Introduction
Lung cancer is the leading cause of cancer-related
mortality in the United States and one of the most common types of
cancer worldwide (1). The average
5-year lung cancer survival is among the poorest (17%) of all types
of cancer (2). The incidence of
lung cancer increases with age as well as with cumulative exposure
to tobacco smoke (2). Smoking is
the most important risk factor, accounting for ~85% of all US lung
cancer mortality (2). Smoking
cessation is clearly the first step in reducing lung cancer risk,
however, former smokers still carry a significant risk and most
lung cancer cases are now diagnosed in former smokers (3). Late intervention by targeting the
process of lung carcinogenesis is one of the few strategies in
chemoprevention studies to reduce the burden of lung cancer in
former smokers or individuals with high cancer risk (4).
Previous findings have shown that nicotine and its
metabolite, the tobacco-specific carcinogen
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), activate
protein kinase B (Akt) and mammalian target of rapamycin (mTOR) in
murine lung lesions and in primary human bronchial and small airway
epithelial cells (5–7). Activation of Akt/mTOR signaling is a
poor prognostic factor in non-small cell lung cancer (NSCLC)
patients. The phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathway
has, thus, become an important target for cancer therapy (8). It has been shown that PI3K activates
Akt, which, in turn, phosphorylates downstream target molecules
that include fork-head transcription factor (FKHR) and mTOR
(9,10). mTOR, a serine-threonine kinase that
regulates many cell functions including proliferation, survival and
protein translation, has emerged as a significant target for
anticancer therapy in various types of cancer (11). mTOR forms two different cell
complexes: mTOR complex 1 (mTORC1) (12–14)
and mTOR complex 2 (mTORC2), which can be activated by various
stimuli (growth factors, hormones and metabolic stress). The
activity of these complexes depends on the phosphorylation status
of mTOR (at Ser-2448). The involvement of PI3K/AKT in the
regulation of mTOR activity was further suggested by findings of
Bjornsti et al and Sekulić et al (9,13) in
in vitro and in vivo studies wherein mTOR was
phosphorylated at specific amino acid residues. Activated mTOR
phosphorylates key translational regulators such as p70 S6 kinase
(p-S6K1), eukaryotic translation initiation factor 4E-binding
protein (4EBP1) and subsequently, ribosomal protein S6 (14). Activated S6K1 stimulates ribosome
biogenesis, which upregulates the translational capacity of the
tumor cell. These downstream molecules, therefore, serve as
rational pharmacological targets; and targeting the Akt/mTOR
pathway may be a viable approach for lung cancer treatment or
chemoprevention. Chemoprevention, with administration of compounds
that inhibit, retard or reverse the process of carcinogenesis, are
an effective approach in reducing the risk for development or
progression of lung cancer (15).
Thus, rapamycin and its analogs (rapalogs), which inhibit mTOR, are
promising cancer chemopreventive agents with demonstrated antitumor
effects in various types of cancer (11).
As a downstream effector of PI3K/mTOR, Akt is
activated constitutively in many types of human tumors, including
lung cancer (8,9). The activation of Akt was associated
with disease progression in tumors and regulates cell survival by
increasing the level of anti-apoptotic proteins including Bcl-2 and
Bcl-xL and the inactivation of pro-apoptotic proteins, such as Bad
and caspase-9 (10). The activation
of Bcl-2 can be regulated by the post-translational phosphorylation
of Akt, mTOR and p70S6K (16,17).
The pro-survival Bcl-2 family members, including the anti-apoptotic
Bcl-2 and Bcl-xL, and pro-apoptotic members such as Bax, are
important regulators of apoptosis (10,18).
Autophagy is another important regulatory mechanism involving the
formation of double-membrane autophagosome vesicles, which engulf
cytoplasmic constituents and their subsequent delivery to the lytic
compartment to potentiate cell death or cell survival mechanisms in
response to several stresses (19).
Apoptosis is considered to be a type of programmed cell death type
I, whereas autophagic cell death is considered to be programmed
cell death type II or non-apoptotic death. Functional cross-talk
between apoptotic and autophagy forms of cell death, determined by
the molecules and pathways activated during the two events have
been previously reported (20,21).
Previous findings suggest that Beclin-1, which is a key regulatory
molecule in autophagy that binds to the Bcl-2 family members, has a
direct role in regulating apoptotic signaling (20,22).
The expression of Beclin-1 is significantly decreased in lung
cancer tissues, suggesting that autophagy is involved in the
pathogenesis of lung cancer (23).
However, the effect of rapamycin on the progression
from lung adenoma to adenocarcinoma and its molecular mechanism(s)
has not been studied sufficiently. The purpose of the present study
was to evaluate whether early or delayed intervention with
rapamycin provided improved chemo-preventive efficacy against
NNK-induced lung adenoma and adenocarcinoma formation in female A/J
mice with favorable pharmaceutical properties and to understand the
molecular mechanisms associated with lung tumor inhibition. The A/J
mouse model that is widely used in evaluating chemopreventive
efficacy was selected (4,24) and the impact of rapamycin during
different stages of NNK-induced lung carcinogenesis was
investigated by assessing MAP LC3α/β to monitor autophagy.
Additionally, the functional relationship between the Bcl-2 family
and mTOR under apoptotic conditions was examined in NNK-induced
lung carcinogenesis.
Materials and methods
Animals, diets, chemopreventive agents,
chemicals and reagents
All animal experiments were performed in accordance
with the National Institute of Health (NIH) guidelines and the
University of Oklahoma Health Sciences Center Institutional Animal
Care and Use Committee approved protocol (IACUC#09-067B). Female
A/J mice were obtained at 6 weeks of age from the Jackson
Laboratory (Bar Harbor, ME, USA). Ingredients for the semi-purified
diets were purchased from Bioserv (Frenchtown, NJ, USA) and stored
at 4°C prior to diet preparation. Diets were based on the modified
American Institute of Nutrition 76A (AIN-76A) diet containing 20%
casein, 52% corn starch, 13% dextrose, 5% corn oil, 5% alphacel,
3.5% AIN mineral mix, 1.2% AIN revised vitamin mix, 0.3%
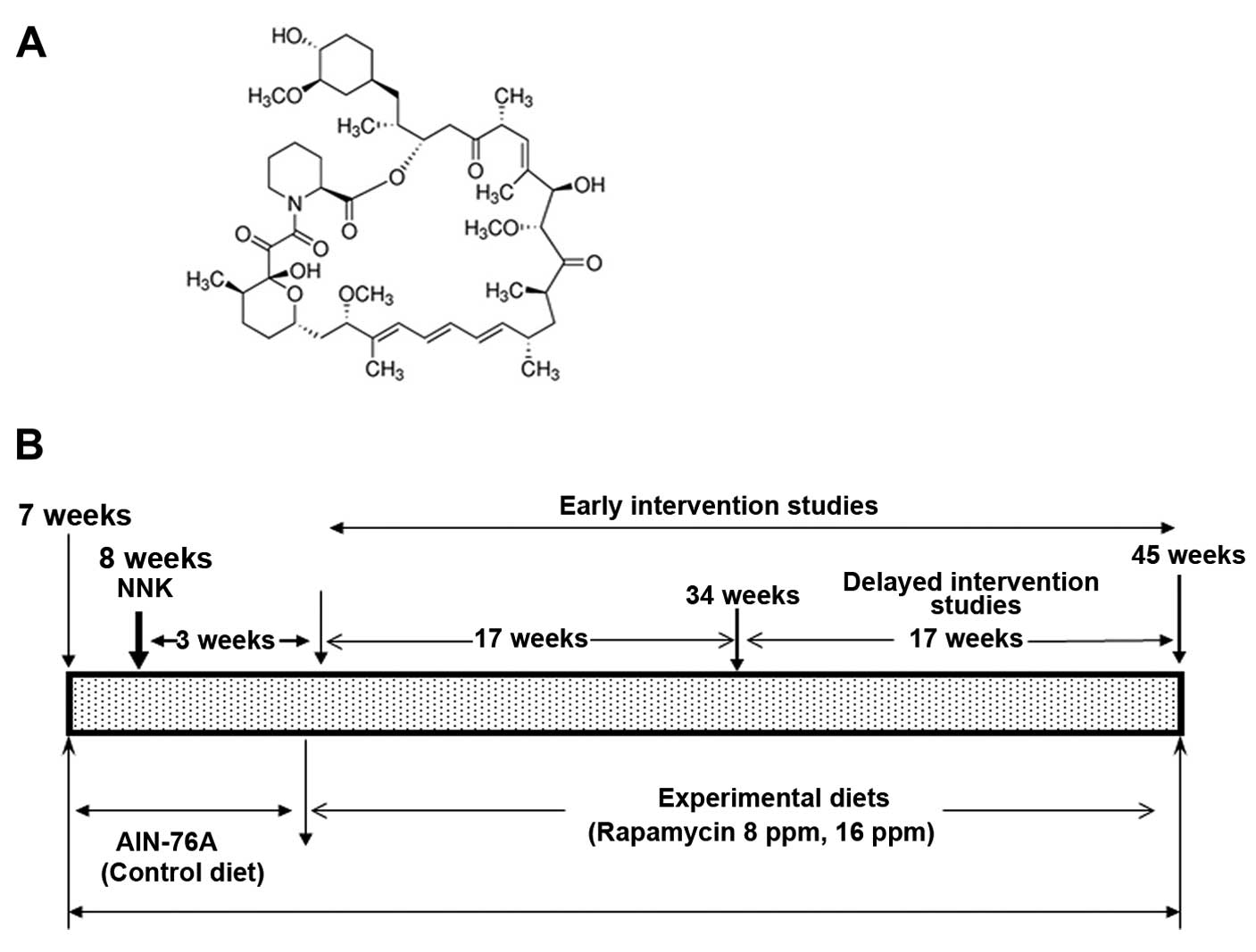
