Introduction
Malignant melanoma is aggressive and
therapy-resistant (1), thus
advanced and metastatic melanoma is almost uniformly fatal.
Patients with advanced melanoma have a short median survival of 6–9
months (2). Melanoma is becoming
more common and its incidence has risen dramatically worldwide in
Caucasian populations (3,4).
It is known that melanoma cells become resistant to
chemotherapeutic agents and escape from death through inactivation
of apoptosis (5). Apoptosis is a
crucial event in the cytotoxicity induced by anticancer agents
(6). The role of apoptosis in the
cytotoxicity of anticancer agents has become more and more clear
(7). Apoptosis occurs via two
pathways, the death receptor-dependent (extrinsic) pathway or the
independent (intrinsic or mitochondrial) pathway (8). The mitochondrial pathway of apoptosis
is initiated by downregulation of anti-apoptotic proteins (Bcl-2
family members) and upregulation of pro-apoptotic proteins (Bax and
Bad). Several changes in mitochondrial biogenesis and function were
found to be associated with the commitment to intrinsic apoptosis.
The loss of mitochondrial transmembrane potential is responsible
for changes in mitochondrial biogenesis and activity resulting in
the opening of mitochondrial permeability transition pores (PTPs)
to release cytochrome c (Cyto c) which eventually
activates caspase-9 and caspase-3 and subsequent cell death.
Translocation of Cyto c has been considered as a crucial
step in the activation of the apoptotic machinery (9,10). On
the other hand, Cyto c release from mitochondria appears to
be largely mediated by direct or indirect reactive oxygen species
(ROS) action (10). The involvement
of calcium in cell death has been recognized very early in the
history of apoptosis. Elevation of cytosolic calcium both occurs at
early and late stages of the apoptotic pathways and is considered
to be the final common pathway of all types of cell death (11).
In previous studies, we reported that
N-acetyl-S-(p-chloro phenylcarbamoyl)cysteine
(NACC) and its analogs are a novel class of anticancer agents
(12). NACC was found to be the
most potent compound of this series against melanoma cells through
inducing apoptosis rather than affecting the cell cycle (12). However, how apoptosis is triggered
by NACC in melanoma cells remains unknown. The aims of the present
study were to investigate the events involved in NACC-induced
apoptosis and to elucidate the possible mechanism of action of NACC
as an apoptosis inducer in melanoma cells. In addition, in
vitro migration assays were performed to determine whether NACC
is able to inhibit the migration of melanoma cells.
Materials and methods
Materials
NACC was synthesized in our laboratory according to
a previously published method (12)
and prepared as a 100 mM stock solution in dimethylsulphoxide
(DMSO) for cell-based assays. Fetal bovine serum (FBS), RPMI-1640
growth medium, phosphate-buffered saline (PBS),
penicillin/streptomycin solution and 0.25% trypsin-EDTA solution
were purchased from Gibco Life Technologies (Grand Island, NY,
USA). Fluo-3AM probe, mitochondria isolating kit, ROS kit, JC-1 kit
and caspase-3, -8 and -9 kits were from Beyotime (Haimen, Jiangsu,
China). EDTA disodium salt, sodium chloride, sodium dodecyl sulfate
(SDS), glycine, 2-amino-2-(hydroxymethyl)propane-1,3-diol
hydrochloride (Tris-HCl), Tween-20 and phenylmethanesulphonyl
fluoride (PMSF) were purchased from Sigma-Aldrich Chemical Co.
(Milwaukee, WI, USA). BD Pharmingen™ fluorescein isothiocyanate
(FITC) Annexin V apoptosis detection kit I was from BD Biosciences
(San Jose, CA, USA). Antibodies against Bcl-2, Bax, Bad, Mcl-1,
Cyto c, p53, β-actin and horseradish peroxidase
(HRP)-conjugated secondary antibodies were purchased from Cell
Signaling Technology, Inc. (Beverly, MA, USA). Pierce®
ECL Plus kit was purchased from Lumigen, Inc. (Southfield, MI,
USA). Other reagents were obtained in their highest purity grade
available commercially.
Cell line and culture conditions
Human melanoma cell line UACC-62 was a kind gift
from Dr Xiangming Guan of South Dakota State University, USA. The
melanoma cells were cultured at 37°C in a humidified atmosphere of
5% CO2 in RPMI-1640 medium supplemented with 10% FBS,
100 U/ml of penicillin and 100 μg/ml of streptomycin.
Apoptotic assay by flow cytometry
UACC-62 cells were seeded at densities of 10,000 or
30,000 cells/well in 6-well plates for the 3- and 5-day treatment,
respectively. After a 24-h attachment at 37°C, UACC-62 cells were
treated with growth medium containing various concentrations of
NACC. After the 3- or 5-day treatment, the treated and untreated
cells were collected by trypsinization. Subsequently, Annexin
V/propidium iodide (PI) staining was performed using the BD
Pharmingen™ FITC Annexin V apoptosis detection kit I according to
the manufacturer's instruction. The samples were analyzed with a
Beckman Coulter flow cytometer.
Drug treatment for western blotting and
caspase activity assays
UACC-62 cells were seeded into 100-mm culture dishes
at densities of 1.5×105/dish and 4.5×105/dish
for the 3- and 5-day treatment, respectively. The cells were
allowed to attach for 24 h followed by treatment with either growth
medium with 0.025% DMSO as control or NACC (1, 5 and 25 μM)
for 3 or 5 days.
Determination of caspase activity
Activities of caspase-3, -8 and -9 were determined
using caspase-3, -8 and -9 colorimetric assay kits from Beyotime as
previously described by Cheng et al (13) with minor modifications. Briefly,
UACC-62 cells were treated with NACC for 3 days as described above.
The cells were collected and washed twice with PBS and then lysed
for 15 min on ice in lysis buffer. The lysates were centrifuged at
16,000 × g for 15 min at 4°C, and the supernatant was collected.
Lysates were incubated with caspase-3, -8 and -9 substrates
separately in reaction buffer at 37°C for 1 h and their absorbance
was recorded at 405 nm using a Multiskan Spectrum microplate reader
(Thermo Scientific). The protein concentrations of the lysates were
determined by the Bradford method.
Western blotting
The UACC-62 cells were treated with NACC as
described above and then lysed in RIPA lysis buffer. The protein
concentrations were determined by bicinchoninic acid (BCA) method.
Aliquots of 25–80 μg protein were fractionated in 12.5 and
15% sodium dodecyl sulfate polyacryl-amide gel (SDS-PAGE) under
reducing conditions and then transferred to polyvinylidene fluoride
(PVDF) membranes. After blocking with 5% non-fat milk, the
membranes were probed with the appropriate dilution of primary
antibodies followed by appropriate HRP conjugated secondary
antibody. The resulting conjugates were visualized using
Pierce® ECL Plus kit in a ChemiDoc XPS system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Release of Cyto c
The cell homogenate containing cytosolic Cyto
c released from mitochondria was generated by removing the
intact mitochondria using a mitochondria isolating kit following
the manufacturer's protocol. In brief, the UACC-62 cells were
treated with or without NACC as described above for western
blotting. The cells were harvested by trypsinization and rinsed
once with cold PBS. The samples were centrifuged at 600 × g for 5
min at 4°C and then the cell pellets were resuspended in
mitochondria isolation buffer containing 1 mM PMSF. The cells were
gently homogenized in a glass homogenizer avoiding breakage of the
mitochondria. The homogenate was centrifuged at 600 × g for 10 min
at 4°C and the supernatants were centrifuged again at 11,000 × g
for 10 min at 4°C. The supernatants containing the cytosolic
fraction were collected, and the protein concentration was
determined by the BCA assay. The release of the cytosolic Cyto
c was tested by western blotting using 25 μg protein
as obtained above.
Flow cytometric analysis of mitochondrial
membrane potential (Δψm)
JC-1 staining was performed to measure the changes
in in Δψm in UACC-62 cells. The cells were seeded at
densities of 16,000 or 48,000 cells/well in 6-well plates for the
3- and 5-day treatment, respectively. After a 24-h attachment, the
cells were treated with NACC (0, 1, 5 and 25 μM) for various
time periods. After the treatment, the cells were harvested by
trypsinization and incubated with JC-1 (5 μg/ml) in JC-1
staining buffer for 20 min at 37°C. Δψm was monitored by
determining the relative amounts of dual emissions from
mitochondrial JC-1 monomers (green) or aggregates (red) using a
Beckman Coulter flow cytometer. At high Δψm, JC-1 forms
aggregates with intense red fluorescence, and JC-1 remains in the
monomeric form at low Δψm, which shows green
fluorescence. For a positive control, the cells were incubated with
50 μM of the protonophore carbonyl cyanide
m-chlorophenylhydrazone (CCCP), for 30 min at 37°C followed
by JC-1 staining as described above.
