Introduction
Worldwide, prostate cancer (PCa) is the second most
frequently diagnosed cancer in men and the fifth leading cause of
cancer-related death (1). Recently,
the incidence of PCa in China has been substantially increasing
with implementation of comprehensive screening programs (2). For the majority of PCa patients,
endocrine therapy is effective at first, but almost all patients
undergo progression from androgen-dependent prostate cancer to
androgen-independent prostate cancer (AIPC) following a median
period of 14–30 months. To date, the specific mechanisms involved
in androgen-independent (AI) transition have not been entirely
clarified.
Lysine-specific demethylase 1 (LSD1) is the first
defined histone demethylase, and specifically demethylates histone
H3 at the lysine K4 locus and remodels chromatin to regulate gene
expression (3). LSD1 protein was
reported to interact with androgen receptor (AR) protein and
demethylate histone H3 at lysine 9 (H3-K9) to repress the AR target
gene (4,5). In addition, LSD1 protein interacts
with and demethylates p53 protein, a non-histone protein, at lysine
K370 to repress its function (6,7). The
biological effects of LSD1 have been studied most extensively, and
evidence demonstrates that LSD1 is aberrantly overexpressed in
various types of human cancers, such as breast, lung, colorectal,
bladder and ovarian cancer, and plays crucial roles in
tumorigenesis and progression (8–13).
LSD1 is significantly upregulated in PCa and may
serve as a predictive biomarker of aggressive PCa (14–16).
Accumulated data imply that LSD1 is closely correlated with cell
proliferation, angiogenesis, migration and invasion in PCa
(17–19). However, the correlation of LSD1 with
AI transition of PCa under androgen-ablated conditions has rarely
been reported.
AR is thought to play a critical role in the
development of AIPC via various mechanisms, including AR
amplification, mutation and activation via ligand-independent
modifications, increased AR sensitivity to low-level androgen, and
bypass of intact AR pathways (20–23).
The transcription factor p53 functions as a tumor-suppressor gene,
and is activated by DNA damage, deficiency of nutrients or growth
factors, and induces apoptosis and cell cycle arrest (24). p53 gene mutation is quite common in
human cancers, such as late-stage PCa (25,26).
Reportedly, LSD1 interacts with AR and p53 and regulates their
biological functions through post-translational demethylation
(4,6).
The present study aimed to ascertain whether LSD1 is
involved in AI transition of human androgen-dependent prostate
cancer LNCaP cells. Our data revealed that over-expression of LSD1
'rescued' LNCaP cells from androgen ablation-induced apoptosis and
G0/G1 cell cycle arrest and promoted
androgen-independent transition via activation of the AR signaling
pathway and suppression of the p53 signaling pathway.
Materials and methods
Patients and specimens
From December 2010 to October 2013, prostate cancer
tissues and paired adjacent non-tumor tissues were collected from
20 PCa patients undergoing radical prostatectomy at the Department
of Urology, Union Hospital, Tongji Medical College, Huazhong
University of Science and Technology, Wuhan, China. Fresh tissues
were used to isolate total RNA for reverse-transcription
quantitative-polymerase chain reaction (RT-qPCR) and total protein
for western blotting. The diagnoses of PCa were confirmed by
histopathological examination. The study involving the use of human
prostate cancer specimens was approved by the Institutional Review
Board of Tongji Medical College of Huazhong University of Science
and Technology. Written informed consents was obtained from each
participant.
Cell culture and establishment of the
LNCaP-AI cell model
The human prostate cancer LNCaP cell line was
purchased from the Shanghai Cell Bank, Chinese Academy of Sciences
(Shanghai, China). LNCaP cells were cultured in Phenol
Red-containing RPMI-1640 medium containing 10% fetal bovine serum
(FBS) (both from Gibco, Carlsbad, CA, USA) at 37°C in a 5%
CO2 incubator. To obtain AI cells, the LNCaP cells were
continuously cultured in Phenol Red-negative RPMI-1640 medium
(Gibco) supplemented with 10% charcoal-stripped FBS (CS-FBS;
Biowest, Nuaillé, France) for three months (27). During the three-month period,
morphological changes in the LNCaP cells were dynamically
observed.
Cell transfection
Overexpression plasmids for LSD1 and NC were
constructed by Shanghai GeneChem Co. (Shanghai, China). sh-LSD1 and
sh-NC were purchased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). LNCaP and LNCaP-AI cells (8×105/well) were seeded
in 6-well plates and maintained in antibiotic-free complete culture
medium for 24 h. At transfection, the medium was replaced with
1,500 µl of antibiotic-free and FBS-free medium per well.