DL-methionine and 0.2% choline bitartrate. Rapamycin (Fig. 1A) was kindly supplied by the
National Cancer Institute, Division of Cancer Prevention Repository
(Bethesda, MD, USA). The test agent was premixed with a small
quantity of casein and then blended into a bulk diet using a Hobart
Mixer. Control and experimental diets were prepared weekly and
stored in a cold room. The content of rapamycin in the experimental
diets was determined periodically in multiple samples taken from
the top, middle and bottom portions of individual diet preparations
to ensure uniform distribution (25).
NNK (>99% purity) was synthesized and provided by
the laboratory of Dr Shantu Amin (Department of Pharmacology, Penn
State Hershey, PA, USA). Protease inhibitor cocktail was purchased
from Sigma (St. Louis, MO, USA). Antibodies to p-S6K1, S6K1, p-ERK,
ERK1, Bcl-xL and actin were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-p-mTOR (Ser2448)
was purchased from Cell Signaling (Danvers, MA, USA).
Assessment of tumor formation
The experiment was designed to evaluate whether
early and delayed intervention with rapamycin provides protection
against the NNK-induced lung carcinogenesis in female A/J mice. The
experimental design is provided in Fig.
1B. Female A/J mice at 6 weeks of age were purchased from the
Jackson Laboratory and were maintained in the pathogen-free Rodent
Barrier Animal Facility at the University of Oklahoma Health
Sciences Center. At 6 weeks of age, the mice were fed a control
irradiated AIN-76A modified diet. At 8 weeks of age, the mice
intended for carcinogen treatment received a single dose of 10
µmol NNK/mouse by i.p. injection. The mice were weighed once
every two weeks until termination of the present study. Three weeks
after NNK treatment, the groups of mice (25 mice/group) intended
for the early, continuous treatment were fed control AIN 76-A or
experimental diets containing 8 or 16 ppm of rapamycin. The mice
were euthanized by CO2 asphyxiation followed by cervical
dislocation after 17 (10 mice/group) or 34 weeks (15 mice/group) of
exposure to test agents. To assess the delayed rapamycin treatment,
two groups (15 mice/group) were fed experimental diets containing 8
or 16 ppm of rapamycin from 20 weeks after NNK treatment until the
end of the experiment. The doses were selected based on the
previous preclinical studies (25,26).
At the time of the sacrifice, the lungs were lavaged, perfused and
washed in PBS and evaluated under a dissecting microscope for the
number of tumors and tumor size. Tumors on the lung surface were
enumerated using a dissecting microscope by at least two
experienced readers who were blinded to sample identifiers. Tumor
diameters were measured using Fisherbrand digital calipers
(Pittsburgh, PA, USA).
Tumor histology
Fixed lung samples were then embedded in paraffin,
sectioned and stained with hematoxylin and eosin (H&E).
H&E-stained lung sections from three predetermined depths were
evaluated by a Board-Certified Pathologist (Dr Stan Lightfoot,
Department of Pathology and VA Hospital, Oklahoma, OK, USA) for the
number of adenomas and adenocarcinomas. The tumors were categorized
according to criteria of the Mouse Models of Human Cancers
Consortium (27).
Immunohistochemistry
The effects of rapamycin on the expression of
proliferating cell nuclear antigen (PCNA), p-S6K1 and MAP LC-3α/β
were evaluated in adenocarcinomas by immunohistochemistry (IHC) as
previously described (28).
Briefly, paraffin sections were deparaffinized in xylene,
rehydrated through graded ethanol solutions and washed in
phosphate-buffered saline (PBS). Antigen retrieval was carried out
by treating sections in 0.01 M citrate buffer (pH 6.0) for 30 min
in a boiling water bath. Endogenous peroxidase activity was
quenched by incubation in 3% H2O2 in PBS for
5 min. Non-specific binding sites were blocked by incubation with
2% BSA, and the sections were incubated overnight at 4°C with a
1:50 dilution of monoclonal antibodies against PCNA, p-S6K1 and MAP
LC-3α/β. After several washes with PBS, the slides were incubated
with the appropriate secondary antibody for 2 h and then washed and
incubated with avidin-biotin complex reagent (Zymed Laboratories,
Camarillo, CA, USA). The slides were rinsed with PBS, incubated
with chromogen 3-diaminobenzidine (DAB) for 3 min, then rinsed and
counterstained with hematoxylin. Non-immune rabbit immunoglobulins
were substituted for primary antibodies as negative controls. The
slides were observed under an Olympus microscope 1X701 and digital
computer images were recorded using an Olympus DP70 camera.
The identity of all the slides was concealed from
the investigators prior to scoring and in all cases, tumors and/or
normal lungs were assessed for 5 mice/group. For PCNA evaluation,
the cells with a brown nucleus in the tumor were considered
positive at a magnification of x400. The proliferative index was
determined by dividing the number of positive cells by the number
of negative cells and multiplying by 100. For p-S6K1 and MAP
LC-3α/β, staining intensity was scored for the whole of the tumor
as absent (0), mild (i), moderate (ii) or strong (iii). For p-S6K1
and MAP LC-3α/β, a staining index was achieved by summing the
products of the fraction of cells stained with a given intensity
times the intensity as just described.
Western blot analysis of the protein
expression
Lung tumors harvested from mice in different
treatment groups were frozen immediately in liquid nitrogen and
stored at −80°C for subsequent analysis. For marker analysis,
larger tumors (heterogeneous population of adenoma and
adenocarcinoma) were excised from the lungs and total cell lysates
were prepared by a previously described cell fractionation
procedure (10). The lysates were
aliquoted, their protein content was determined and they were
stored at −80°C. An aliquot (50 µg protein/lane) of the
total protein was separated via 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to nitrocellulose membranes. After blocking the
membranes with 5% milk powder, membranes were probed for the
expression of p-mTOR (Ser 2448), p-S6K1, total S6K1, p-ERK, ERK and
β-actin in hybridizing solution [1:500 in Tris-buffered saline
(TBS)-Tween-20] using the respective primary antibodies and then
probed with their appropriate horseradish peroxidase
(HRP)-conjugated secondary antibodies. Detection was performed
using the SuperSignal West Pico chemiluminescence reagent (Pierce,
Rockford, IL, USA). The bands were captured on Ewen Parker Blue
sensitive X-ray film and quantified by densitometry. Each blot was
re-probed for β-actin, although only one β-actin blot of the test
agent exposure at 34 weeks was presented.