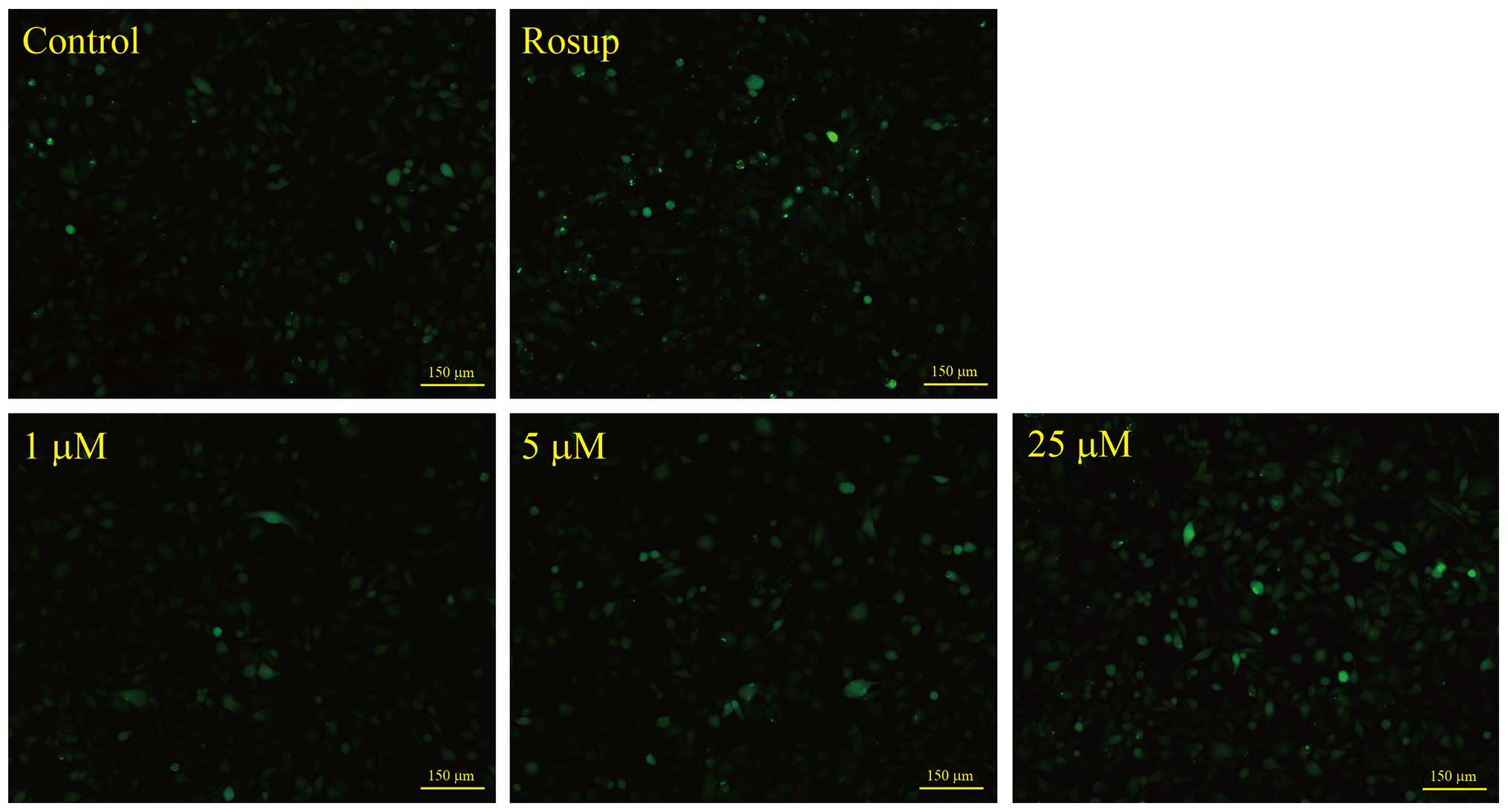
ROS production
ROS production induced by NACC was evaluated using
the cell-permeative probe DCFH-DA. Upon entry into the cytoplasm,
this probe is cleaved by cellular esterase and oxidized by ROS to
yield green fluorescence. Briefly, UACC-62 cells were plated in
12-well plates at a density of 60,000 cells/well. After a 24-h
attachment, the cells were treated with NACC at concentrations of
0, 1, 5 and 25 μM for 4 h followed by incubation with 10
μM DCFH-DA in FBS-free growth medium at 37°C for 20 min. For
a positive control, the cells were treated with 50 μM of
Rosup at 37°C for 20 min followed by DCFH-DA staining. The cells
were washed with PBS three times and then the green fluorescence
intensity was compared by the fluorescent images captured with a
Nikon Ti-S fluorescent microscope.
Microscopy analysis of cytosolic calcium
concentration in the UACC-62 cells
The cytosolic calcium concentrations were monitored
using Fluo-3 acetoxymethyl (AM) probe as described by Gong et
al (14). In brief, the
cultured UACC-62 cells at a density of 80,000 cells/well in a
12-well plate were treated with various concentrations of NACC for
6 h. The cells were then incubated in full growth medium containing
5 μM Fluo-3 AM at 37°C for 45 min. The probe-loaded cells
were rinsed with PBS for 3 times and the green fluorescent images
were captured using a Nikon Ti-S fluorescence microscope.
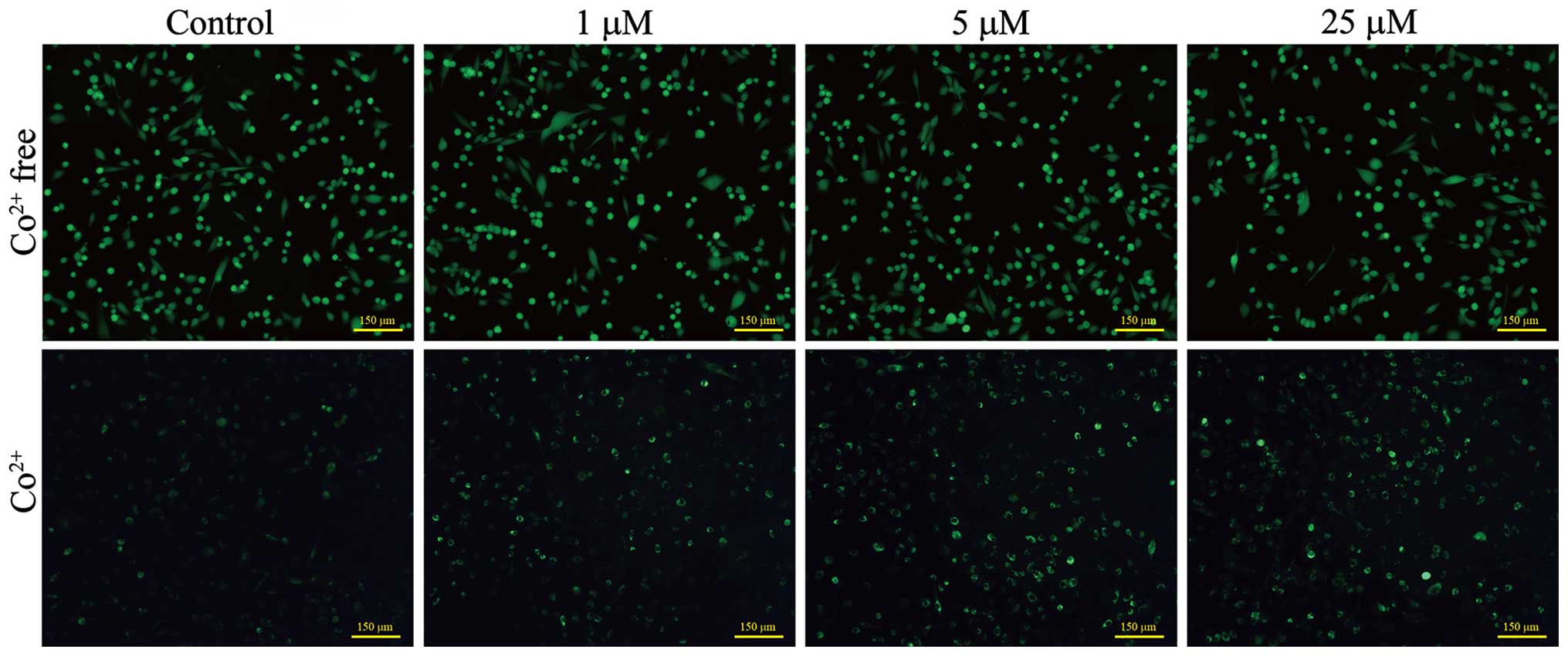
Mitochondrial PTP opening assay
PTP opening assay was performed using calcein-AM dye
as described by Gautier et al (15). In brief, UACC-62 cells were seeded
at densities of 8,000 or 24,000 cells/well in 12-well plates for
the 3- and 5-day treatment, respectively. After a 24-h attachment
at 37°C, the cells were treated with growth medium containing
various concentrations of NACC. After the 3- or 5-day treatment,
the cells were washed with Hank's Balanced Salt Solution (HBSS) 1X
and then loaded with calcein-AM (1 μM) with or without
CoCl2 (1 mM) both in HBSS 1X at 37°C for 20 min. The
cells were washed three times with HBSS 1X and the fluorescent
images were captured with a Nikon Ti-S fluorescence microscope.
