(38.9–85.0) |
3.48×10−1b |
| T2 | 30 | 71.0
(36.7–92.5) | |
| T3-4 | 18 | 60.2
(38.2–105.7) | |
| Nodal status |
| N0 | 60 | 60.2
(41.5–93.6) |
2.81×10−1b |
| N1 | 14 | 60.5
(34.7–89.6) | |
| N2-3 | 15 | 47.3
(35.3–87.5) | |
| Metastatic
status |
| M0 | 87 | 56.8
(37.7–87.5) |
7.63×10−2b |
| M1 | 2 | 119.4
(114.3–124.5) | |
| Pathological |
| TNM stage |
| I | 50 | 57.8
(38.9–92.5) |
8.72×10−1b |
| II | 12 | 62.0
(44.8–83.9) | |
| III–IV | 27 | 59.9
(35.3–114.3) | |
| EGFR
mutation |
| Wild-type | 63 | 63.7
(43.8–92.5) |
1.56×10−1b |
| Mutant | 26 | 46.1
(35.9–84.2) | |
| K-ras
mutation |
| Wild type | 84 | 58.2
(38.6–89.8) |
7.91×10−1b |
| Mutant | 5 | 59.9
(30.6–84.9) | |
PTPRH DNA methylation is reduced and
inversely correlated with mRNA expression in LADC
Based on the hypothesis that altered mRNA expression
may be caused by aberrant DNA methylation, we investigated the DNA
methylation status of PTPRH in LC-C2 samples for which
genome-wide DNA methylation profiles were available. As there was
no significant correlation between PTPRH mRNA expression
levels and histological type, we used LC-C2 samples and focused on
LADC, a major histological type in LC-C1, for further analysis of
the epigenetic regulation of PTPRH. DNA methylation levels
of the CpG site cg11261264, located in PTPRH intron 1, were
significantly decreased in tumorous compared with non-cancerous
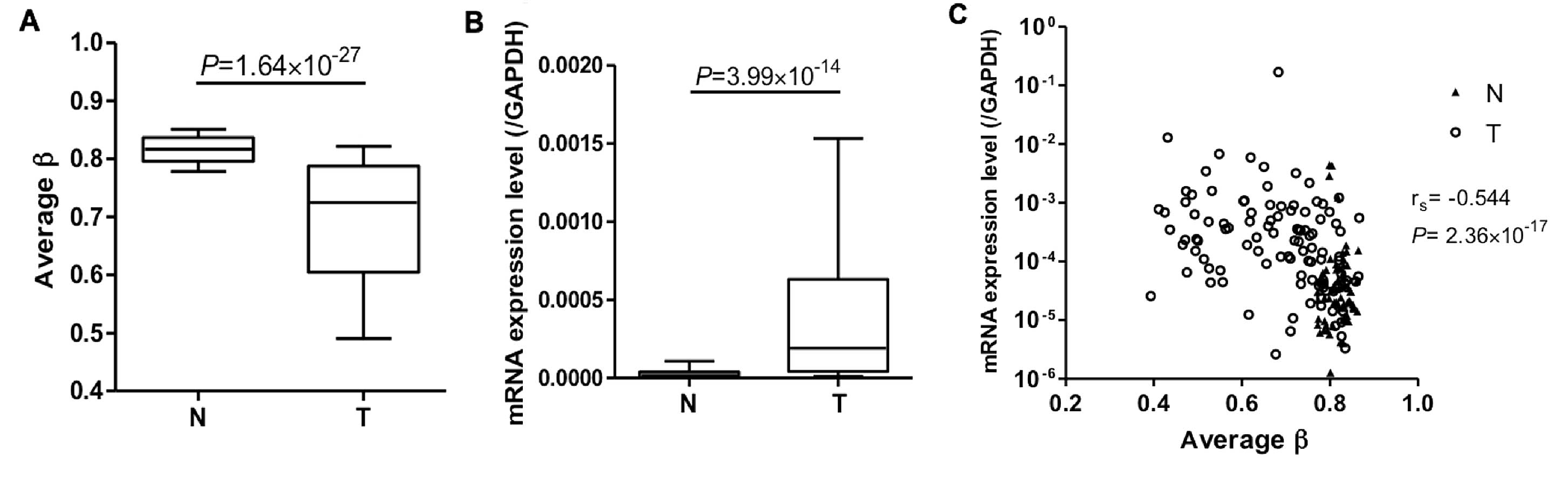
tissues (Fig. 2A). We also examined
PTPRH mRNA expression levels in LC-C2 samples by qRT-PCR.