Statistical analysis
Differences in body weights among groups were
analyzed by analysis of variance (ANOVA). Adenoma and
adenocarcinoma multiplicities (number of tumors/mouse), were
presented as means ± SD. Protein expression and proliferative
indices were presented as means ± SEM and analyzed using the
unpaired t-test with Welch's correction. Dose-response effects were
analyzed by linear regression analysis. Differences were considered
statistically significant at p<0.05.
Results
Evaluation of rapamycin toxicity
In the present study, we evaluated the efficacy of
rapamycin as an inhibitor of NNK-induced lung tumors in A/J mice.
We applied 25 and 50% of the maximally tolerated dose (MTD) of
rapamycin to assess the chemopreventive efficacy. Administration of
8 or 16 ppm rapamycin for 34 (early intervention) or 17 weeks
(delayed intervention) did not cause any body weight loss or any
other histological toxicity in major organ sites (data not shown)
and did not induce any overt toxicities in female A/J mice when
administered orally.
Effect of early stage intervention with
dietary rapamycin on lung tumor formation
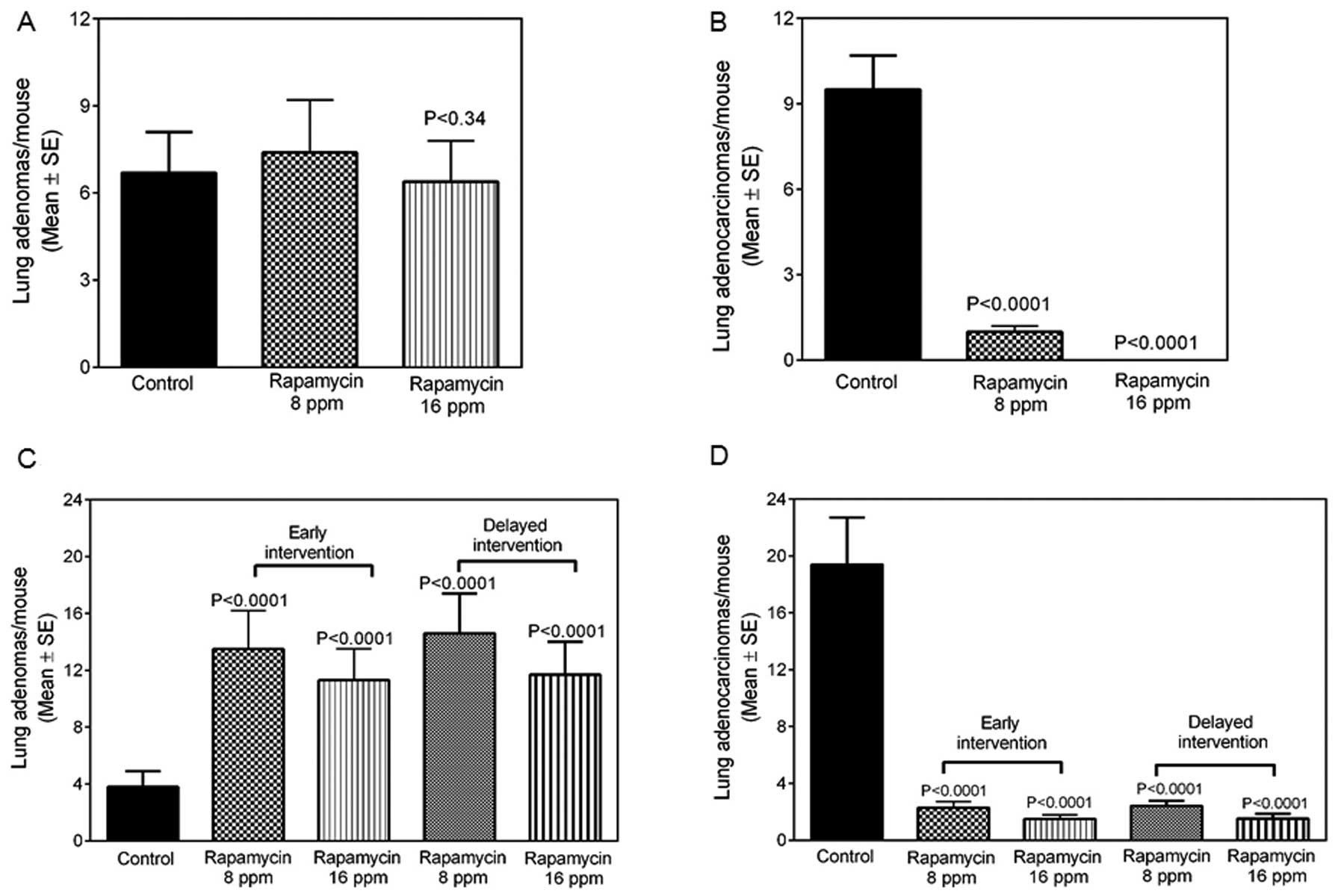
The effects of rapamycin on lung tumor multiplicity
are summarized in Table I and
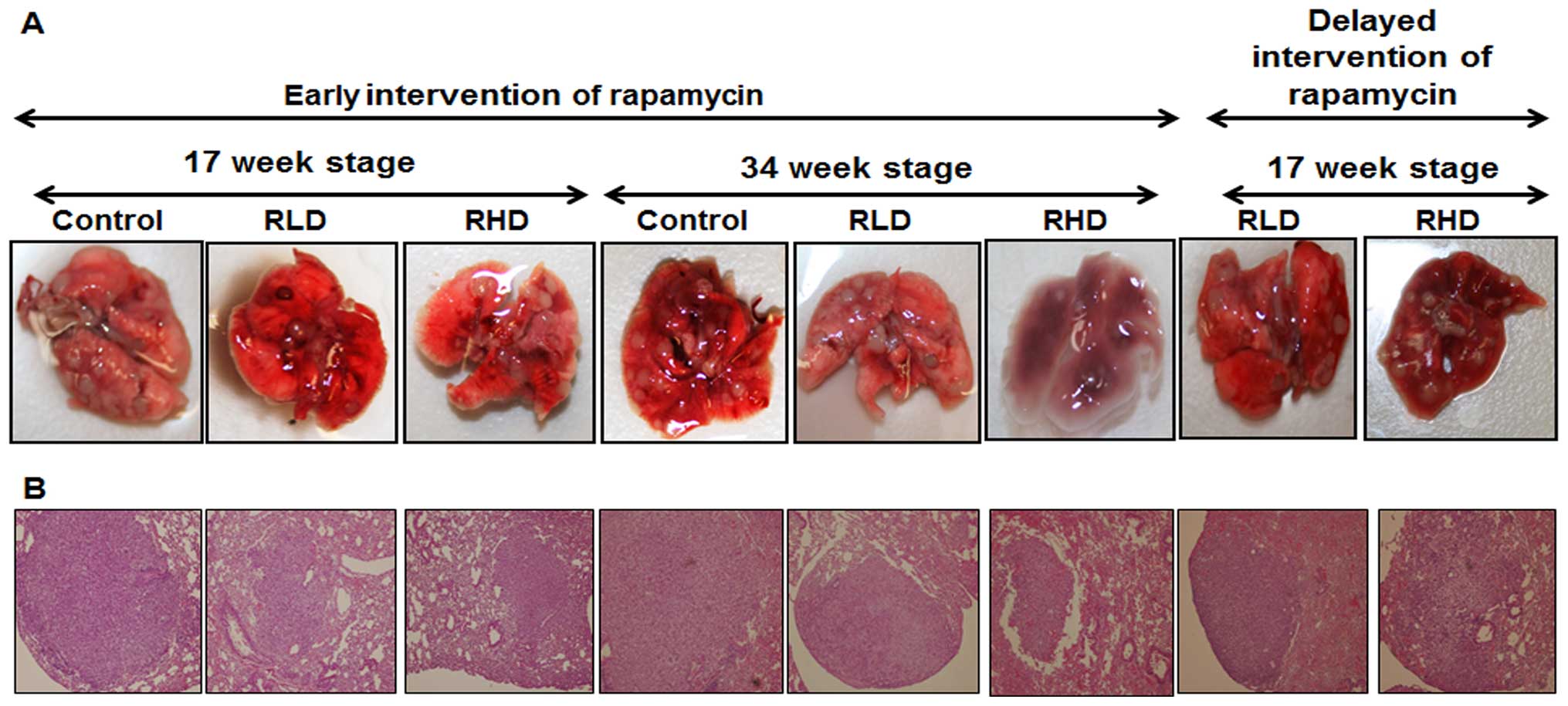
Fig. 2A. Tumors were classified by
a pathologist into adenoma or adenocarcinoma, based on previously
established criteria (27). Both
adenomas and adenocarcinomas were observed in NNK-treated mice.