Real-time monitoring of cellular
migration
The migration assays were performed on an
xCELLigence system from ACEA Biosciences Inc. (San Diego, CA, USA)
using CIM-16 plates. The experiments were conducted as described by
Eisenberg et al (16) with
minor modifications. In brief, the wells of the bottom chamber were
filled with 165 μl of medium containing 10% serum and then
the top and bottom portions of the plates were assembled together.
Serum-free media (30 μl) was added to the top chamber wells
and the CIM-16 plates were allowed to equilibrate for 1 h at 37°C
in a 5% CO2 incubator. The CIM-16 plates were then
placed into the RTCA-DP instrument for baseline test. The UACC-62
cells were harvested by trypsinization and seeded to the top
chamber wells at a density of 2,000 cells/well in 50 μl of
serum-free medium. Another 50 μl of serum-free medium
containing various concentrations of NACC was then added to the top
chamber wells. The CIM-16 plates were allowed to incubate at room
temperature for 30 min and placed into the RTCA-DP instrument for
data collection. In addition, the cytotoxicity of NACC in UACC-62
cells was determined by the same xCEL-Ligence system. In brief, the
UACC-62 cells were seeded into E-plate 16 plates at a density of
2,000 cells/well followed by NACC treatment. The xCELLigence
software was set to collect impedance data (cell index) every 15
min for 96 h.
Statistical analysis
Each experiment was performed in at least
triplicate. Data were analyzed with INSTAT software (Graph Pad, San
Diego, CA, USA). ANOVA followed by Tukey's post test was applied to
compare the statistical difference between the NACC-treated groups
and the untreated controls. P<0.05 was considered to indicate a
statistically significant difference. Values are expressed as mean
± the standard deviation of the mean.
Results
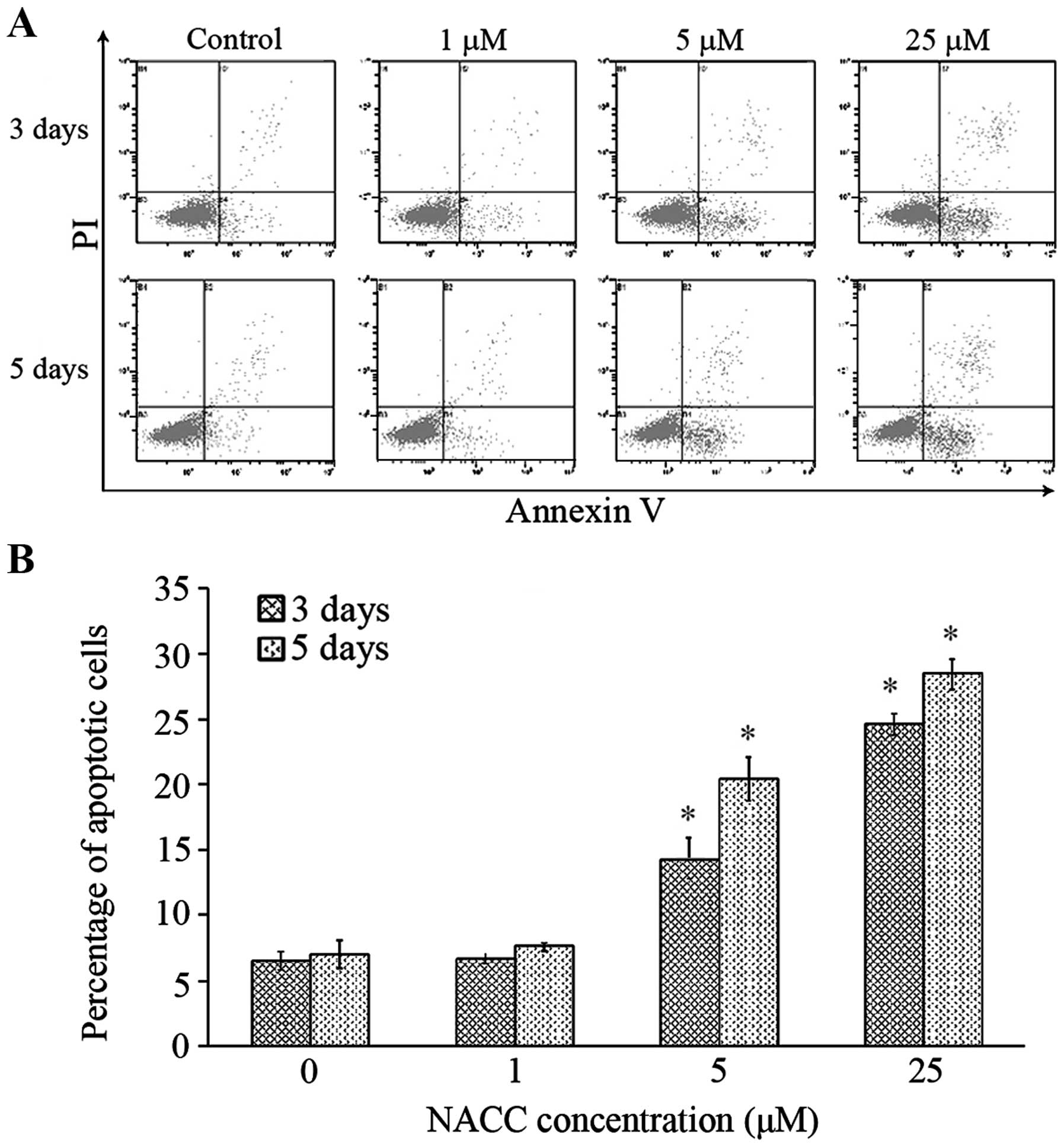
NACC induces apoptosis in UACC-62
cells
NACC was previously found to be able to induce
apoptosis in melanoma cells (12).
In the present study, NACC-induced apoptosis was investigated at
earlier treatment time points. FITC-conjugated Annexin V was
utilized to detected the externalization of phosphatidylserine
which is a marker of apoptosis. Combined with PI, an indicator of
cell membrane destruction, the cell population undergoing apoptosis
was quantified using flow cytometry. UACC-62 cells were treated
with different concentrations of NACC for 3 or 5 days, and then the
untreated and treated cells were collected followed by Annexin V/PI
staining. Flow cytometric analysis was performed to detect the
apoptosis in the UACC-62 cells induced by NACC. As shown in
Fig. 1, NACC induced significant
apoptosis in the UACC-62 cells following treatment with moderate (5
μM) and high (25 μM) concentrations of NACC in a
dose- and time-dependent manner. NACC at 5 μM and 25
μM concentrations induced 14.2 and 24.6% and 20.4 and 28.5%
apoptotic cells compared to that of 6.5 and 7.0% in the control
samples following the 3- and 5-day treatment, respectively. The
majority of the apoptotic cells were in the early stage of
apoptosis.