Lipofectamine 2000 reagent (6 µl) (Invitrogen, Carlsbad, CA,
USA) and 3 µg of plasmid were diluted in 250 µl
Opti-MEM Reduced Serum Medium (Gibco), respectively, and incubated
for 5 min at room temperature. Diluted plasmid was added into the
diluted Lipofectamine 2000 reagent and incubated for 20 min at room
temperature. A plasmid-lipid complex (500 µl) was added to
each well. Stably transfected cells were selected by G418 (EMD
Chemicals, Inc., San Diego, CA, USA) or puromycin (Santa Cruz
Biotechnology).
Cell Counting Kit-8 (CCK-8)
Cells (1×103/well) were seeded into
96-well plates and cultured in Phenol Red-containing medium with
10% FBS or Phenol Red-negative medium with 10% CS-FBS for 0, 2, 4,
6 and 8 days, respectively. CCK-8 solution (10 µl) (Dojindo,
Kumamoto, Japan) was added into each well and incubated for 2.5 h.
Optical density (OD) values were measured at 450 nm on a microplate
reader (Tecan, Männedorf, Switzerland).
Enzyme-linked immunosorbent assay
(ELISA)
LNCaP-AI and LNCaP cells
(1×104cells/well) were plated into 24-well plates and
cultured in Phenol Red-containing medium with 10% FBS or Phenol
Red-negative medium with 10% CS-FBS for 2, 4, 6 and 8 days,
respectively. Culture supernatants were harvested, and the protein
levels of prostate-specific antigen (PSA) were determined using the
Human PSA Immunoassay (R&D, Minneapolis, MN, USA) according to
the manufacturer's instructions.
Cell apoptosis analysis via flow
cytometry
Cell apoptosis was detected using the Annexin V-FITC
apoptosis detection kit (Beyotime Biotechnology, Jiangsu, China).
Cells (5×105 cells/flask) were seeded into a 25
cm2 culture flask and maintained in Phenol Red-negative
medium with 10% CS-FBS for 96 h. Cells were collected, suspended in
Annexin V-FITC binding buffer, incubated with Annexin V-FITC and PI
for 20 min at room temperature in the dark and detected by flow
cytometry.
Cell cycle analysis by flow
cytometry
The cell cycle was analyzed using a cell cycle and
apoptosis analysis kit (Beyotime). Cells (5×105
cells/flask) were seeded into a 25 cm2 culture flask and
maintained in Phenol Red-negative medium with 10% CS-FBS for 48 h.
Cells were collected, fixed with ice-cold 70% ethanol for 24 h at
4°C, washed, stained with PI staining solution for 30 min at room
temperature in dark, and detected via flow cytometry.
RNA extraction and RT-qPCR
Total RNA was isolated from tissues or cells with
RNAiso Plus (Takara Biotechnology Co., Ltd., Dalian, China)
following the manufacturer's instructions. Total RNA was
retro-transcribed into cDNA using the RevertAid™ First Strand cDNA
synthesis kit (Thermo Fisher Scientific Inc., Beijing, China)
following the manufacturer's protocols. RT-qPCR was carried out
with Thermo Scientific Maxima SYBR-Green/ROX qPCR Master Mix (2X)
(Thermo Fisher Scientific Inc.) on the StepOne™ real-time PCR
system (Life Technologies, Grand Island, NY, USA). The primer pairs
were searched from PrimerBank and synthesized by Life Technologies.
PCR was performed as follows: initial denaturation for 10 min at
95°C, and then amplification of 40 cycles at 95°C for 15 sec and
60°C for 60 sec. GAPDH was used as endogenous control. The results
were analyzed by the 2−ΔΔCT method. The primer pairs
were as follows: LSD1, 5′-TGACCG GATGACTTCTCAAGA-3′ (sense) and
5′-GTTGGAGAGTA GCCTCAAATGTC-3′ (antisense); AR, 5′-CCAGGGACCAT
GTTTTGCC-3′ (sense) and 5′-TCTGGGGTGGAAAGTAAT AGTCA-3′ (antisense);
p53, 5′-TTTGCGTGTGGAGTATTTG GAT-3′ (sense) and
5′-CAACCTCAGGCGGCTCATA-3′ (anti-sense); GAPDH,
5′-AAGGTGAAGGTCGGAGTCAAC-3′ (sense) and
5′-GGGGTCATTGATGGCAACAATA-3′ (antisense).