Adenomas were generally <2 mm in diameter, and
well-circumscribed areas of proliferative cuboidal to columnar
cells lining an alveolus (Fig. 2B).
Adenocarcinomas were typically >2 mm in diameter and showed
invasion and loss in alveolar architecture, increased
nuclear/cytoplasmic ratio, cellular atypia, a large mass of
undifferentiated cells and nuclear pleomorphism. Administration of
rapamycin at 8 or 16 ppm significantly suppressed NNK-induced total
lung tumor formation by 48.14 or 60.49% (p<0.0001),
respectively, at the 17-week stage (Table I) and by 31.89 or 44.82%,
respectively, after 34 weeks in the early intervention studies
(p<0.0001; Table I).
Administration of rapamycin at 8 or 16 ppm after 17 weeks of tumor
formation significantly suppressed NNK-induced total lung tumor
formation by 26.72 or 42.24% (p<0.001–0.0001) in the late
intervention studies (Table I). In
addition to reducing tumor multiplicity, tumors arising in the
presence of rapamycin were much smaller than their untreated
counterparts. NNK-treated mice fed the control diet developed 100%
tumor incidence at 34 weeks after the carcinogen treatment. Control
diet mice sacrificed at 17 weeks after NNK-treatment showed an
average of 6.7±1.4 lung adenomas and 9.22±2.2 lung adenocarcinomas
(means ± SEM) or 41.4% adenomas and 58.6% adenocarcinomas (Table I). Mice fed the control diet and
sacrificed at 34 weeks after NNK treatment showed 3.8±1.1 adenomas
and 19.4±3.1 (16.4% of the tumors were adenomas and 83.6% were
adenocarcinomas; Table II), reflecting an increased progression of
adenomas to adenocarcinomas. The effect of rapamycin on total lung
tumor multiplicities is provided in Table I. Mice administered 8 ppm of
rapamycin for 17 weeks showed a slightly higher number of lung
adenomas (Fig. 3A), whereas lung
adenocarcinomas were significantly (p<0.0001) suppressed by 89
or 99%, respectively, at the 8 or 16 ppm doses (Fig. 3B). A similar trend was observed in
the mice exposed to rapamycin for 34 weeks. NNK-induced
adenocarcinomas were inhibited by 88 or 92% at the two doses
(p<0.0001) at the early intervention stage (Table I, Fig.
3D) and NNK-induced adenocarcinomas were inhibited by 87 or 91%
at the two doses (p<0.0001) in delayed intervention (Table I, Fig.
3D). However, we observed a slight increase in the lung
adenomas (Fig. 3C), reflecting some
delay in tumor progression from adenoma to adenocarcinoma in
rapamycin-treated mice at the two interventions time points.
Irrespective of the time of intervention with rapamycin, tumor size
was significantly reduced (by 50%) in all of the rapamycin-treated
groups as compared with the control untreated counterparts. The
size of NNK-induced tumors decreased from 2.45 mm2 in
control to 0.76 mm2 in the early intervention
rapamycin-treated groups (p=0.005; Fig.
2A) and to 0.86 mm2 (p=0.0043; Fig. 2A and B) in the late intervention
rapamycin groups. We did not find any lung tumor incidence
differences between the control and treated groups. However, our
long-term administration of 8 and 16 ppm rapamycin was effective in
suppressing adenocarcinoma incidence by 46.66 (p<0.006) and
60.0% (p<0.0007), respectively, as compared to the 100%
adenocarcinoma incidence in NNK-treated and control diet fed mice.
Thus, rapamycin prevented significant tumor growth and the
progression of adenoma to adenocarcinoma.
 | Table IEffect of rapamycin against
NNK-induced lung tumorigenesis in female A/J mice. |
Table I
Effect of rapamycin against
NNK-induced lung tumorigenesis in female A/J mice.
| Treatment
group | Early stage
intervention
| Late stage
intervention
|
|---|
17 weeks after NNK
treatment
| 34 weeks after NNK
treatment
| 34 weeks after NNK
treatment
|
|---|
| Adenoma | ADCA | Total | Adenoma | ADCA | Total | Adenoma | ADCA | Total |
|---|
| NNK + control
diet | 6.7±1.4 | 9.5±2.2 | 16.2±2.9 | 3.8±1.1 | 19.4±3.1 | 23.2±3.7 | 3.8±1.1 | 19.4±3.1 | 23.2±3.7 |
| Rapamycin | 7.4±1.8 | 1.0±0.5 | 8.4±1.9 | 13.5±2.7 | 2.3±0.8 | 15.8±3.1 | 14.6±2.8 | 2.4±0.8 | 17.0±3.1 |
| (8 ppm diet) | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | |
| Rapamycin | 6.4±1.4 | 0±0.0 | 6.4±1.4 | 11.3±2.2 | 1.5±0.6 | 12.8±2.4 | 11.7±2.3 | 1.7±0.8 | 13.4±2.4 |
| (16 ppm diet) | p<0.34 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 | p<0.0001 |
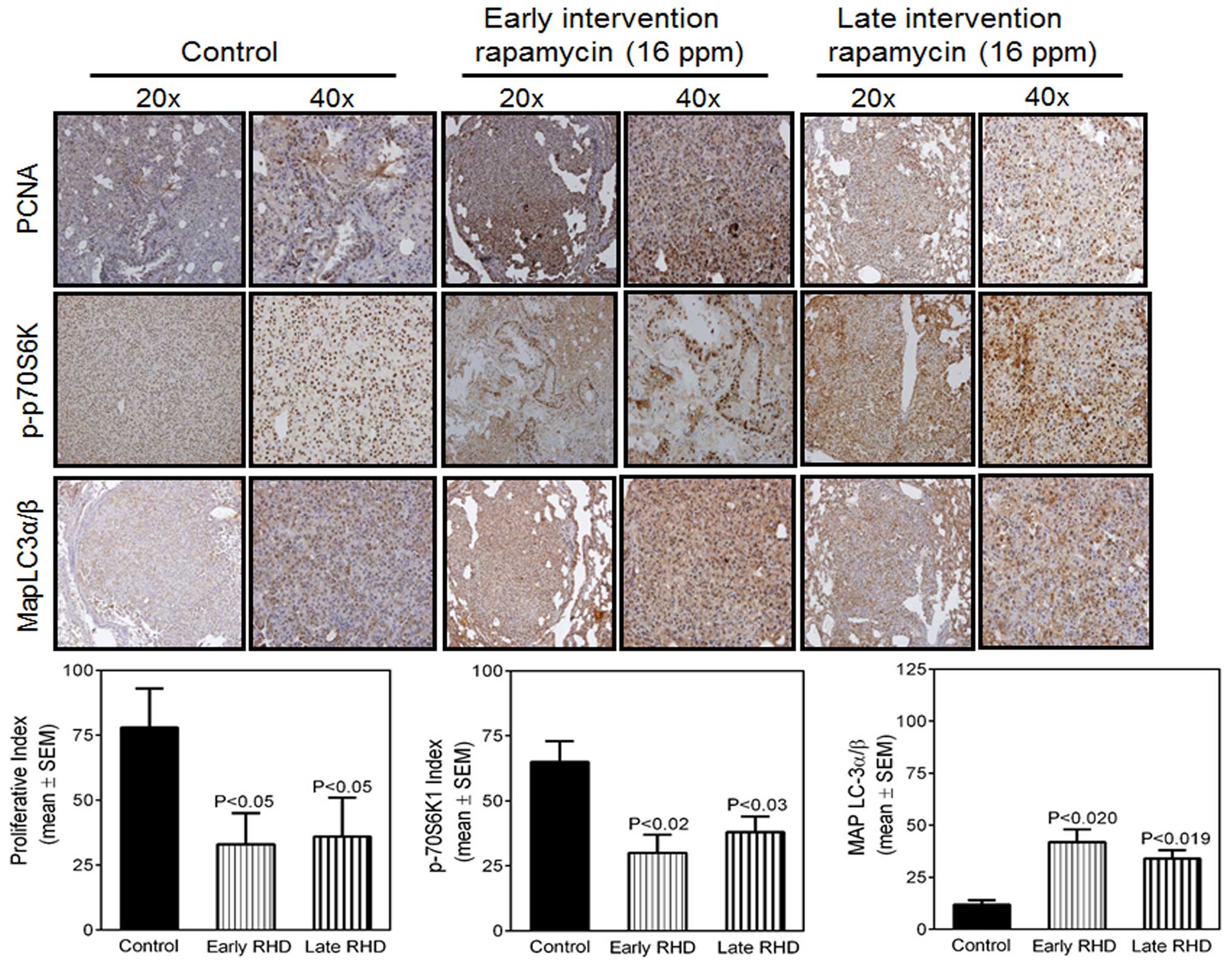
Effect of rapamycin on
immunohistochemical staining of PCNA, p-S6K1 and MAP LCα/β and in
NNK-induced lung tumors