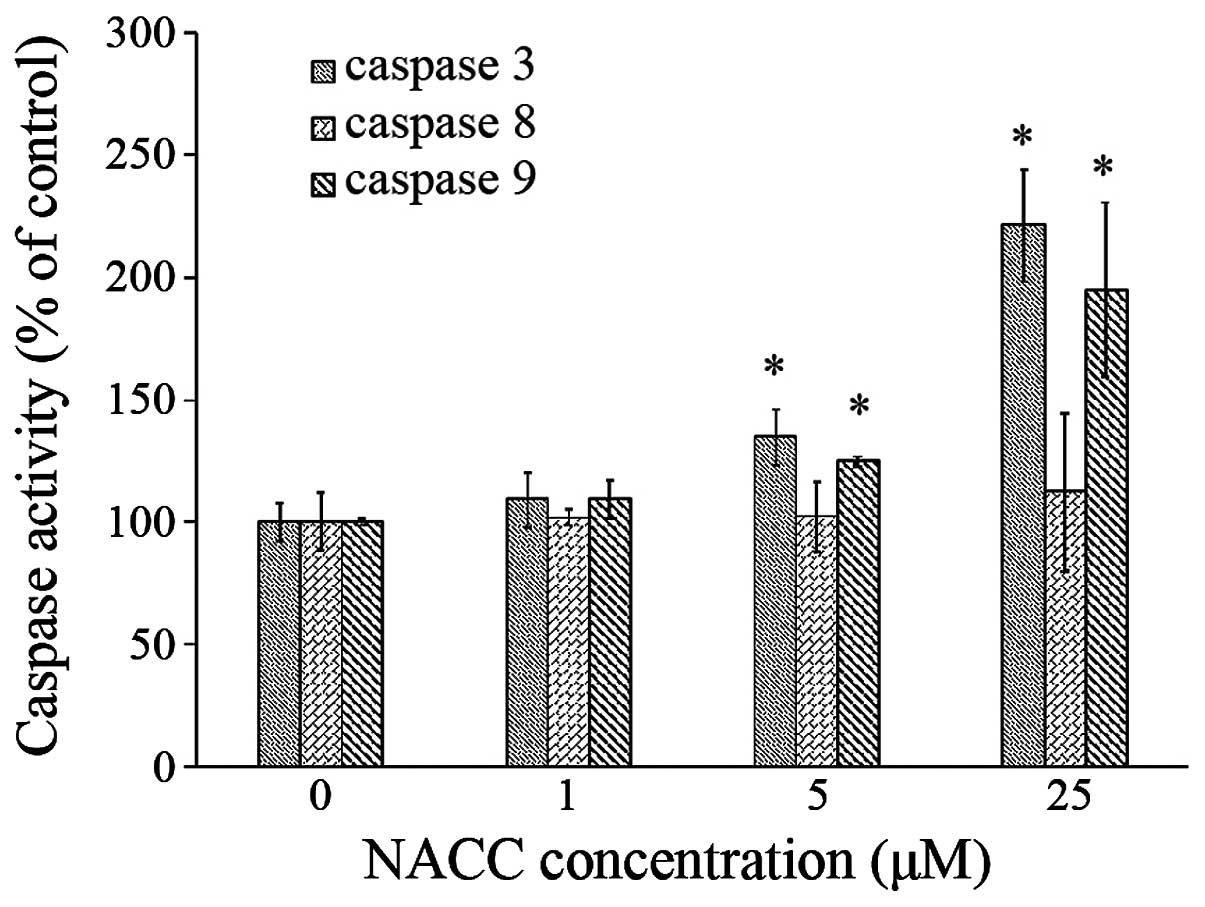
Caspase activity is affected by NACC
To examine whether NACC plays a role in the
activation of caspases and which apoptotic pathway is activated, we
performed spectrophotometric analysis of the UACC-62 cell lysate to
detect the activities of caspase-3, -8 and -9. The results showed
that NACC significantly increased the activities of caspase-3 and
caspase-9 in the melanoma cells in a dose-dependent manner but had
no effect on the activity of caspase-8 (Fig. 2). These results demonstrated that
the caspase cascade was activated during NACC-induced apoptosis and
only the intrinsic pathway was involved in the NACC-induced
apoptosis.
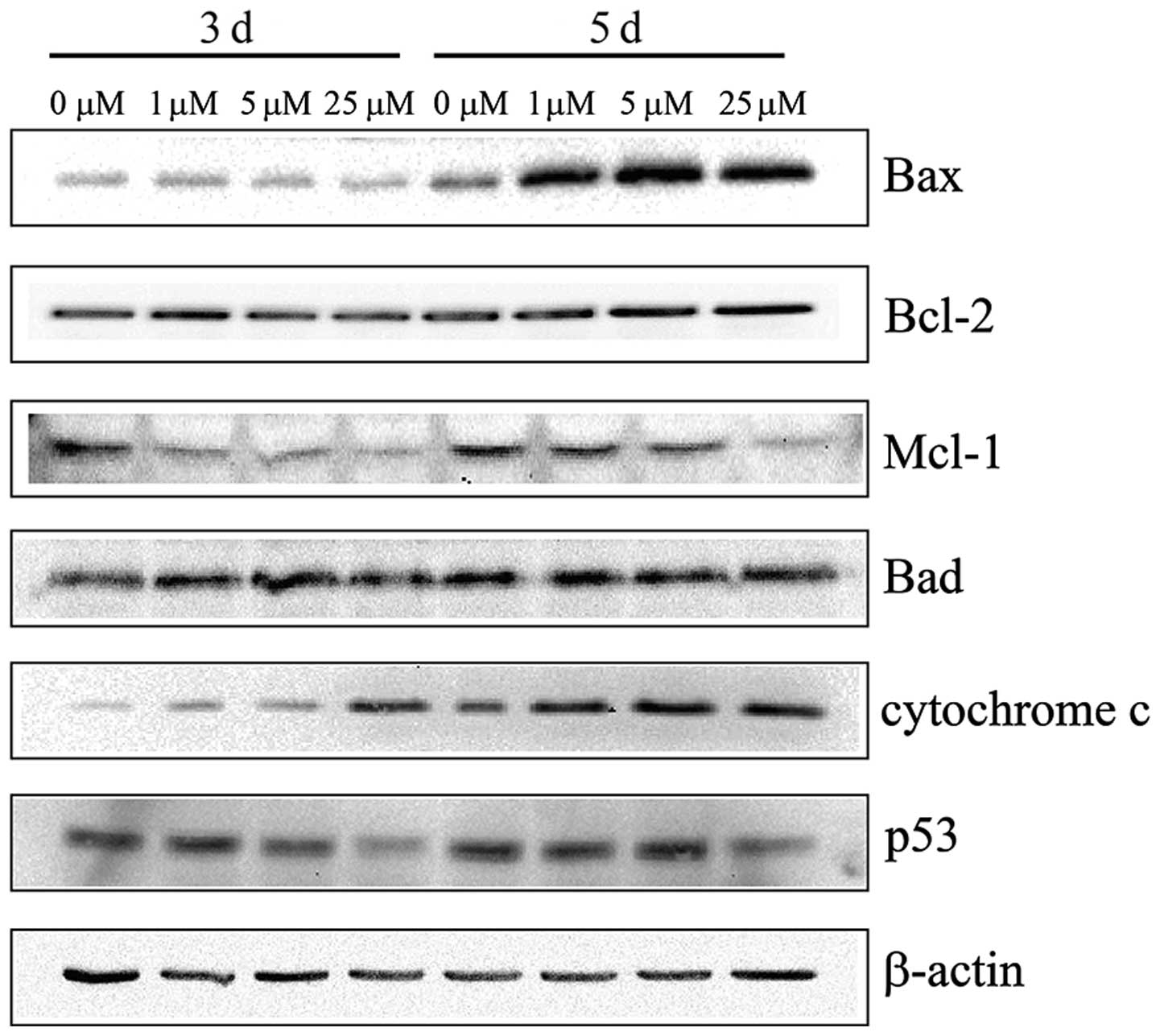
Western blotting
The apoptotic parameters, including protein levels
of Bax, Bcl-2, Bad, Mcl-1 and Cyto c were investigated by
western blotting to determine whether or not the intrinsic pathway
is involved in the apoptosis induction in UACC-62 cells by NACC
(Fig. 3). Bax and Bad, the
pro-apoptotic members of the Bcl-2 protein family, were
investigated in this study. The results showed that Bax expression
was upregulated following the 5-day treatment and that of Bad
remained unchanged. NACC treatment showed downregulation of Mcl-1
expression and did not affect the Bcl-2 level. The results also
showed that the level of cytosolic Cyto c increased in the
NACC-treated melanoma cells (Fig.