This showed that PTPRH mRNA expression levels were again
higher in tumorous tissues compared with the corresponding
non-cancerous (Fig. 2B) and
inversely correlated with DNA methylation of this single CpG site
(Fig. 2C). These data suggested
that PTPRH DNA hypomethylation may result in increased mRNA
expression in tissue samples from the same cohort.
PTPRH gene expression is regulated by DNA
hypomethylation
To improve understanding of the influence of DNA
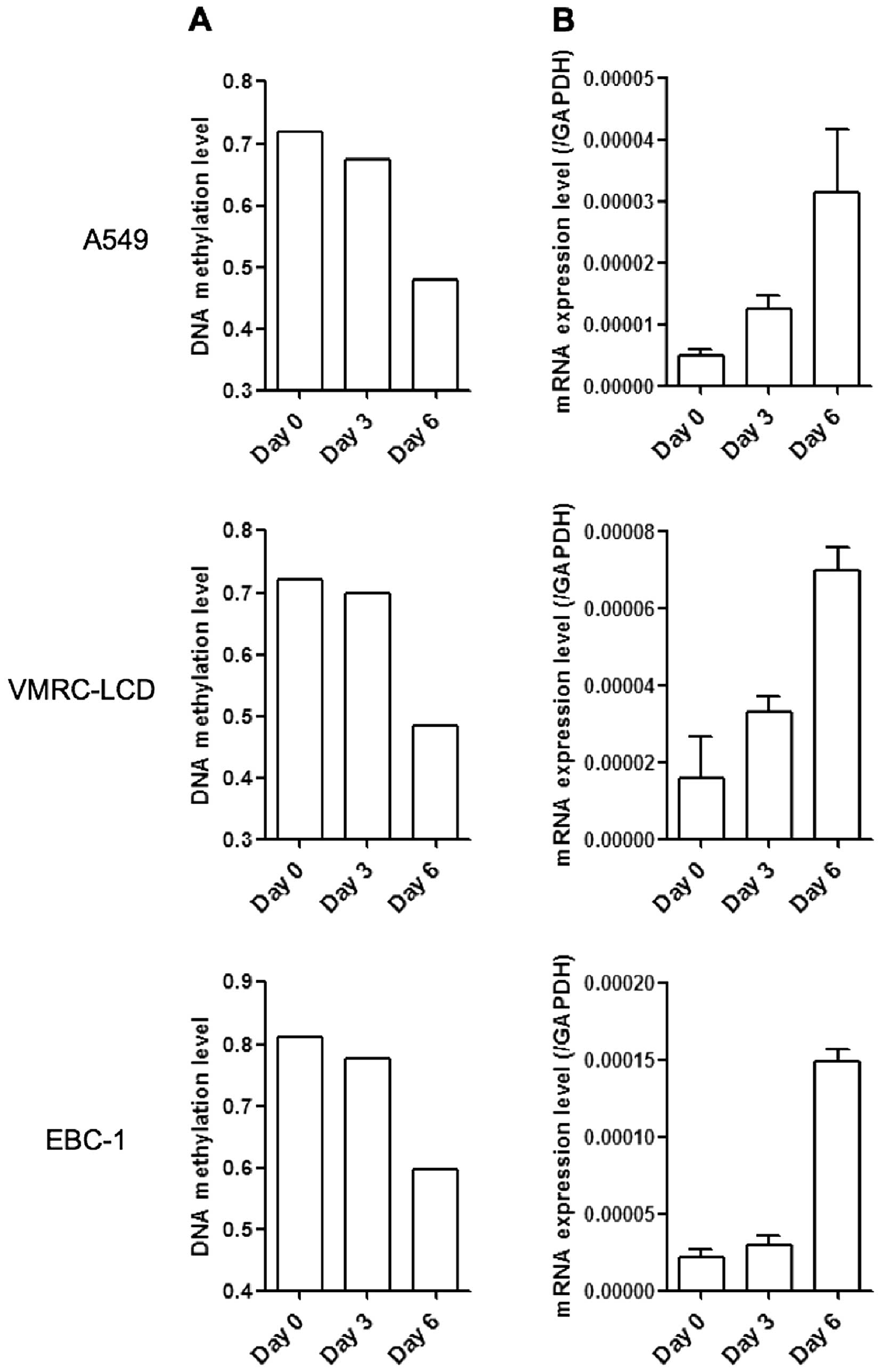
methylation on PTPRH gene expression, lung cancer cell lines
were treated with the DNA methylation inhibitor, 5-aza-dC. In three
lung cancer cell lines A549, VMRC-LCD, and EBC-1 with low
PTPRH mRNA expression, 5-aza-dC treatment induced a marked
reduction of DNA methylation and restored PTPRH mRNA
expression levels (Fig. 3). These
data suggested that increased expression of PTPRH was primarily
regulated by DNA hypomethylation in LADC.
DNA hypomethylation of PTPRH as a
prognostic factor
Because investigation of DNA methylation by the