The effects of rapamycin on immunohistochemical
staining for PCNA, p-S6K1 and MAP LCα/β in NNK-induced lung tumors
derived from the A/J strain of mice were determined (Fig. 4). The effect of rapamycin on lung
adenocarcinoma cell proliferation was assessed through labeling of
PCNA-positive cells (Fig. 4A). The
PCNA labeling index was reduced significantly in tumors from
rapamycin-treated mice (p=0.05) compared with tumors from mice fed
a control diet. We observed a decrease in p-S6K1 expression in the
tumors from rapamycin-treated animals as compared with the control.
Staining varied from high levels in tumor tissues from control
animals, although little staining was detected in lung tumor
tissues from the rapamycin-treated groups (p=0.02 and p=0.03)
(Fig. 4B) after the two
intervention time points. Previous studies have also supported a
requirement for mTOR activity in the maintenance of established
NNK-induced tumors and suggested that rapamycin retards tumor
growth by inhibiting S6 phosphorylation and cell proliferation
(29). We investigated changes in
the expression of MAP LC3α/β, which also regulates cell death
through autophagy and observed moderate induction with rapamycin
treatment as compared with the control (p=0.02 or 0.01 at the two
times; Fig. 4C). The difference in
the treatment groups in the intervention studies was not
statistically significant.
Rapamycin decreases the expression of
p-mTOR and p-S6-kinase
The expression of p-mTOR (Ser 2448), p-ERK, ERK1,
p-S6K1, total S6K1, Bcl-xL and actin were subsequently analyzed by
western blot analysis. Rapamycin caused significant changes in
these biomarkers after the early intervention as compared with the
late intervention studies (Fig. 5).
As shown in Fig. 5A, the early
intervention with rapamycin significantly inhibited the
phosphorylation of mTOR at Ser-2448 in lung tumors as compared with
that in mice fed the control diet. mTORC1 promotes cancer cell
survival and growth by stimulation of protein synthesis, which is
achieved through phosphorylation of ribosomal S6K1. S6K1 is a
substrate for mTOR and its phosphorylation is dependent on mTOR
activity. We observed a decreased phosphorylation of S6K1 in mice
fed rapamycin diets compared with control diets, irrespective of
whether the intervention was early (p=0.01) or late (0.04).
However, the total S6 kinase protein levels were unaltered. NNK can
stimulate the growth of lung cancer cells, suppress apoptosis and
increase p-ERK (30,31). Rapamycin decreased the levels of ERK
phosphorylation in lung tumors while total ERK protein levels
remained unaltered. Fig. 5B shows
the optical density (OD) for different protein markers.
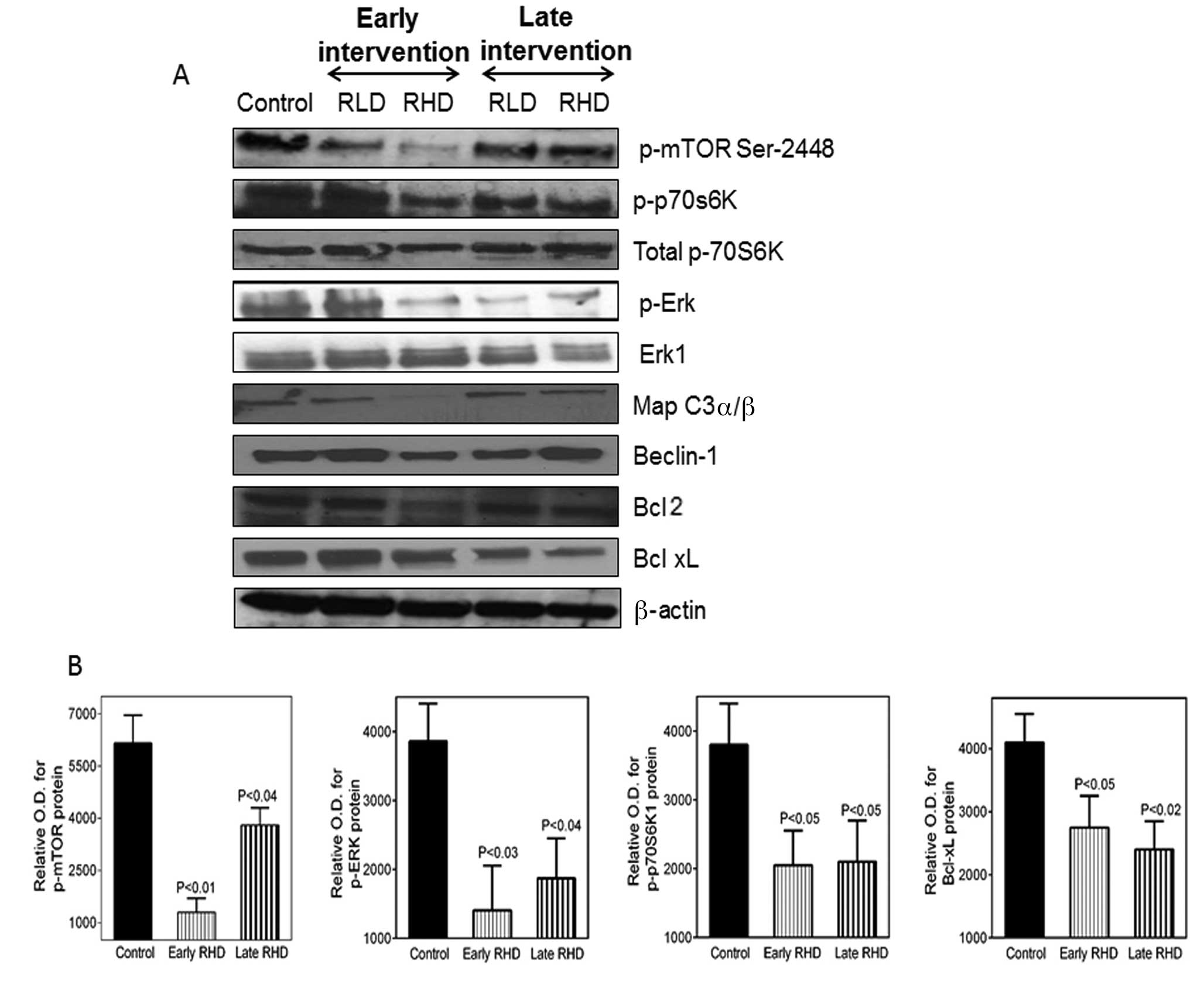 | Figure 5(A) Immunoblotting analyses of lung
tumor lysates derived from A/J mice treated with NNK control (C), 8
ppm of rapamycin (RLD) or 16 ppm of rapamycin (RHD) at the 36-week
stage for the early and delayed intervention studies. Expression
levels for p-mTor, p-S6K10 and western blot analysis for total S6
kinase, Beclin-1, MAP LC3α/β, Bcl-2, caspase-9,Bcl-xL, p-ERK, ERK-1
and β-actin are shown for lung tumor samples at the 36-week stage
from A/J mice administered 8 or 16 ppm of rapamycin. Lung tumor
tissue lysates were homogenized in lysis buffer and were subjected
to SDS-PAGE followed by western blot analysis as described in
Materials and methods. Membranes were probed with specific primary
antibodies followed by appropriate peroxidase-conjugated secondary
antibodies. Proteins were visualized with an enhanced
chemilumniscence detection system. (B) Band intensity was
calculated by densitometry using the ImageJ software provided by
NIH. Results are expressed as OD. The significance of differences
between the control and treatment groups was analyzed by the
one-tailed t-test with Welch's correction. OD, optical density. |
Treatment of lung cancer cells with
rapamycin results in the induction of autophagy in NNK-induced lung
tumors
We investigated the effect of rapamycin on autophagy
and the induction of apoptosis. mTORC1 inhibition led to the
induction of autophagy and formation of autophagosome
membrane-bound vacuoles. One of the ways to measure autophagy is by
examining MAP LC3α/β and measuring the MAP LC3α and MAP-light chain
3β subunits. MAP LC3α is involved in the formation of
autophagosomal vacuoles. MAP LC3β is associated with autophagosome
membranes after processing and is essential for autophagy.