Western blot analysis
Total protein was extracted using cell lysis buffer
(Beyotime). Proteins were separated on SDS-PAGE gels and
transferred onto PVDF membranes (Millipore, Billerica, MA, USA).
After blocking with 5% non-fat milk, the membranes were incubated
with the primary antibodies at 4°C overnight, followed by
incubation with peroxidase-conjugated secondary antibodies
(Proteintech, Wuhan, China). Protein bands were developed with
BeyoECL Plus (Beyotime) on a Kodak Image Station 4000MM (Eastman
Kodak Company, Rochester, NY, USA). The primary antibodies against
rabbit polyclonal LSD1 (1:1,000; Abcam, Cambridge, MA, USA), mouse
monoclonal AR (1:1,000), rabbit polyclonal p53 (1:500) (both from
Santa Cruz), rabbit polyclonal H3 mono-methyl K9 (1:500) and mouse
monoclonal H3 di-methyl K9 (1:500) (both from Abcam), rabbit
polyclonal p53 di-methyl K370 (1:500; Ameritech Biomedicines,
Houston, TX, USA), rabbit monoclonal caspase-9 (1:500; Beyotime),
mouse monoclonal caspase-8 (1:500; Proteintech), rabbit polyclonal
caspase-3 (1:500; Beyotime), rabbit polyclonal Bax (1:1,000),
rabbit polyclonal PUMA (1:1,000), rabbit polyclonal Bcl-2 (1:1,000;
all from Proteintech), rabbit monoclonal Bcl-xL (1:1,000;
Beyotime), rabbit polyclonal cyclin A (1:1,000; Santa Cruz), rabbit
polyclonal cyclin E1 (1:1,000; Proteintech), mouse monoclonal CDK2
(1:1,000; Santa Cruz), mouse monoclonal cyclin D1 (1:1,000;
Proteintech), rabbit polyclonal CDK4 (1:1,000) and rabbit
polyclonal p21 (1:1000) (both from Santa Cruz) and mouse monoclonal
GAPDH (1:4,000; Proteintech) were used. GAPDH was used as the
loading control.
Co-immunoprecipitation (Co-IP)
Co-IP was performed using Pierce
Co-immunoprecipitation kit (Pierce Biotechnology, Rockford, IL,
USA) following the manufacturer's protocols. Antibodies (LSD1, AR
and p53) were immobilized onto the resin in different spin columns,
respectively. Cells were lysed with ice-cold Pierce IP lysis. Each
lysate was pre-cleared using Pierce control Agarose Resin slurry at
4°C for 1 h with gentle end-over-end mixing. Each sample of total
protein was added into a spin column containing the
antibody-coupled resin, and incubated with gentle rocking at 4°C
overnight, followed by centrifugation. Elution buffer were added,
incubated at room temperature for 5 min and centrifuged.
Flow-throughs were collected for SDS-PAGE analysis.
Statistical analysis
All experiments were repeated twice. Data analysis
was carried out with the Statistical Package for the Social
Sciences (SPSS), version 17.0 (SPSS Inc., Chicago, IL, USA). Data
are expressed as the mean ± standard deviation and P<0.05 was
considered to indicate a statistically significant difference.