MassARRAY system allowed a comprehensive coverage of CpG sites, we
assessed DNA methylation levels at the relatively CpG-rich region
containing the CpG site cg11261264 in the same LC-C2 samples using
the MassARRAY system (Genomic positions of the CpG sites are shown
in Table III). DNA methylation
levels obtained by the MassARRAY system and Infinium assay
correlated well (r=0.952, P=1.44×10−73), confirming the
reliability of the latter assay. DNA methylation patterns
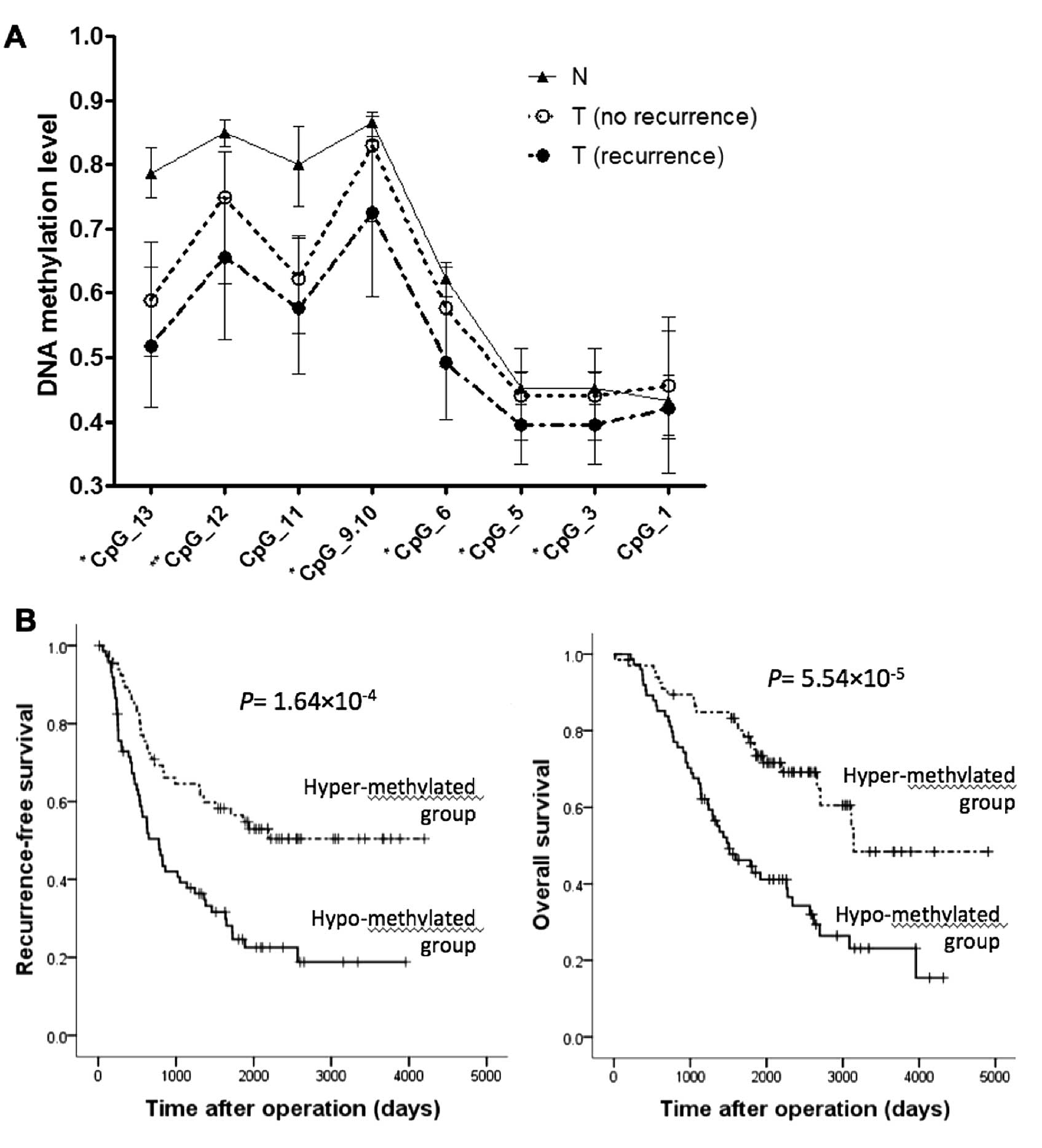
quantified at each analyzed CpG site in non-cancerous, tumorous
tissue with no recurrence and those with recurrence are shown in
Fig. 4A. The consecutive CpG sites
CpG_9.10 provided one measured value as a 'CpG unit̓ by the
massARRAy system. DNA methylation levels at most CpG sites showed a
significant decrease in tumorous tissues with recurrence compared
to those with no recurrence. The correlation between the DNA
methylation levels of cancerous tissues at CpG_9.10 and
clinicopathological parameters are summarized in Table IV. DNA hypomethylation at this CpG