3). In addition, the protein expression of p53 was investigated
and the result showed that the p53 level remained unchanged
following both 3- and 5-day treatment with low (1 μM) and
medium (5 μM) concentrations of NACC. However, 25 μM
of NACC slightly downregulated p53 in the UACC-62 cells. Thus, the
increased Bax/Bcl-2 ratio and the downregulation of Mcl-1 were
associated with the induction of apoptosis in the NACC-treated
UACC-62 cells. As a result, Cyto c was released from the
mitochondria to the cytosol.
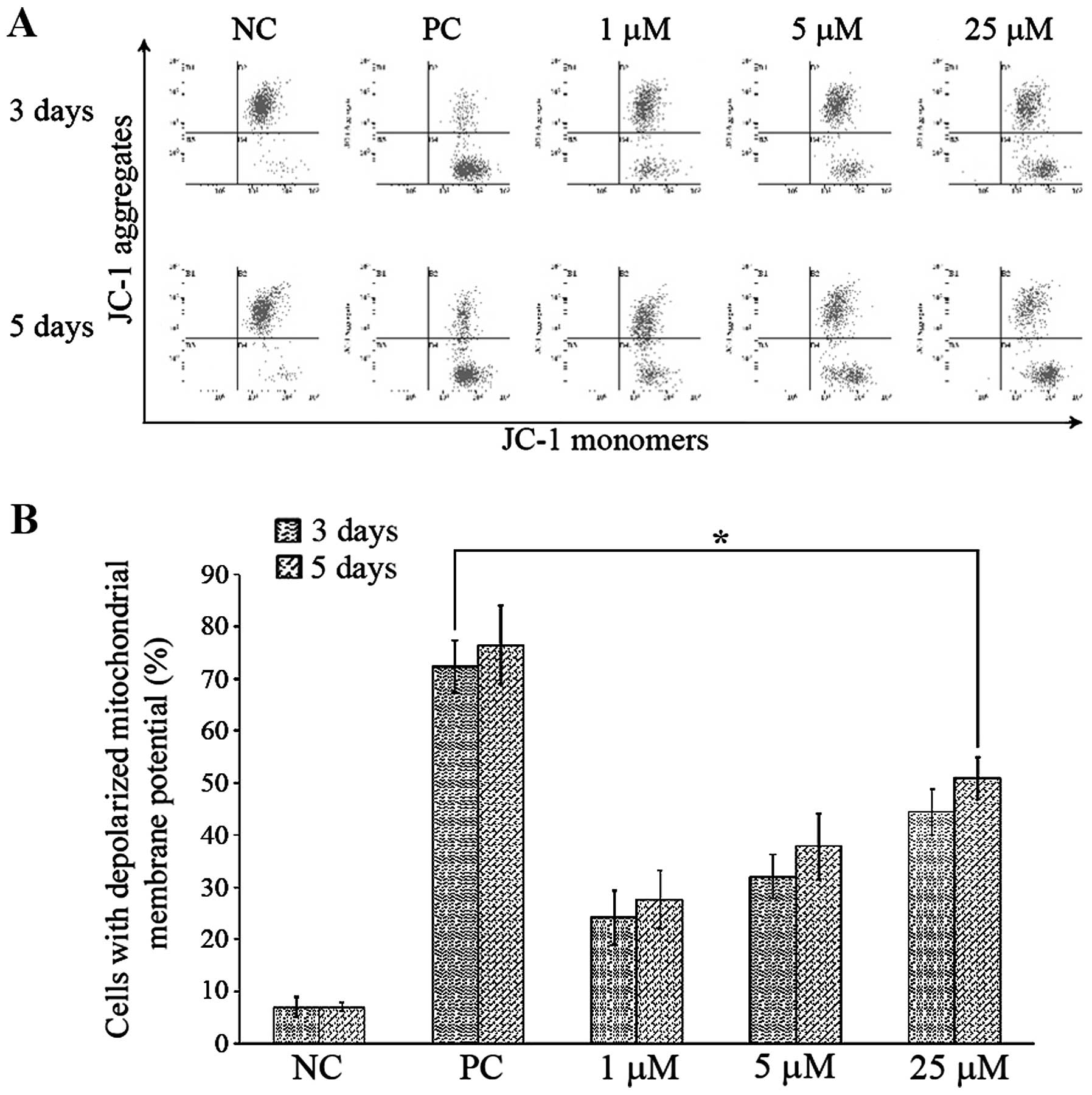
NACC induces mitochondrial membrane
potential (Δψm) depletion
A decline in Δψm may be an early event in
the process of cell death (6). JC-1
staining was performed to determine the changes in Δψm
induced by NACC. JC-1 is a cationic dye that exhibits
potential-dependent accumulation in mitochondria. During the loss
of Δψm, the fluorescence of the JC-1 dye shifts from red
to green. The increase in green fluorescence indicates the loss of
Δψm in the treated cells. Therefore, we studied the
status of Δψm in the NACC-treated UACC-62 cells by JC-1
with flow cytometric analysis. As shown in Fig. 4, NACC treatment induced a dose- and
time-dependent increase in the depletion of Δψm.
Concentrations of 1, 5 and 25 μM of NACC increased the
percentage of cells undergoing Δψm depolarization from
~7.0% in the negative controls to 24.2, 32.0 and 44.4%,
respectively following the 3-day treatment and 27.6, 38.8 and
50.9%, respectively, following the 5-day treatment. Positive
controls treated with 50 μM of CCCP were conducted to ensure
reliability of the analysis.
NACC increases ROS production in UACC-62
cells
To investigate whether ROS production is involved in
NACC-induced apoptosis in UACC-62 cells, the generation of ROS in
NACC-treated cells was probed by DCFH-DA fluorescent dye. The
intensity of the green fluorescence which reflected the
intracellular concentration of ROS was imaged by a fluorescence
microscope. As shown in Fig. 5,
NACC led to an increase in green fluorescence in NACC-treated
UACC-62 cells indicating an increased ROS generation by NACC. Based
on these observations, we concluded that NACC-induced apoptosis in
part involved the production of ROS in UACC-62 cells.
Cytosolic calcium elevation in
NACC-treated UACC-62 cells
To investigate whether NACC affects the cytosolic
calcium concentration in UACC-62 cells, the intracellular calcium
levels were probed by Fluo-3 AM. The cells were treated with NACC
for 6 h and then stained with Fluo-3 AM fluorescent dye. The
intensity of the green fluorescence which reflected the
intracellular free calcium concentrations was compared by the
images captured by a fluorescence microscope. As shown in Fig. 6 NACC led to an increase in the green
fluorescence in the NACC-treated UACC-62 cells indicating marked
elevation of cytosolic calcium concentration induced by NACC.
Mitochondrial PTP opening
Since Cyto c release and a loss of
mitochondrial membrane potential were observed in the NACC-treated
melanoma cells, we evaluated opening of the mitochondrial PTP,
which may regulate the Cyto c release across the
mitochondrial inner membrane (17,18).
We compared the opening of PTP in NACC-treated and untreated
melanoma cells using the calcein AM fluorescence assay. Calcein-AM
is a membrane permeable fluorophore that diffuses freely into all
subcellular compartments including mitochondria. The AM group of
the fluorophore is cleaved by ubiquitous intracellular esterase.
The hydrophilic calcein is then trapped within all subcellular
compartments. The cells were then loaded with Co2+,
which quenches calcein fluorescence in all subcellular compartments
except the mitochondrial matrix, since the inner mitochondrial
membrane is the only Co2+-impermeable intracellular
membrane (15,19). During the opening of the PTP,
Co2+ enters into mitochondria and quenches mitochondrial
calcein fluorescence (20).
Fig. 7 shows that the NACC
treatment did not induce a decrease in green fluorescence which
indicated that NACC did not trigger PTP opening in the UACC-62
cells.