Rapamycin (16 ppm) moderately induced MAP protein expression in the
NNK-induced lung tumors as compared with that in mice fed the
control diet (Fig. 5A); however, we
were not able to observe MAP LC3-β in tumor tissues. We also
analyzed the expression of Beclin-1, which is an essential
autophagic gene that contributes to initial vesicle nucleation and
the formation of the autophagosome (32). There was no significant change in
Beclin-1 protein expression in the rapamycin-treated groups as
compared with those fed the control diet.
Rapamycin suppresses Bcl-2, Bcl-xL
expression and promotes caspase-9 activation in NNK-induced lung
cancer
Another mechanism for lung cancer cell survival is
through the upregulation of anti-apoptotic proteins that inhibit
apoptosis and promote cell division. Increasing evidence suggests
that there is a cross-talk between apoptotic and autophagic
processes, which share common regulatory molecules such as Bcl-2
family members (22,33). To examine the mechanism by which
rapamycin induces apoptosis in NNK-induced lung tumors, we examined
the expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL. As
shown in Fig. 5A, early
intervention with rapamycin significantly inhibited the Bcl-2 and
Bcl-xL protein expression in lung tumors as compared with that in
mice fed the control diet. These data suggest that rapamycin
induces apoptosis by reducing the anti-apoptotic Bcl-2 and Bcl-xL
protein expression.
Discussion
Although cessation of cigarette smoking is one of
the best prevention approaches to reducing lung cancer mortality,
chemoprevention may also be useful to decreasing lung cancer in
high-risk populations. Previous results suggest that tobacco
components activate the Akt pathway, which leads to tumor cell
survival, proliferation and an increase in cell size (34,35).
The aberrant expression and activation of Akt leads to poor
prognosis in NSCLC (36). Akt
contributes to proliferation through mTOR activation in NSCLC
(37,38). Our findings that p-mTOR and
activated p-S6K1 are overexpressed in NNK-induced lung tumors in
A/J mice suggest that these proteins are required in the
development of the tumors. Preclinical studies have reported the
ability of rapamycin to inhibit experimentally induced lung
tumorigenesis (39,40). Our results show that rapamycin is
effective in preventing tumor growth in a mouse model of tobacco
carcinogen-induced lung tumorigenesis. Administration of rapamycin
in the diet early or late during carcinogenesis prevented the
development (88–92%) of NNK-induced tumors and decreased tumor size
as compared with mice fed the control diet. Yan et al
(40) and Granville et al
(39) also showed that treatment
with rapamycin reduced tumor size; however, they did not observe an
effect on tumor multiplicity. The differing results may be due to
different routes of rapamycin administration, different dosing
schedules as well as to differing duration of the studies.
To identify specific molecular alterations that may
contribute to the protective effect of rapamycin at different times
during NNK-induced lung carcinogenesis, we examined rapamycin
effects on the expression of p-mTOR and its downstream kinase
p-S6K1 that modulate tumor cell proliferation. Activated Akt
phosphorylates downstream target molecules, including FKHR and mTOR
(9). However, it has been suggested
that the tuberous sclerosis complex may mediate S6K1 activation
independently of mTOR or that S6K1 may directly phosphorylate mTOR
(41). In vitro studies
suggest that NNK activates mTORC1, as assessed by the increased
phosphorylation of S6K, 4E-BP1 and increasing the expression of the
matrix glycoprotein fibronectin (41). In the present study, we observed
that p-mTOR was overexpressed in tumors of mice fed a control diet,
whereas its expression was downregulated in mice fed a rapamycin
diet. We also found decreased levels of p-S6K1 in mice fed
rapamycin as compared with those in mice fed the control diet.
These results are in agreement with those of earlier studies
showing that, activation of p-S6K1 is dependent on mTOR activation
(13). The extent of p-mTOR
inhibition is greater in mice receiving the early as compared with
the later rapamycin intervention, as is the decrease in tumor size,
possibly because the tumors in the early intervention group were
exposed to rapamycin for a longer period of time. Since rapamycin
administration in two different dosing schedules inhibited mTOR and
p-S6K, our data suggest that NNK activation of mTORC1 is dependent
on the Akt/mTOR pathway. Pharmacologic targeting of p-mTOR may
provide an improved strategy to prevent the development and
progression of tobacco carcinogen-induced lung tumors.
Carcinomas of the lung have a very high labeling
index for PCNA, which is required by DNA polymerase for DNA
replication and thus serves as a common marker for proliferation.
An increase in PCNA-positive cells is a general characteristic of
tumor progression. PCNA is highly expressed in the bronchial
epithelial cells of smokers (42).
The present study demonstrates that the dietary administration of
rapamycin decreased tumor size as well as the PCNA labeling index
as compared with those in mice fed the control diet. The mechanism
may involve suppression by rapamycin of the phosphorylation of S6
kinase downstream of MTORC1 leading to disruption of cell cycle
regulation and inhibition of cell proliferation. MTORC1 activation
promotes cell prolife ration via its downstream targets, including
EIF4BP1 (43,44). Rapamycin suppressed cell
hyper-proliferation when administered at an early stage of
NNK-induced carcinogenesis and it inhibited the progression from
adenoma to adenocarcinoma in mouse lung tumorigenesis. To
investigate whether rapamycin inhibits tumor cell proliferation
in vivo, we examined endo genous p-ERK, which is regulated
by growth factors and nutrient conditions. We observed a 40%
decrease in p-ERK in tumors from rapamycin-treated animals,
suggesting that rapamycin may control the interaction between
endogenous mTOR and p-ERK.
Failure of many chemotherapeutic drugs often has
been associated with the overexpression of anti-apoptotic proteins
in cancer cells. Increasing evidence suggests that the cross-talk
between apoptosis and autophagy is important and that these two
death processes share many common regulatory molecules such as
Bcl-2, Beclin-1 and mTOR signaling pathway members (38). Earlier studies reported that
inhibition of mTOR signaling causes cell death that is associated
with apoptosis and autophagy (45).