unit was significantly correlated with male gender, heavy smoking
status, advanced pathological stage and wild-type EGFR.
| Table IIICpG sites analyzed by the MassARRAY
system. |
Table III
CpG sites analyzed by the MassARRAY
system.
| CpG site | Positiona |
|---|
| CpG_1 | Chromosome 19:
55,720,456 |
| CpG_2 (not
covered) | Chromosome 19:
55,720,467 |
| CpG_3 | Chromosome 19:
55,720,485 |
| CpG_4 (not
covered) | Chromosome 19:
55,720,516 |
| CpG_5 | Chromosome 19:
55,720,534 |
| CpG_6 | Chromosome 19:
55,720,554 |
| CpG_7 (not
covered) | Chromosome 19:
55,720,565 |
| CpG_8 (not
covered) | Chromosome 19:
55,720,668 |
| CpG_9.10b | Chromosome 19:
55,720,728. 55,720,737 |
| CpG_11 | Chromosome 19:
55,720,777 |
| CpG_12 | Chromosome 19:
55,720,786 |
| CpG_13 | Chromosome 19:
55,720,817 |
| Table IVCorrelation between DNA methylation
levels of PTPRH and clinicopathological parameters of patients with
lung adenocarcinomas. |
Table IV
Correlation between DNA methylation
levels of PTPRH and clinicopathological parameters of patients with
lung adenocarcinomas.