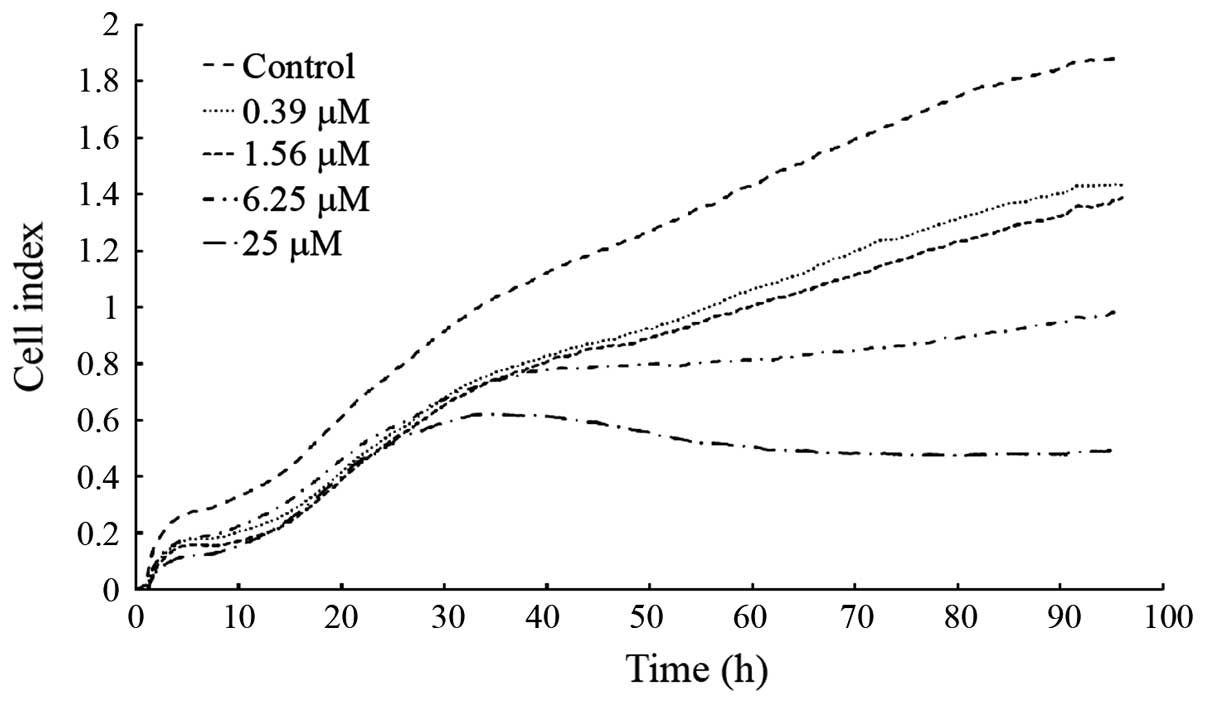
NACC suppresses the migration of UACC-62
cells in vitro
The migration assay in UACC-62 cells was performed
using an xCELLigence system. Cells seeded in the top chamber move
through the microporous membrane into the bottom chamber containing
FBS. Then the cells are able to adhere to the micro-electrode
sensors leading to an increase in cell index. The instrument
provides real-time and successive detection of the total migrated
cells. To ensure that the NACC-induced migration suppression was
not caused by the cytotoxicity of NACC, parallel experiments were
conducted in E-plate 16 plates instead of CIM-16 plates to
determine the cytotoxicity in the UACC-62 cells. The results showed
that the migratory ability of UACC-62 cells was significantly
suppressed in a dose- and time-dependent manner (Fig. 8). The cell index curves of the
control and NACC-treated cells separated at the very beginning of
the experiments. Among the curves of migration, 25 μM NACC
started to show inhibitory effects on migration at 25 h, and 6.25
μM NACC started to flatten the curve from ~40 h. The curves
for low concentrations (1.56 and 0.39 μM) started to
separate at ~50 h. The IC50 value of NACC on UACC-62
cell migration following the 96-h treatment was determined to be
4.93±2.64 μM based on three independent experiments. The
cytotoxicity experiments showed that NACC at 0.39, 1.56, 6.25 and
25 μM inhibited 2.89±3.9, 7.35±1.75, 13.38±0.58 and
19.94±0.8% cell growth compared to the control following the 96-h
treatment, respectively. Obviously, the cell growth inhibition
rates were much lower than that of the migration inhibition rates
induced by the same concentration of NACC, indicating that the
suppression of NACC-induced migration was not caused by
NACC-induced cytotoxicity.
Discussion
Malignant melanoma is well-known for its high
therapeutic resistance resulting in failed clinical trials. At
present, to the best of our knowledge, there are no efficacious
therapies for malignant melanoma, and defective apoptotic pathways
are thought to be one of the most important barriers in the
treatment of malignant melanoma (21,22).
NACC was found to be an apoptosis-inducer in melanoma cells. Flow
cytometric analysis revealed that NACC induced UACC-62 cell
apoptosis in a dose- and time-dependent manner (Fig. 1). To shed more light on the
apoptotic mechanism triggered by NACC, we investigated the
underlying molecular mechanisms of NACC-induced apoptosis in
melanoma cells.
It is well-known that the complex cascade of
caspases is involved in the process of apoptotic cell death
(23). Since caspases play
important roles in the apoptotic process, involvement of the
caspase cascade in NACC-induced UACC-62 cell apoptosis was
investigated. There are at least two major pathways, intrinsic and
extrinsic, initiated by caspase-8 and caspase-9, respectively. They
in turn activate the caspase cascade in cells (23). The activities of caspase-3, an
important effector caspase, and two initiator caspases, caspase-8
and caspase-9, were determined by photometric assays. The results
showed that caspase-3 and -9 were significantly activated in the
NACC-treated UACC-62 cells, however, the activity of caspase-8
remained unchanged. This indicated that only the intrinsic pathway
of apoptosis was activated by NACC.
The intrinsic apoptotic pathway also called the
mitochondrial apoptotic pathway is mediated by the interplay
between anti-apoptotic and pro-apoptotic members of the Bcl-2
family (24,25). Bcl-2 family proteins are thought to
play essential roles in the regulation of apoptotic execution of
cells (26). In this study, we
investigated the expression of Bcl-2 family proteins in UACC-62
cells treated with NACC. First of all, the expression levels of
Bcl-2 and Bax, the most important members of the Bcl-2 family, were
determined by western blotting in UACC-62 cells. The results showed
that NACC upregulated Bax, a representative of the pro-apoptotic
members (27), but did not affect
the expression of Bcl-2. Mcl-1, an anti-apoptotic member of the
Bcl-2 family which is extensively expressed in melanoma and
contributes to melanoma's well-documented drug resistance and poor
prognosis (28), was found to be
downregulated in the NACC-treated melanoma cells. p53, a tumor
suppressor, is at the center of an intricate protein network
determining important cellular responses including apoptosis
(29). The western blotting results
showed that the wild-type p53 carried in the UACC-62 cells
(30) was not found to be
upregulated by NACC treatment indicating that NACC-induced
apoptosis was p53-independent. All of these results suggest that
NACC-induced apoptosis was initiated by an increased ratio of
Bax/Bcl-2 and downregulation of Mcl-1. Moreover, another
pro-apoptotic member Bad was found unaltered in the NACC-induced
apoptosis. Accumulation of pro-apoptotic proteins on the
mitochondrial outer membrane changes the mitochondrial membrane
permeability and release of Cyto c from the mitochondria to
the cytoplasm. Cyto c is a small hemeprotein normally
absorbed at the outer aspect of the inner membrane of mitochondria
and is an essential component of the electron transport chain. Cyto
c activates the caspase-dependent or -independent apoptotic
pathways and plays a central role in the caspase-dependent
mitochondrial-mediated apoptotic pathway (17,31,32).
The results of western blotting (Fig.
3) showed that Cyto c was released to the cytosol in the
UACC-62 cells treated with NACC as a consequence of the regulation
of Bax and Mcl-1 expression by NACC.