Results
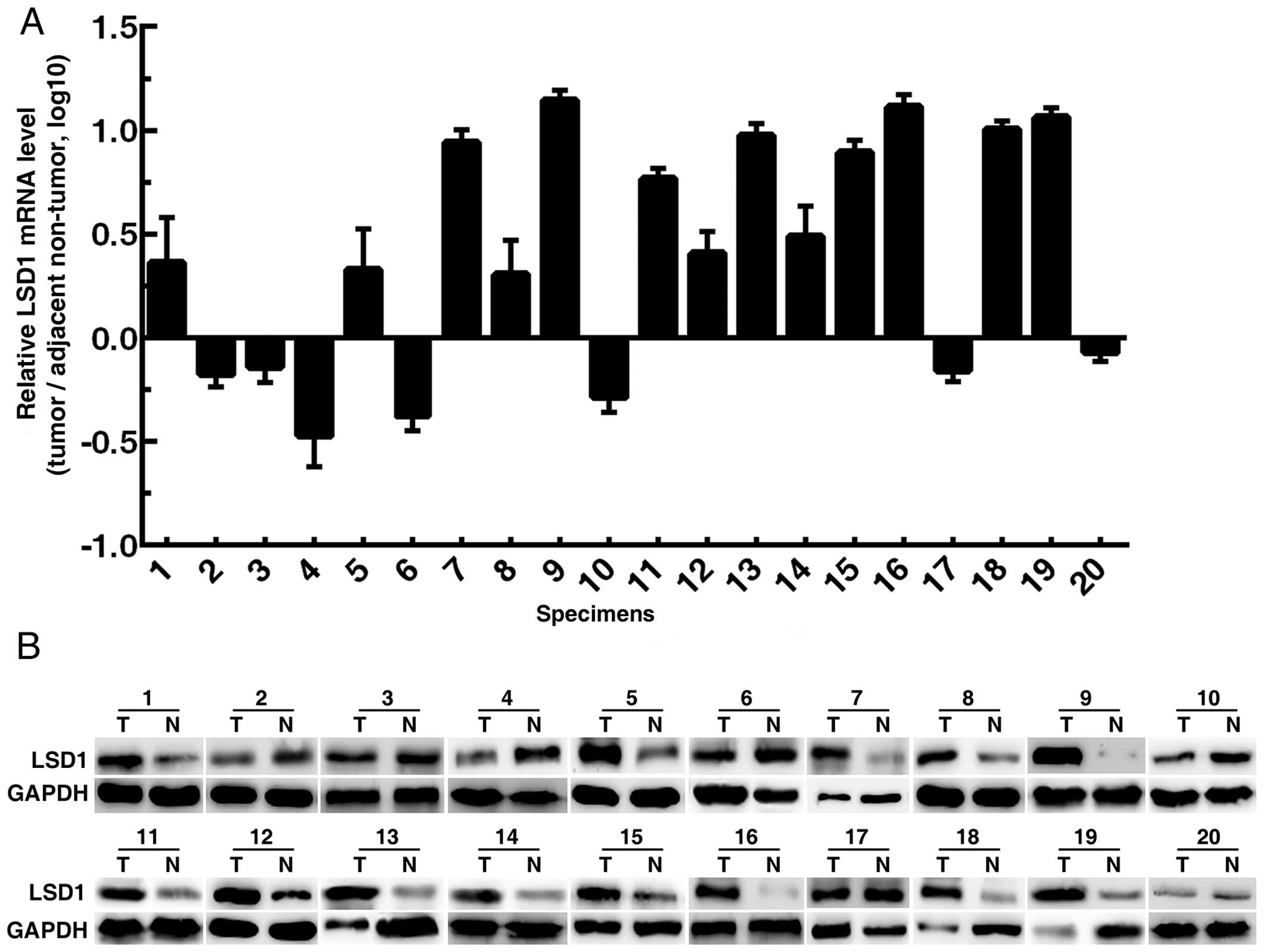
Expression of LSD1 in human PCa
specimens
Twenty pairs of PCa specimens (tumor tissues and
adjacent non-tumor tissues) were collected to perform RT-qPCR and
western blotting. The results showed that LSD1 expression in tumor
tissues was upregulated in 65% (13/20) of the specimens (Fig. 1).
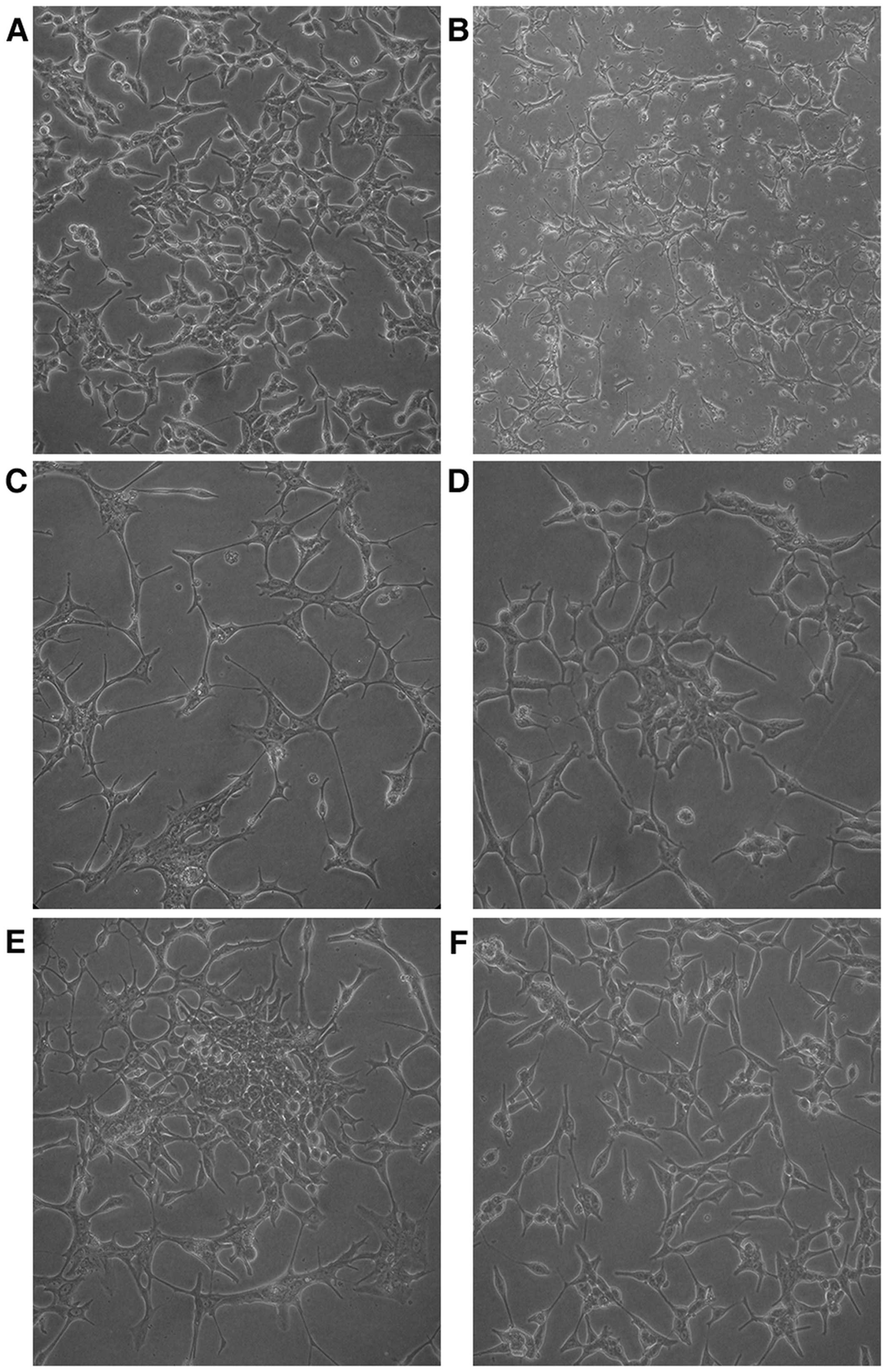
Establishment of the LNCaP-AI cell
model
In androgen-containing medium, the LNCaP cells grew
well and showed an epithelial morphology. The cell body was large
and cell processes were small and short (Fig. 2A). In androgen-deprived medium, the
LNCaP cells barely proliferated, and showed a neuroendocrine-like
morphology for a long period. The cell body turned small, and cells
were connected with each other via increased and elongated
processes (Fig. 2B and C). After 2
months, the LNCaP cells began to grow and form cell colonies
slowly, and elimination of the neuroendocrine-like state occurred
gradually (Fig. 2D and E). At the
end of the third month, cells proliferated rapidly and the
neuroendocrine-like state was not observed (Fig. 2F). At this point, the obtained cells
were androgen-independent and were named as LNCaP-AI cells.