| Clinicopathological
parameters | No. | Median DNA
methylation level (interquartile range) | P-valuea |
|---|
| Years of age | | |
7.01×10−2b (rs=0.153) |
| Gender |
| Male | 81 | 0.723
(0.597–0.830) |
3.05×10−3c |
| Female | 64 | 0.833
(0.690–0.870) | |
| Smoking status
(pack-year) | | |
3.51×10−3b (rs=−0.244) |
| Tumor stage |
| T1 | 64 | 0.793
(0.672–0.850) |
3.15×10−1b |
| T2 | 63 | 0.727
(0.593–0.850) | |
| T3–4 | 18 | .0.737
(0.627–0.872) | |
| Nodal status |
| N0 | 94 | 0.793
(0.627–0.860) |
1.96×10−1b |
| N1 | 24 | 0.768
(0.602–0.0.845) | |
| N2-3 | 27 | 0.713
(0.643–0.797) | |
| Pathological TNM
stage |
| I | 83 | 0.807
(0.627–0.867) |
1.57×10−2b |
| II | 31 | 0.767
(0.653–0.853) | |
| III | 31 | 0.703
(0.600–0.790) | |
| EGFR
mutation |
| Wild-type | 73 | 0.717
(0.547–0.843) |
7.28×10−3b |
| Mutant | 48 | 0.820
(0.647–0.873) | |
| K-ras
mutation |
| Wild-type | 109 | 0.767
(0.617–0.857) |
9.39×10−2b |
| Mutant | 12 | 0.665
(0.505–0.755) | |
Kaplan-Meier analysis based on the optimal cutoff
determined by ROC curve analysis showed that patients with
hypomethylation at the CpG unit had a shorter recurrence-free and
overall survival compared with patients with hypermethylation
(Fig. 4B; P=1.64×10−4
and P=5.54×10−5, respectively). Since multiple
covariates can affect patient survival, we performed multivariate
analysis to confirm that the DNA methylation status of PTPRH
is an independent prognostic factor for LADC patients. In
multivariate Cox proportional hazards regression analysis,
PTPRH DNA methylation, together with gender and pathological
TNM status, emerged as an independent prognostic factor for
recurrence-free survival (Table
V).
| Table VMultivariate analysis of predictive
factors for recurrence-free survival in patients with LADCs (Cox
proportional hazard model). |
Table V
Multivariate analysis of predictive
factors for recurrence-free survival in patients with LADCs (Cox
proportional hazard model).
| Variables | Multivariate
analysis Hazard ratio (95% confidence interval) |
|---|
| Gender | 1.801
(1.012–3.205) |
4.53×10−2 |
| Smoking status
(pack-year) | 1.000
(1.000–1.001) |
1.72×10−1 |
| Pathological TNM
stage | 1.468
(1.276–1.690) |
8.69×10−8 |
| EGFR
mutation | 1.490
(0.923–2.407) |
1.03×10−1 |
| PTPRH DNA
methylation | 0.134
(0.031–0.576) |
6.88×10−3 |
Discussion
Although some studies have demonstrated the
involvement of PTPRH in cancer, especially in intestinal
tumorigenesis (4,12,17),
its role in lung cancer and the molecular mechanisms underlying its
regulation have not been clarified. In the present study, we
examined PTPRH expression in NSCLC and focused on its
regulation by DNA methylation and its clinicopathological
implications.
First, we showed that PTPRH expression is
increased in NSCLC using cDNA microarray analysis. mRNA expression
levels obtained from qRT-PCR confirmed the microarray data. While
PTPRH expression was found to be increased in human colon
and pancreatic cancer (4,17) and reduced in advanced human
hepatocellular carcinoma (7), to
our knowledge no previous study has examined its expression in lung
cancer. Our data from LC-C1 samples suggested that PTPRH may
have a significant role in NSCLC and be associated with its
clinicopathological features. In fact, PTPRH expression was
associated with smoking status that is a well-known risk factor for
NSCLC. However, no other association was observed at a
statistically significant level.