Previous studies have shown that mTOR and the anti-apoptotic
proteins Bcl-xL and Bcl2 are able to compete for FKBP38 binding
(46–48) and thereby influence each other's
function. Dietary rapamycin significantly decreased the expression
of Bcl-2 and Bcl-xL in the treatment groups as compared with that
in the mice fed the control diet. Thus, the rapamycin induction of
apoptosis may be mediated, in part, through the inhibition of Bcl-2
and Bcl-xL expression and consequent reduction of their binding to
FKBP38 (46,48), which complexes act as inhibitors of
mTORC1. However, the molecular mechanism underlying the inhibition
of Bcl-2 and Bcl-xL expression by rapamycin remains to be
identified. Decreased Bcl-2 and Bcl-xL may result from inhibition
of the PI3K/Akt/mTOR pathway, which is known to stimulate certain
transcription factors, such as E2F and nuclear factor κB (49), or alternative mechanisms may be
involved that remain to be determined. Anti-apoptotic proteins
released from the Bcl-2 and Bcl-xL complex can directly induce
mitochondrial permeabilization and subsequent apoptosis.
Proteins of the Bcl-2 family regulate the apoptotic
pathway by associating with pro-apoptotic family members, including
Bax, via BH3 domains and inhibiting autophagy by antagonizing the
BH3-only protein Beclin-1, an essential inducer of autophagy. In
the present study, we demonstrated that dietary rapamycin regulates
the mTOR pathway by decreasing the phosphorylation of p-mTOR and
p-70S6K (a substrate of mTOR). This process is accompanied by cell
death by induction of apoptosis and initiation of autophagy in
NNK-induced lung tumors fed a rapamycin diet. As the levels of
Bcl-2 decrease, the Bcl-2-Beclin-1 complexes may become disrupted,
releasing Beclin-1 and inducing autophagy or apoptosis. Thus,
during this processes, Beclin-1 may contribute to the
rapamycin-induced cell death, although the expression of Beclin-1
levels do not greatly alter (Fig.
5A). The conditional knockdown of Beclin-1 in the A549 cell
line promotes cell growth and inhibits apoptosis (47). Our results corroborate these results
(50) suggesting that Beclin-1
plays a direct role in apoptosis. In the present study, we observed
an increased expression of autophagy-associated proteins MAPLC3-β
in rapamycin-treated NSCLC lines as compared with that in untreated
cells.
In conclusion, our results reveal the potent
inhibitory effects of rapamycin intervention during NNK-induced
lung carcinogenesis in the A/J mouse strain. In two dosing
schedules, rapamycin was able to decrease tumor multiplicity and
size in association with the downregulation of p-mTOR and p-S6K1 at
the 36-week stage after early or delayed intervention times. Our
results also show that rapamycin exhibits strong anti-proliferative
and pro-apoptotic activities by inducing autophagy.
Abbreviations:
|
NNK
|
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
|
|
AIN-76A
|
American Institute of Nutrition 76A
diet
|
|
FBS
|
fetal bovine serum
|
|
H&E
|
hematoxylin and eosin
|
|
IHC
|
immunohistochemistry
|
|
PBS
|
phosphate-buffered saline
|
|
BSA
|
bovine serum albumin
|
|
DAB
|
3-diaminobenzidine
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
SDS-PAGE
|
sodium dodecyl sulfate polyacrylamide
gel electrophoresis
|
|
TBS
|
Tris-buffered saline
|
|
HRP
|
horseradish peroxidase
|
|
RT-qPCR
|
reverse transcriptase-quantitative
polymerase chain reaction
|
|
mTOR
|
mammalian target of rapamycin
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
OD
|
optical density
|
|
p-S6K1
|
phospho-S6 kinase 1
|
|
4-EBP1
|
eukaryotic translation initiation
factor 4E-binding protein
|
|
p-ERK
|
phosphorylated extracellular
signal-regulated kinase
|
|
PI3K
|
phosphoinositide-3-kinase
|
|
Akt
|
protein kinase B
|
|
FKHR
|
forkhead transcription factor
|
|
NSCLC
|
non-small cell lung cancer
|
|
FKBP38
|
FK506 binding protein 38
|
|
RLD
|
rapamycin low dose (8 ppm)
|
|
RHD
|
rapamycin high dose (16 ppm)
|
|
ADCA
|
adenocarcinoma
|
|
MAP LC-3α/β
|
microtubule-associated light chain
3α/β
|
|
MTD
|
maximally tolerated dose
|
Acknowledgments
The authors would like to thank the University of
Oklahoma Health Sciences Center Rodent Barrier Facility Staff and
Dr Julie Sando for their assistance in the preparation of the
manuscript and for editing. We would like to thank Dr Shantu Amin
and Dr Dhimant Desai, from the Department of Pharmacology (Penn
State Hershey, PA, USA) for providing NNK carcinogen to carry out
these experiments. Additionally, the authors thank the Peggy and
Charles Stephenson Cancer Center, Histology tissue core facility
for IHC staining.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Humphrey L, Deffebach M, Pappas M, Baumann
C, Artis K, Mitchell JP, Zakher B, Fu R and Slatore C: Screening
for Lung Cancer: Systematic Review to Update the U.S. Preventive
Services Task Force Recommendation. Rockville, MD: 2013
|
|
3
|
American Cancer Society: Cancer Facts
& Figures 2013. American Cancer Society; Atlanta, GA: 2013
|
|
4
|
Hecht SS, Kassie F and Hatsukami DK:
Chemoprevention of lung carcinogenesis in addicted smokers and
ex-smokers. Nat Rev Cancer. 9:476–488. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsurutani J, Castillo SS, Brognard J,
Granville CA, Zhang C, Gills JJ, Sayyah J and Dennis PA: Tobacco
components stimulate Akt-dependent proliferation and
NFkappaB-dependent survival in lung cancer cells. Carcinogenesis.
26:1182–1195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
West KA, Brognard J, Clark AS, Linnoila
IR, Yang X, Swain SM, Harris C, Belinsky S and Dennis PA: Rapid Akt
activation by nicotine and a tobacco carcinogen modulates the
phenotype of normal human airway epithelial cells. J Clin Invest.
111:81–90. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
West KA, Linnoila IR, Belinsky SA, Harris
CC and Dennis PA: Tobacco carcinogen-induced cellular
transformation increases activation of the phosphatidylinositol
3′-kinase/Akt pathway in vitro and in vivo. Cancer Res. 64:446–451.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Markman B, Dienstmann R and Tabernero J:
Targeting the PI3K/Akt/mTOR pathway - beyond rapalogs. Oncotarget.
1:530–543. 2010. View Article : Google Scholar
|
|
9
|
Bjornsti MA and Houghton PJ: The TOR
pathway: A target for cancer therapy. Nat Rev Cancer. 4:335–348.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar G, Dange P, Kailaje V, Vaidya MM,
Ramchandani AG and Maru GB: Polymeric black tea polyphenols
modulate the localization and activity of
12-O-tetradecanoylphorbol-13-a cetate-mediated kinases in mouse
skin: Mechanisms of their anti-tumor-promoting action. Free Radic
Biol Med. 53:1358–1370. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Foster KG and Fingar DC: Mammalian target
of rapamycin (mTOR): Conducting the cellular signaling symphony. J
Biol Chem. 285:14071–14077. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim DH, Sarbassov DD, Ali SM, King JE,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: mTOR
interacts with raptor to form a nutrient-sensitive complex that
signals to the cell growth machinery. Cell. 110:163–175. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sekulić A, Hudson CC, Homme JL, Yin P,
Otterness DM, Karnitz LM and Abraham RT: A direct linkage between
the phosphoinositide 3-kinase-AKT signaling pathway and the
mammalian target of rapamycin in mitogen-stimulated and transformed
cells. Cancer Res. 60:3504–3513. 2000.
|
|
14
|
Hsu PP, Kang SA, Rameseder J, Zhang Y,
Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al:
The mTOR-regulated phosphoproteome reveals a mechanism of
mTORC1-mediated inhibition of growth factor signaling. Science.