Mitochondria play a critical role in the regulation
of apoptosis. The disruption of Δψm can mediate
liberation of Cyto c into the cytosol (33). As shown in Fig. 4, NACC treatment caused depletion of
Δψm, and the percentage of the cells with depolarized
mitochondria exposed to 1, 5 and 25 μM of NACC increased
from 7.0 to 24.2%, 32.0 and 44.4% following a 3-day treatment and
from 6.9 to 27.6%, 37.8 and 50.9% following a 5-day treatment,
respectively. Mitochondria are also known as the major
intracellular source of ROS. ROS participate as effective factors
in apoptosis by disruption of mitochondrial membrane potential
(34). When the generation of ROS
is out of control, ROS are capable of inducing apoptosis (35). Increased ROS have been found to be
implicated in the induction of apoptosis by promoting the release
of Cyto c to facilitate caspase cascade activation (36). Changes in the level of ROS in
UACC-62 cells were further assessed. The results showed that NACC
generated ROS in UACC-62 cells. ROS generation could precede the
depolarization of the mitochondrial membrane (37), which is one of the earliest events
of apoptosis. We observed an increase in ROS production after a 4-h
exposure of melanoma cells to NACC suggesting that the generation
of ROS is an early event in NACC activity in treated melanoma
cells. ROS generation by NACC could be one of the factors
responsible for the disruption of mitochondrial membrane potential
in the melanoma cells. Recently, we found that NACC is an
irreversible inhibitor which is capable of inhibiting intracellular
thioredoxin reductase (TrxR) in UACC-62 cells (38). It was reported by Fang et al
(39) that TrxR inhibitor is able
to induce ROS production in the presence of oxygen. Therefore, the
observed ROS generation by NACC might be a consequence of the
inhibition of intracellular TrxR in UACC-62 cells.
Sulofenur, the parent compound of NACC (40), was reported to be able to alter the
mitochondrial membrane potential by elevation of cytosolic calcium
concentration in human colon cancer cells (41). The cytosolic calcium level was
assessed in the melanoma cells exposed to NACC for 6 h. A dramatic
increase in cytosolic calcium was observed in the UACC-62 cells
indicating that NACC had the same ability as sulofenur in the
elevation of cytosolic calcium (Fig.
6). Mitochondrial membrane permeabilization is considerate as
an important hallmark of early apoptosis. Elevation of the
cytosolic calcium concentration may play a role in induction of PTP
(42). Mitochondrial permeability
transition is thought to be regulated by the opening of the PTP
(43). However, the role of PTP in
the mitochondrial pathway of apoptosis is still controversial
(42,44). In the present study, opening of the
PTP was assessed in the UACC-62 cells treated with NACC. Notably,
the results showed that NACC did not induce the opening of the PTP
(Fig. 7). PTP putatively consists
of the voltage dependent anion channel (VDAC) in the outer
mitochondrial membrane (OMM), the adenine nucleotide translocase
(ANT) in the inner mitochondrial membrane (IMM) and a mitochondrial
matrix protein cyclophilin D (CyP-D) (44). It has been reported that the
pro-apoptotic protein Bax is able to interact with VDAC and ANT to
accelerate the opening of PTP (42,44,45).
Yet, the PTP opening was not triggered by Bax upregulation in the
UACC-62 cells. It is known that Bax can be activated to form pores
in the OMM and dictate mitochondrial outer membrane
permeabilization (MOMP) which triggers Cyto c release from
the intramembrane space and subsequent apoptosis (46). Our results showed that in the
UACC-62 cells, NACC induced Cyto c release via MOMP by
upregulated Bax and was not PTP-dependent. The results were
consistent with a previous report which found that the regulatory
release of Cyto c from mitochondria could occur
independently through regulation of PTP opening (47).
Melanomas are highly metastatic resulting in poor
therapeutic outcomes and a low survival rate (48). Thus, suppression of cancer cell
metastasis is one of the key factors in cancer therapy. Migration
is one of the key steps of metastasis. In the present study, we
investigated the suppressive ability of NACC against the migration
of the UACC-62 cell line. The IC50 value of NACC on the
migration in UACC-62 cells following a 96-h treatment was
determined to be 4.93±2.64 μM implying that NACC has
promising anti-metastatic activity against melanoma cells. The
thioredoxin system has been shown to mediate the ability of cell
migration and tissue invasion of melanoma (49,50).
As previously mentioned, NACC is an irreversible inhibitor of TrxR
and is capable of inhibiting intracellular TrxR. Therefore,
inhibition of the activity of TrxR which is the major component of
the thioredoxin system, may contribute to the suppressive effect of
NACC on the migration in UACC-62 cells. How the thioredoxin system
is involved in tumor cell migration and invasion is not yet clear.
The impacts of NACC on other steps of metastasis and its mechanism
of action will be further investigated in future studies.
In conclusion, NACC has the capability of inducing
mitochondrial-mediated apoptosis, which is well regulated by the
caspase enzymes. Moreover, the active role of mitochondria in
NACC-induced programmed cell death was confirmed by depletion of
the mitochondrial membrane potential, release of Cyto c,
generation of ROS, cytosolic calcium elevation and regulation of
pro-apoptotic and anti-apoptotic proteins (Bax/Bcl-2 and Mcl-1).
The results suggest that NACC is a promising anti-melanoma agent
that is not only capable of triggering apoptosis via the
mitochondrial-dependent pathway, but is also able to suppress the
migration of melanoma cells.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (81202553), Medical and Health
Science Foundation of Zhejiang Province (2012KYA034) and Zhejiang
Key Laboratory of Diagnosis and Treatment Technology on Thoracic
Oncology (Lung and Esophagus).
References
|
1
|
Markovic SN, Erickson LA, Rao RD, Weenig
RH, Pockaj BA, Bardia A, Vachon CM, Schild SE, McWilliams RR, Hand
JL, et al Melanoma Study Group of the Mayo Clinic Cancer Center:
Malignant melanoma in the 21st century, part 1: Epidemiology, risk
factors, screening, prevention, and diagnosis. Mayo Clin Proc.
82:364–380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gogas HJ, Kirkwood JM and Sondak VK:
Chemotherapy for metastatic melanoma: Time for a change? Cancer.
109:455–464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bergomi M, Pellacani G, Vinceti M,
Bassissi S, Malagoli C, Alber D, Sieri S, Vescovi L, Seidenari S
and Vivoli R: Trace elements and melanoma. J Trace Elem Med Biol.
19:69–73. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Vries E, Houterman S, Janssen-Heijnen
ML, Nijsten T, van de Schans SA, Eggermont AM and Coebergh JW:
Up-to-date survival estimates and historical trends of cutaneous
malignant melanoma in the south-east of The Netherlands. Ann Oncol.
18:1110–1116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang J, Amiri KI, Burke JR, Schmid JA and
Richmond A: BMS-345541 targets inhibitor of kappaB kinase and
induces apoptosis in melanoma: Involvement of nuclear factor kappaB
and mitochondria pathways. Clin Cancer Res. 12:950–960. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu WP, Yu HS, Sung PJ, Tsai FY, Shen YK,
Chang LS and Wang JJ: DC-81-Indole conjugate agent induces
mitochondria mediated apoptosis in human melanoma A375 cells. Chem
Res Toxicol. 20:905–912. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim R: Recent advances in understanding
the cell death pathways activated by anticancer therapy. Cancer.
103:1551–1560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jana S, Paul S and Swarnakar S: Curcumin
as anti-endometriotic agent: Implication of MMP-3 and intrinsic
apoptotic pathway. Biochem Pharmacol. 83:797–804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang X and Wang X: Cytochrome c-mediated
apoptosis. Annu Rev Biochem. 73:87–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rizzuto R, Pinton P, Ferrari D, Chami M,
Szabadkai G, Magalhães PJ, Di Virgilio F and Pozzan T: Calcium and
apoptosis: Facts and hypotheses. Oncogene. 22:8619–8627. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen W, Seefeldt T, Young A, Zhang X and
Guan X: Design, synthesis and biological evaluation of
N-acetyl-S-(p-chloro phenylcarbamoyl)cysteine and its analogs as a
novel class of anticancer agents. Bioorg Med Chem. 19:287–294.
2011. View Article : Google Scholar :
|
|
13
|
Cheng J, Tang W, Su Z, Guo J, Tong L and
Wei Q: Calcineurin subunit B promotes TNF-alpha-induced apoptosis
by binding to mitochondria and causing mitochondrial
Ca2+ overload. Cancer Lett. 321:169–178. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gong YY, Si XM, Lin L and Lu J: Mechanisms
of cholecystokinin-induced calcium mobilization in gastric antral
interstitial cells of Cajal. World J Gastroenterol. 18:7184–7193.