As PTPRH was upregulated in NSCLC, we became
interested in the molecular mechanisms underlying its increased
expression. Recent studies analyzed the genome-wide DNA methylation
profiles of LADC (13,14). We investigated the DNA methylation
levels of LC-C2 samples using data from the Infinium assay. As
LC-C2 consists of patients with LADC only, we focused on
PTPRH in LADC, a major subtype of NSCLC, to concentrate on
the correlation between measured values and clinicopathological
parameters and patient survival. This showed that PTPRH DNA
methylation was reduced in LADC and inversely correlated with mRNA
expression. Several studies identified tumor-specific
hypermethylation of other types of PTPs, such as PTPRO
(18,19), PTPRD (20), PTPRG (21,22),
PTPN6 (23–25) and PTPN13 (26), reviewed by Jacob and Motiwala
(27), and Julien et al
(3). However, tumor-specific
hypomethylation of PTP has not been reported. 5-aza-dC treatment of
human lung cancer cell lines with low PTPRH expression
restored PTPRH mRNA expression levels indicating that
PTPRH is reactivated by DNA hypomethylation during lung
tumorigenesis.
We speculated that PTPRH may play an oncogenic role
in lung tumorigenesis because PTPRH is thought to promote
intestinal tumorigenesis (6). We
attempted a transient knockdown of PTPRH using siRNA in lung cancer
cell lines. However, we were unable to find any lung cancer cell
lines where cell proliferation was inhibited by its knockdown (data
not shown). Although PTPRH was shown to have a capacity to activate
Src (28), Src kinase may not be
responsible for its oncogenic properties. Sadakata et al
reported that ablation of PTPRH did not reduce the levels of c-Src
activity in Apcmin/+ mice (6) and we did not observe changes in Src
phosphorylation (Y416) in PTPRH-knockdown cells (data not shown).
Recently, the substrate specificity of the R3 subtype of
receptor-type PTPs toward receptor tyrosine kinases was described
(29). Further studies are needed
to clarify the mechanisms downstream of PTPRH.
We further confirmed the data from the Infinium
assay by using the MassARRAY platform and investigated the
clinicopathological and prognostic significance of PTPRH DNA
methylation in LADC. PTPRH DNA hypomethylation was
significantly correlated with male gender, heavy smoking status,
pathological stage and wild-type EGFR. Seo et al described
the correlation between PTPRH expression and K-ras
mutations in colorectal cancer (17). However, no correlation was found in
the present study, probably due to differences in histology.
Although the underlying significance of these results and the
precise function of PTPRH were not clarified in the present study,
our analysis explored PTPRH hypomethylation as a novel
prognostic factor. To the best of our knowledge, this is the first
study to show that PTPRH is regulated by DNA methylation and
its hypomethylation is related to poor prognosis in LADC.
In conclusion, PTPRH is upregulated in NSCLC
and it is regulated by DNA hypomethylation in LADC. Moreover, the
DNA methylation status of PTPRH has been identified as a
novel prognostic factor for LADC. Further studies are warranted to
clarify its molecular role in the development of NSCLC.
Acknowledgments
We would like to thank Ms. Mikiko Shibuya for her
excellent technical assistance. The present study was supported in
part by the Grants-in-Aid for Scientific Research from the Japan
Society for the Promotion of Science to T.S. (Grant no. 26870569)
and K.S. (Grant no. 22590870) and the Program for Promotion of
Fundamental Studies in Health Sciences (10–42) of the National
Institute of Biomedical Innovation (NiBio), Japan. T.S. is an
awardee of a research resident fellowship from the Foundation for
Promotion of Cancer Research in Japan.
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
PTK
|
protein tyrosine kinase
|
|
EGFR
|
epidermal growth factor receptor
|
|
PTP
|
protein tyrosine phosphatase
|
|
SAP-1
|
stomach cancer-associated protein
tyrosine phosphatase-1
|
|
LADC
|
lung adenocarcinoma
|
|
qRT-PcR
|
quantitative real-time reverse
transcription-poly-merase chain reaction
|
|
MALDI-TOF MS
|
matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry
|
|
5-aza-dC
|
5-aza-2′-deoxycytidine
|
|
ROC
|
receiver operating characteristic
|
|
TNM
|
tumor-node-metastasis
|
|
UICC
|
Union Internationale Contre le
Cancer
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu L, Zhao E, Li C, Huang L, Xiao L,
Cheng L, Huang X, Song Y and Xu D: TRIm28, a new molecular marker
predicting metastasis and survival in early-stage non-small cell
lung cancer. Cancer Epidemiol. 37:71–78. 2013. View Article : Google Scholar
|
|
3
|
Julien SG, Dubé N, Hardy S and Tremblay
ML: Inside the human cancer tyrosine phosphatome. Nat Rev Cancer.