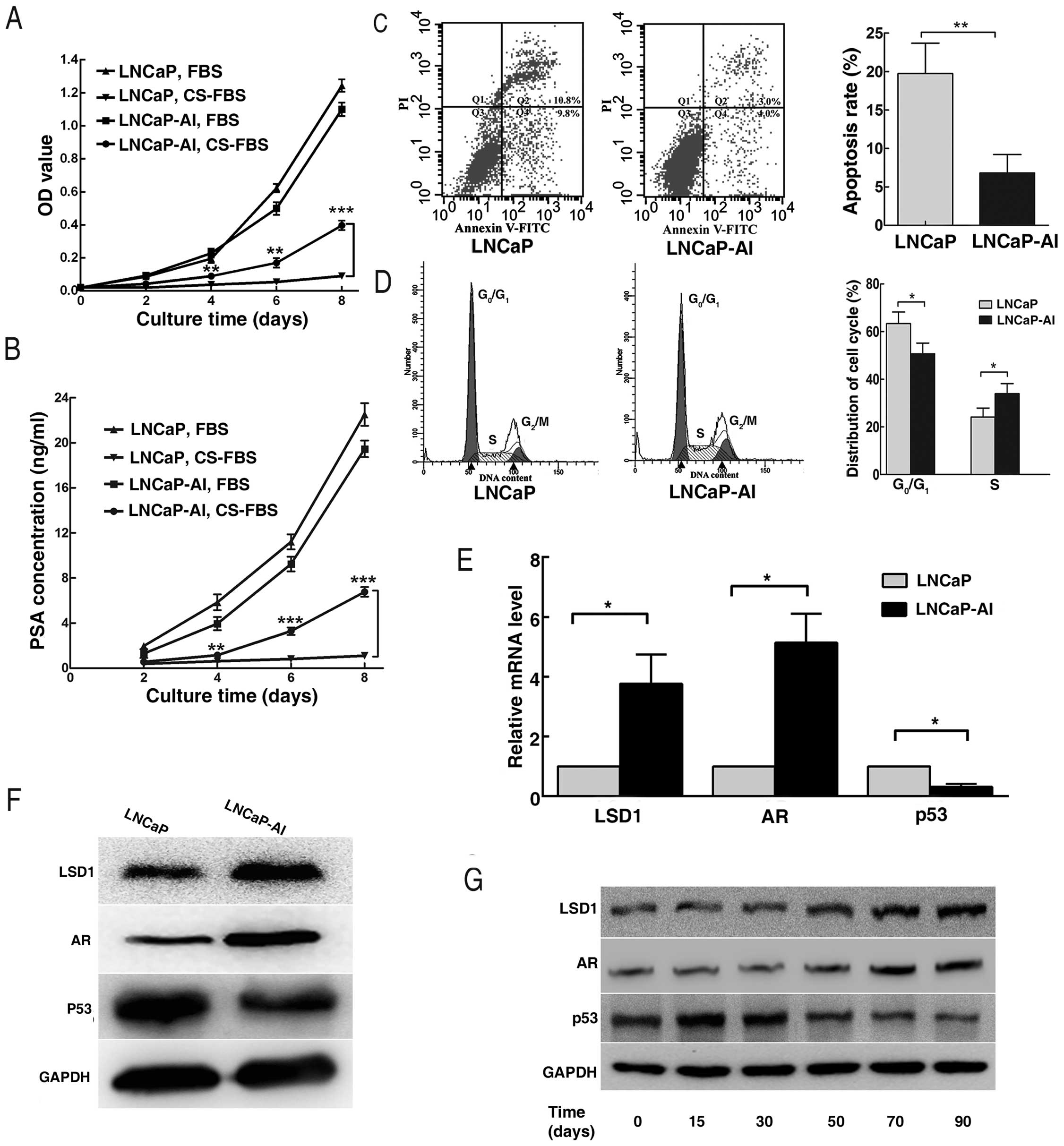
Biological characteristics of the LNCaP
and LNCaP-AI cells
CCK-8 assay showed that proliferation of the
LNCaP-AI cells was unimpeded, while growth of the parental LNCaP
cells was obviously inhibited in the androgen-deprived medium. Both
LNCaP-AI and LNCaP cells proliferated rapidly in the
androgen-containing medium (Fig.
3A). In line with this, the PSA concentration did not alter
distinctly with time in the LNCaP cell supernatant in
androgen-deprived medium, but increased in the LNCaP-AI cell
supernatant. Meanwhile, PSA concentrations in the LNCaP-AI and
LNCaP cell supernatants were both elevated continuously in the
androgen-containing-medium (Fig.
3B). Flow cytometry revealed that the LNCaP cells showed a high
rate of apoptosis and G0/G1 cell cycle arrest
in androgen-deprived medium, compared with the LNCaP-AI cells
(Fig. 3C and D). RT-qPCR and
western blotting analysis indicated that expression levels of LSD1
and AR in the LNCaP-AI cells were upregulated, while expression of
p53 was downregulated (Fig. 3E and
F). Moreover, expression levels of LSD1, AR and p53 in the
LNCaP cells at the indicated time points during androgen ablation
displayed the same trends as described above (Fig. 3G).