2012. View Article : Google Scholar
|
|
15
|
Gautier CA, Giaime E, Caballero E, Núñez
L, Song Z, Chan D, Villalobos C and Shen J: Regulation of
mitochondrial permeability transition pore by PINK1. Mol
Neurodegener. 7:222012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eisenberg MC, Kim Y, Li R, Ackerman WE,
Kniss DA and Friedman A: Mechanistic modeling of the effects of
myoferlin on tumor cell invasion. Proc Natl Acad Sci USA.
108:20078–20083. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ichas F and Mazat JP: From calcium
signaling to cell death: Two conformations for the mitochondrial
permeability transition pore. Switching from low- to
high-conductance state. Biochim Biophys Acta. 1366:33–50. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garrido C, Galluzzi L, Brunet M, Puig PE,
Didelot C and Kroemer G: Mechanisms of cytochrome c release from
mitochondria. Cell Death Differ. 13:1423–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Petronilli V, Miotto G, Canton M, Colonna
R, Bernardi P and Di Lisa F: Imaging the mitochondrial permeability
transition pore in intact cells. Biofactors. 8:263–272. 1998.
View Article : Google Scholar
|
|
20
|
Hüser J, Rechenmacher CE and Blatter LA:
Imaging the permeability pore transition in single mitochondria.
Biophys J. 74:2129–2137. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsao H, Chin L, Garraway LA and Fisher DE:
Melanoma: From mutations to medicine. Genes Dev. 26:1131–1155.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Wu D, Shen J, Zhou M and Lu Y:
Rapamycin induces autophagy in the melanoma cell line M14 via
regulation of the expression levels of Bcl-2 and Bax. Oncol Lett.
5:167–172. 2013.
|
|
23
|
Sun J, Yu CH, Zhao XL, Wang Y, Jiang SG
and Gong XF: Econazole nitrate induces apoptosis in MCF-7 cells via
mitochondrial and caspase pathways. Iran J Pharm Res. 13:1327–1334.
2014.
|
|
24
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar
|
|
25
|
Bi MC, Rosen R, Zha RY, McCormick SA, Song
E and Hu DN: Zeaxanthin induces apoptosis in human uveal melanoma
cells through Bcl-2 family proteins and intrinsic apoptosis
pathway. Evid Based Complement Alternat Med. 2013:2050822013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsumoto A, Isomoto H, Nakayama M,
Hisatsune J, Nishi Y, Nakashima Y, Matsushima K, Kurazono H, Nakao
K, Hirayama T, et al: Helicobacter pylori VacA reduces the cellular
expression of STAT3 and pro-survival Bcl-2 family proteins, Bcl-2
and Bcl-XL, leading to apoptosis in gastric epithelial cells. Dig
Dis Sci. 56:999–1006. 2011. View Article : Google Scholar
|
|
27
|
Robertson JD and Orrenius S: Molecular
mechanisms of apoptosis induced by cytotoxic chemicals. Crit Rev
Toxicol. 30:609–627. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Y, Xie M, Song T, Sheng H, Yu X and
Zhang Z: A novel BH3 mimetic efficiently induces apoptosis in
melanoma cells through direct binding to anti-apoptotic Bcl-2
family proteins, including phosphorylated Mcl-1. Pigment Cell
Melanoma Res. 28:161–170. 2015. View Article : Google Scholar
|
|
29
|
Amaral JD, Xavier JM, Steer CJ and
Rodrigues CM: The role of p53 in apoptosis. Discov Med. 9:145–152.
2010.PubMed/NCBI
|
|
30
|
Bykov VJ, Issaeva N, Selivanova G and
Wiman KG: Mutant p53-dependent growth suppression distinguishes
PRIMA-1 from known anticancer drugs: A statistical analysis of
information in the National Cancer Institute database.
Carcinogenesis. 23:2011–2018. 2002. View Article : Google Scholar
|
|
31
|
Pradelli LA, Bénéteau M and Ricci JE:
Mitochondrial control of caspase-dependent and -independent cell
death. Cell Mol Life Sci. 67:1589–1597. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luo Y, Li X, Huang X, Wong YS, Chen T,
Zhang Y and Zheng W: 1,4-Diselenophene-1,4-diketone triggers
caspase-dependent apoptosis in human melanoma A375 cells through
induction of mitochondrial dysfunction. Chem Pharm Bull (Tokyo).
59:1227–1232. 2011. View Article : Google Scholar
|
|
34
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Su YT, Chang HL, Shyue SK and Hsu SL:
Emodin induces apoptosis in human lung adenocarcinoma cells through
a reactive oxygen species-dependent mitochondrial signaling
pathway. Biochem Pharmacol. 70:229–241. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kagan VE, Tyurin VA, Jiang J, Tyurina YY,
Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V,
et al: Cytochrome c acts as a cardiolipin oxygenase required for
release of proapoptotic factors. Nat Chem Biol. 1:223–232. 2005.
View Article : Google Scholar
|
|
37
|
Rogalska A, Koceva-Chyła A and Jóźwiak Z:
Aclarubicin-induced ROS generation and collapse of mitochondrial
membrane potential in human cancer cell lines. Chem Biol Interact.
176:58–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen W, Jiang Z, Lin N, Zheng Z, Chen Z,
Zhang X and Guan X: Evaluation of
N-acetyl-S-(p-chlorophenylcarbamoyl)cysteine as an irreversible
inhibitor of mammalian thioredoxin reductase1. J Enzyme Inhib Med
Chem. 17:1–7. 2015.
|
|
39
|
Fang J, Lu J and Holmgren A: Thioredoxin
reductase is irreversibly modified by curcumin: A novel molecular
mechanism for its anticancer activity. J Biol Chem.
280:25284–25290. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guan X, Hoffman BN, McFarland DC,
Gilkerson KK, Dwivedi C, Erickson AK, Bebensee S and Pellegrini J:
Glutathione and mercapturic acid conjugates of sulofenur and their
activity against a human colon cancer cell line. Drug Metab Dispos.
30:331–335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Phelps PC, Jain PT, Berezesky IK, Boder GB
and Trump BF: Sulofenur cytotoxicity and changes in cytosolic
calcium and mitochondrial membrane potential in human colon
adenocarcinoma cell lines. Cancer Lett. 88:27–35. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gerasimenko JV, Gerasimenko OV, Palejwala
A, Tepikin AV, Petersen OH and Watson AJ: Menadione-induced
apoptosis: Roles of cytosolic Ca(2+) elevations and the
mitochondrial permeability transition pore. J Cell Sci.
115:485–497. 2002.PubMed/NCBI
|
|
43
|
Nakagawa T, Shimizu S, Watanabe T,
Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T and Tsujimoto Y:
Cyclophilin D-dependent mitochondrial permeability transition
regulates some necrotic but not apoptotic cell death. Nature.
434:652–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Suh DH, Kim MK, Kim HS, Chung HH and Song
YS: Mitochondrial permeability transition pore as a selective
target for anti-cancer therapy. Front Oncol. 3:412013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marzo I, Brenner C, Zamzami N,
Jürgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama
S, Reed JC, et al: Bax and adenine nucleotide translocator
cooperate in the mitochondrial control of apoptosis. Science.
281:2027–2031. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chipuk JE, Bouchier-Hayes L and Green DR:
Mitochondrial outer membrane permeabilization during apoptosis: The
innocent bystander scenario. Cell Death Differ. 13:1396–1402. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Toriumi K, Oma Y, Mimoto A, Futai E,
Sasagawa N, Turk B and Ishiura S: Polyalanine tracts directly
induce the release of cytochrome c, independently of the
mitochondrial permeability transition pore, leading to apoptosis.
Genes Cells. 14:751–757. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bonaventure J, Domingues MJ and Larue L:
Cellular and molecular mechanisms controlling the migration of
melanocytes and melanoma cells. Pigment Cell Melanoma Res.
26:316–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mahmood DF, Abderrazak A, El Hadri K,
Simmet T and Rouis M: The thioredoxin system as a therapeutic
target in human health and disease. Antioxid Redox Signal.
19:1266–1303. 2013. View Article : Google Scholar
|
|
50
|
Arnér ES and Holmgren A: The thioredoxin
system in cancer. Semin Cancer Biol. 16:420–426. 2006. View Article : Google Scholar : PubMed/NCBI
|