11:35–49. 2011. View
Article : Google Scholar
|
|
4
|
Matozaki T, Suzuki T, Uchida T, Inazawa J,
Ariyama T, Matsuda K, Horita K, Noguchi H, Mizuno H, Sakamoto C, et
al: Molecular cloning of a human transmembrane-type protein
tyrosine phosphatase and its expression in gastrointestinal
cancers. J Biol Chem. 269:2075–2081. 1994.PubMed/NCBI
|
|
5
|
Matozaki T, Murata Y, Mori M, Kotani T,
Okazawa H and Ohnishi H: Expression, localization, and biological
function of the R3 subtype of receptor-type protein tyrosine
phosphatases in mammals. Cell Signal. 22:1811–1817. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sadakata H, Okazawa H, Sato T, Supriatna
Y, Ohnishi H, Kusakari S, Murata Y, Ito T, Nishiyama U, Minegishi
T, et al: SAP-1 is a microvillus-specific protein tyrosine
phosphatase that modulates intestinal tumorigenesis. Genes Cells.
14:295–308. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nagano H, Noguchi T, Inagaki K, Yoon S,
Matozaki T, Itoh H, Kasuga M and Hayashi Y: Downregulation of
stomach cancer-associated protein tyrosine phosphatase-1 (SAP-1) in
advanced human hepatocellular carcinoma. Oncogene. 22:4656–4663.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Noguchi T, Tsuda M, Takeda H, Takada T,
Inagaki K, Yamao T, Fukunaga K, Matozaki T and Kasuga M: Inhibition
of cell growth and spreading by stomach cancer-associated
protein-tyrosine phosphatase-1 (SAP-1) through dephosphorylation of
p130cas. J Biol Chem. 276:15216–15224. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takada T, Noguchi T, Inagaki K, Hosooka T,
Fukunaga K, Yamao T, Ogawa W, Matozaki T and Kasuga M: Induction of
apoptosis by stomach cancer-associated protein-tyrosine
phosphatase-1. J Biol Chem. 277:34359–34366. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heller G, Zielinski CC and
Zöchbauer-Müller S: Lung cancer: From single-gene methylation to
methylome profiling. Cancer Metastasis Rev. 29:95–107. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arai E, Chiku S, Mori T, Gotoh M, Nakagawa
T, Fujimoto H and Kanai Y: Single-CpG-resolution methylome analysis
identifies clinicopathologically aggressive CpG island methylator
phenotype clear cell renal cell carcinomas. Carcinogenesis.
33:1487–1493. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sato T, Arai E, Kohno T, Tsuta K, Watanabe
S, Soejima K, Betsuyaku T and Kanai Y: DNA methylation profiles at
precancerous stages associated with recurrence of lung
adenocarcinoma. PLoS One. 8:e594442013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sato T, Arai E, Kohno T, Takahashi Y,
Miyata S, Tsuta K, Watanabe S, Soejima K, Betsuyaku T and Kanai Y:
Epigenetic clustering of lung adenocarcinomas based on DNA
methylation profiles in adjacent lung tissue: Its correlation with
smoking history and chronic obstructive pulmonary disease. Int J
Cancer. 135:319–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jurinke C, Denissenko MF, Oeth P, Ehrich
M, van den Boom D and Cantor CR: A single nucleotide polymorphism
based approach for the identification and characterization of gene
expression modulation using MassARRAY. Mutat Res. 573:83–95. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan J, Upadhye S and Worster A:
Understanding receiver operating characteristic (ROC) curves. CJEM.