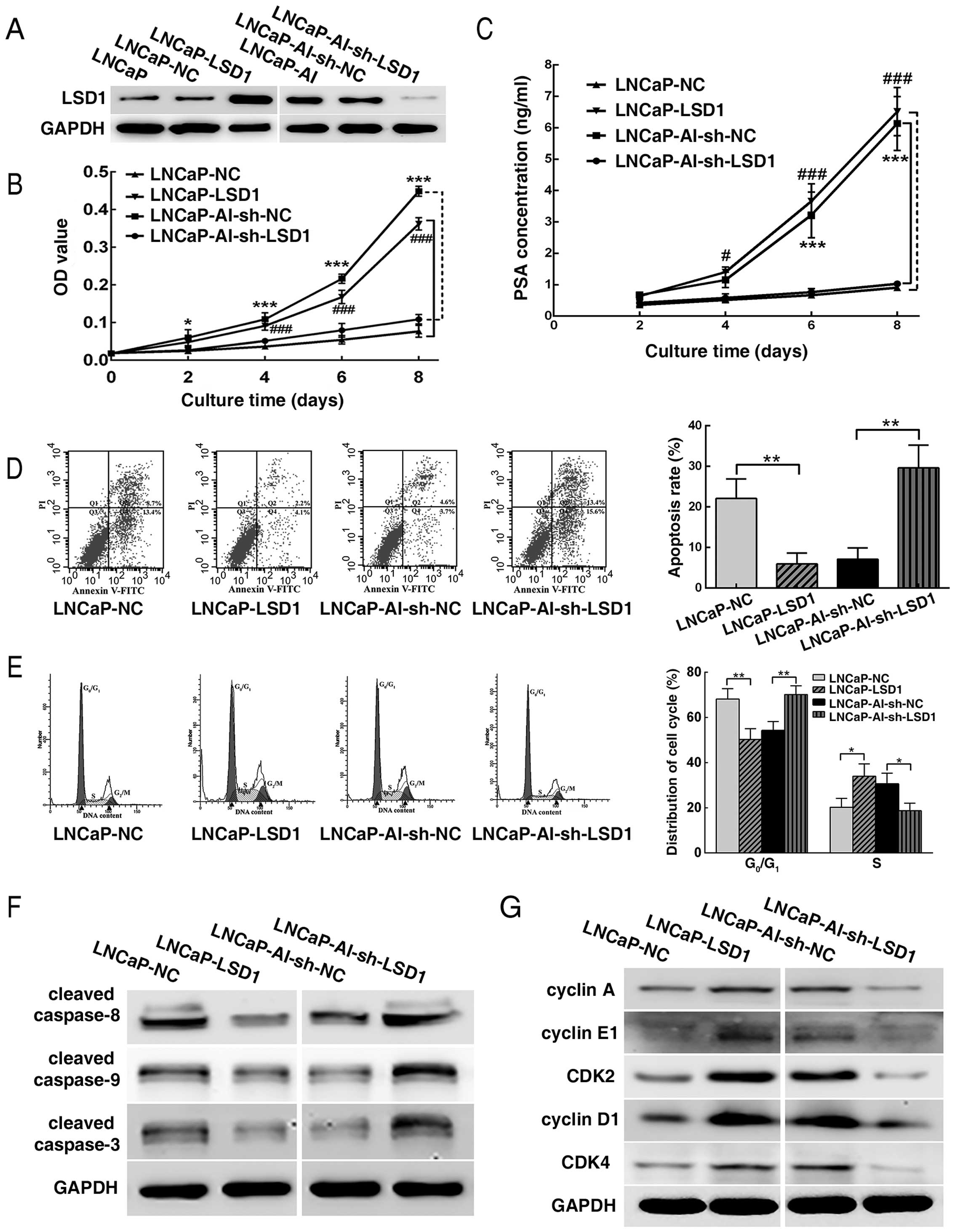
Overexpression of LSD1 promotes LNCaP
cell survival under androgen-deprived conditions
LSD1 was overexpressed in the LNCaP cells and
knocked down in the LNCaP-AI cells by plasmid transfection.
Overexpression and knockdown efficiencies of LSD1 were verified by
western blotting (Fig. 4A). CCK-8
and flow cytometry revealed that the LNCaP-LSD1 cells displayed
more rapid proliferation, a higher PSA concentration in the culture
supernatant and a lower apoptosis rate and
G0/G1 phase arrest in androgen-ablated
medium, in contrast to the LNCaP-NC cells, while LNCaP-AI-sh-LSD1
cells showed a slower growth, lower PSA concentration in the
culture supernatant and a higher apoptosis rate and
G0/G1 arrest, compared with the
LNCaP-AI-sh-NC cells (Fig. 4B–E).