332:1317–1322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kelloff GJ: Perspectives on cancer
chemoprevention research and drug development. Adva Cancer Res.
78:199–334. 2000. View Article : Google Scholar
|
|
16
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Malaguarnera L: Implications of apoptosis
regulators in tumorigenesis. Cancer Metastasis Rev. 23:367–387.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumar G, Tajpara P and Maru G: Dietary
turmeric post-treatment decreases DMBA-induced hamster buccal pouch
tumor growth by altering cell proliferation and apoptosis-related
markers. J Environ Pathol Toxicol Oncol. 31:295–312. 2012.
View Article : Google Scholar
|
|
19
|
Bareford MD, Park MA, Yacoub A, Hamed HA,
Tang Y, Cruickshanks N, Eulitt P, Hubbard N, Tye G, Burow ME, et
al: Sorafenib enhances pemetrexed cytotoxicity through an
autophagy-dependent mechanism in cancer cells. Cancer Res.
71:4955–4967. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thorburn A: Apoptosis and autophagy:
Regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar :
|
|
22
|
Maiuri MC, Le Toumelin G, Criollo A, Rain
JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K,
Tavernarakis N, et al: Functional and physical interaction between
Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 26:2527–2539.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang ZF, Shao LJ, Wang WM, Yan XB and Liu
RY: Decreased expression of Beclin-1 and LC3 in human lung cancer.
Mol Biol Rep. 39:259–267. 2012. View Article : Google Scholar
|
|
24
|
Stoner GD, Adam-Rodwell G and Morse MA:
Lung tumors in strain A mice: Application for studies in cancer
chemoprevention. J Cell Biochem Suppl. 17F:95–103. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harrison DE, Strong R, Sharp ZD, Nelson
JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter
CS, et al: Rapamycin fed late in life extends lifespan in
genetically heterogeneous mice. Nature. 460:392–395.
2009.PubMed/NCBI
|
|
26
|
Nadon NL, Strong R, Miller RA, Nelson J,
Javors M, Sharp ZD, Peralba JM and Harrison DE: Design of aging
intervention studies: The NIA interventions testing program. Age
(Dordr). 30:187–199. 2008. View Article : Google Scholar
|
|
27
|
Nikitin AY, Alcaraz A, Anver MR, Bronson
RT, Cardiff RD, Dixon D, Fraire AE, Gabrielson EW, Gunning WT,
Haines DC, et al: Classification of proliferative pulmonary lesions
of the mouse: Recommendations of the mouse models of human cancers
consortium. Cancer Res. 64:2307–2316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar G, Tajpara P, Bukhari AB,
Ramchandani AG, De A and Maru GB: Dietary curcumin post-treatment
enhances the disappearance of B(a)P-derived DNA adducts in mouse
liver and lungs. Toxicol Rep. 1:1181–1194. 2014. View Article : Google Scholar
|
|
29
|
Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue
P, Fu H and Khuri FR: Activation of Akt and eIF4E survival pathways
by rapamycin-mediated mammalian target of rapamycin inhibition.
Cancer Res. 65:7052–7058. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laag E, Majidi M, Cekanova M, Masi T,
Takahashi T and Schuller HM: NNK activates ERK1/2 and CREB/ATF-1
via beta-1-AR and EGFR signaling in human lung adenocarcinoma and
small airway epithelial cells. Int J Cancer. 119:1547–1552. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vicent S, López-Picazo JM, Toledo G,
Lozano MD, Torre W, Garcia-Corchón C, Quero C, Soria JC,
Martín-Algarra S, Manzano RG, et al: ERK1/2 is activated in
non-small-cell lung cancer and associated with advanced tumours. Br
J Cancer. 90:1047–1052. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tooze SA and Yoshimori T: The origin of
the autophagosomal membrane. Nat Cell Biol. 12:831–835. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oberstein A, Jeffrey PD and Shi Y: Crystal
structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a
novel BH3-only protein. J Biol Chem. 282:13123–13132. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Memmott RM and Dennis PA: The role of the
Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis.
Clin Cancer Res. 16:4–10. 2010. View Article : Google Scholar :
|
|
35
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsurutani J, Fukuoka J, Tsurutani H, Shih
JH, Hewitt SM, Travis WD, Jen J and Dennis PA: Evaluation of two
phosphorylation sites improves the prognostic significance of Akt
activation in non-small-cell lung cancer tumors. J Clinical Oncol.
24:306–314. 2006. View Article : Google Scholar
|
|
37
|
Balsara BR, Pei J, Mitsuuchi Y, Page R,
Klein-Szanto A, Wang H, Unger M and Testa JR: Frequent activation
of AKT in non-small cell lung carcinomas and preneoplastic
bronchial lesions. Carcinogenesis. 25:2053–2059. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han S, Khuri FR and Roman J: Fibronectin
stimulates non-small cell lung carcinoma cell growth through
activation of Akt/mammalian target of rapamycin/S6 kinase and
inactivation of LKB1/AMP-activated protein kinase signal pathways.
Cancer Res. 66:315–323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Granville CA, Warfel N, Tsurutani J,
Hollander MC, Robertson M, Fox SD, Veenstra TD, Issaq HJ, Linnoila
RI and Dennis PA: Identification of a highly effective rapamycin
schedule that markedly reduces the size, multiplicity, and
phenotypic progression of tobacco carcinogen-induced murine lung
tumors. Clin Cancer Res. 13:2281–2289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yan Y, Wang Y, Tan Q, Hara Y, Yun TK,
Lubet RA and You M: Efficacy of polyphenon E, red ginseng, and
rapamycin on benzo(a)pyrene-induced lung tumorigenesis in A/J mice.
Neoplasia. 8:52–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jaeschke A, Hartkamp J, Saitoh M, Roworth
W, Nobukuni T, Hodges A, Sampson J, Thomas G and Lamb R: Tuberous
sclerosis complex tumor suppressor-mediated S6 kinase inhibition by
phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol.
159:217–224. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Khuri FR, Lee JS, Lippman SM, Lee JJ,
Kalapurakal S, Yu R, Ro JY, Morice RC, Hong WK and Hittelman WN:
Modulation of proliferating cell nuclear antigen in the bronchial
epithelium of smokers. Cancer Epidemiol Biomarkers Prev.
10:311–318. 2001.PubMed/NCBI
|
|
43
|
Fingar DC and Blenis J: Target of
rapamycin (TOR): An integrator of nutrient and growth factor
signals and coordinator of cell growth and cell cycle progression.
Oncogene. 23:3151–3171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dowling RJ, Topisirovic I, Alain T,
Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj
A, Liu Y, et al: mTORC1-mediated cell proliferation, but not cell
growth, controlled by the 4E-BPs. Science. 328:1172–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Degtyarev M, De Mazière A, Orr C, Lin J,
Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al: Akt
inhibition promotes autophagy and sensitizes PTEN-null tumors to
lysosomotropic agents. J Cell Biol. 183:101–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y
and Jiang Y: Rheb activates mTOR by antagonizing its endogenous
inhibitor, FKBP38. Science. 318:977–980. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ma D, Bai X, Zou H, Lai Y and Jiang Y:
Rheb GTPase controls apoptosis by regulating interaction of FKBP38
with Bcl-2 and Bcl-XL. J Biol Chem. 285:8621–8627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shirane M and Nakayama KI: Inherent
calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and
inhibits apoptosis. Nat Cell Biol. 5:28–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chun KH, Kosmeder JW II, Sun S, Pezzuto
JM, Lotan R, Hong WK and Lee HY: Effects of deguelin on the
phosphatidylinositol 3-kinase/Akt pathway and apoptosis in
premalignant human bronchial epithelial cells. J Natl Cancer Inst.
95:291–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang W, Fan H, Zhou Y, Duan P, Zhao G and
Wu G: Knockdown of autophagy-related gene BECLIN1 promotes cell
growth and inhibits apoptosis in the A549 human lung cancer cell
line. Mol Med Rep. 7:1501–1505. 2013.PubMed/NCBI
|