8:19–20. 2006.PubMed/NCBI
|
|
17
|
Seo Y, Matozaki T, Tsuda M, Hayashi Y,
Itoh H and Kasuga M: Overexpression of SAP-1, a transmembrane-type
protein tyrosine phosphatase, in human colorectal cancers. Biochem
Biophys Res Commun. 231:705–711. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Motiwala T, Majumder S, Kutay H, Smith DS,
Neuberg DS, Lucas DM, Byrd JC, Grever M and Jacob ST: Methylation
and silencing of protein tyrosine phosphatase receptor type O in
chronic lymphocytic leukemia. Clin Cancer Res. 13:3174–3181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Motiwala T, Kutay H, Ghoshal K, Bai S,
Seimiya H, Tsuruo T, Suster S, Morrison C and Jacob ST: Protein
tyrosine phosphatase receptor-type O (PTPRO) exhibits
characteristics of a candidate tumor suppressor in human lung
cancer. Proc Natl Acad Sci USA. 101:13844–13849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Veeriah S, Brennan C, Meng S, Singh B,
Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, et
al: The tyrosine phosphatase PTPRD is a tumor suppressor that is
frequently inactivated and mutated in glioblastoma and other human
cancers. Proc Natl Acad Sci USA. 106:9435–9440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van Doorn R, Zoutman WH, Dijkman R, de
Menezes RX, Commandeur S, Mulder AA, van der Velden PA, Vermeer MH,
Willemze R, Yan PS, et al: Epigenetic profiling of cutaneous T-cell
lymphoma: Promoter hypermethylation of multiple tumor suppressor
genes including BCL7a, PTPRG, and p73. J Clin Oncol. 23:3886–3896.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang JF and Dai DQ: metastatic suppressor
genes inactivated by aberrant methylation in gastric cancer. World
J Gastroenterol. 13:5692–5698. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oka T, Ouchida M, Koyama M, Ogama Y,
Takada S, Nakatani Y, Tanaka T, Yoshino T, Hayashi K, Ohara N, et
al: Gene silencing of the tyrosine phosphatase SHP1 gene by
aberrant methylation in leukemias/lymphomas. Cancer Res.
62:6390–6394. 2002.PubMed/NCBI
|
|
24
|
Koyama M, Oka T, Ouchida M, Nakatani Y,
Nishiuchi R, Yoshino T, Hayashi K, Akagi T and Seino Y: Activated
proliferation of B-cell lymphomas/leukemias with the SHP1 gene
silencing by aberrant CpG methylation. Lab Invest. 83:1849–1858.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Reddy J, Shivapurkar N, Takahashi T,
Parikh G, Stastny V, Echebiri C, Crumrine K, Zöchbauer-Müller S,
Drach J, Zheng Y, et al: Differential methylation of genes that
regulate cytokine signaling in lymphoid and hematopoietic tumors.
Oncogene. 24:732–736. 2005. View Article : Google Scholar
|
|
26
|
Yeh SH, Wu DC, Tsai CY, Kuo TJ, Yu WC,
Chang YS, Chen CL, Chang CF, Chen DS and Chen PJ: Genetic
characterization of fas-associated phosphatase-1 as a putative
tumor suppressor gene on chromosome 4q21.3 in hepatocellular
carcinoma. Clin Cancer Res. 12:1097–1108. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jacob ST and Motiwala T: Epigenetic
regulation of protein tyrosine phosphatases: Potential molecular
targets for cancer therapy. Cancer Gene Ther. 12:665–672. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wälchli S, Espanel X and Hooft van
Huijsduijnen R: Sap-1/PTPRH activity is regulated by reversible
dimerization. Biochem Biophys Res Commun. 331:497–502. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sakuraba J, Shintani T, Tani S and Noda M:
Substrate specificity of R3 receptor-like protein-tyrosine
phosphatase subfamily toward receptor protein-tyrosine kinases. J
Biol Chem. 288:23421–23431. 2013. View Article : Google Scholar : PubMed/NCBI
|