Furthermore, western blot analysis revealed that cleaved caspase-8,
cleaved caspase-9 and cleaved caspase-3 were downregulated in the
LNCaP-LSD1 cells when compared to the LNCaP-NC cells. Expectedly,
the opposite results occurred in the LNCaP-AI-sh-LSD1 cells vs. the
LNCaP-AI-sh-NC cells (Fig. 4F). In
a similar manner, expression levels of cyclin A, cyclin E1, CDK2,
cyclin D1 and CDK4 were upregulated in the LNCaP-LSD1 cells vs. the
LNCaP-NC cells, while the opposite results appeared in the
LNCaP-AI-sh-LSD1 cells vs. the LNCaP-AI-sh-NC cells (Fig. 4G).
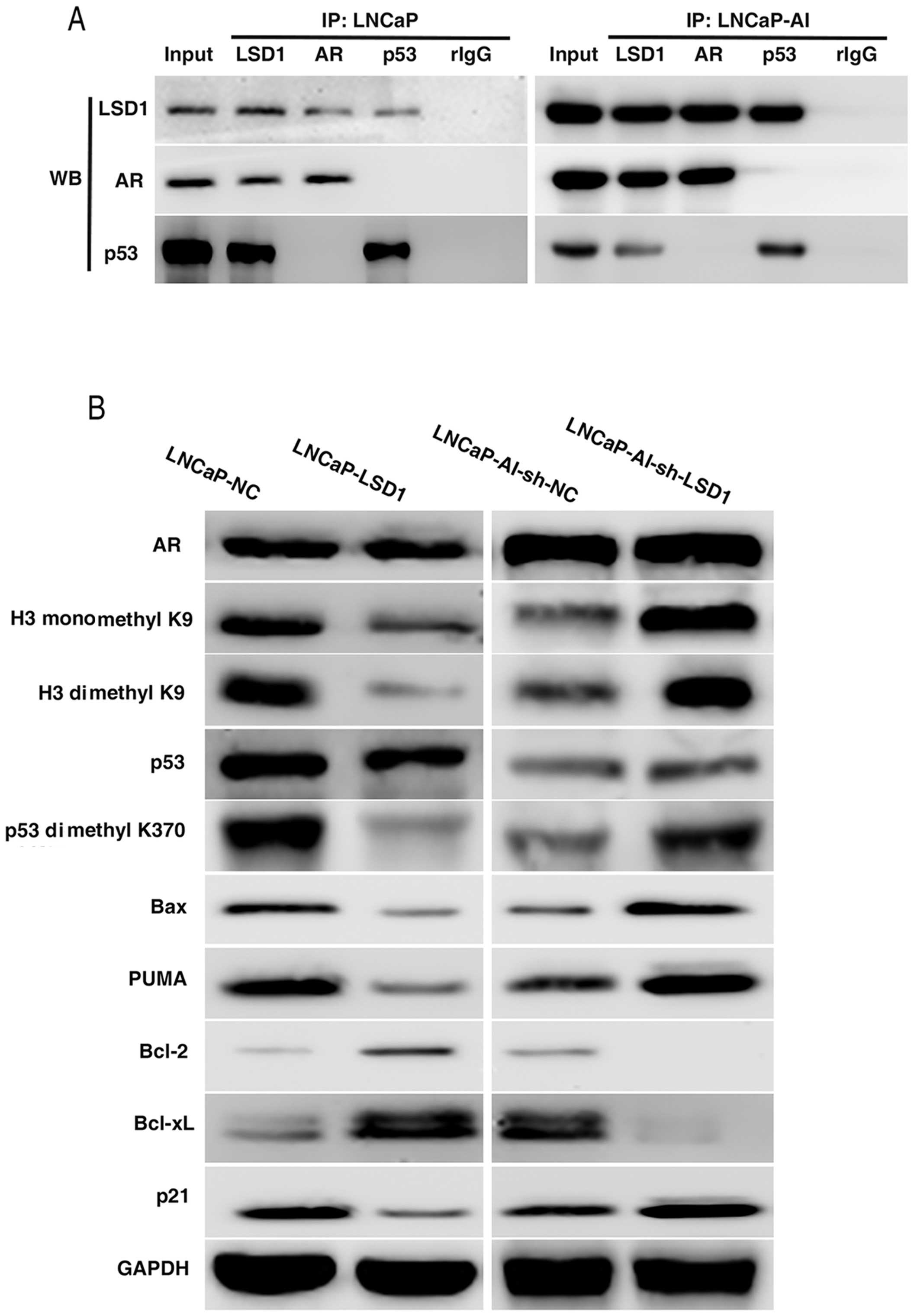
Overexpression of LSD1 activates AR and
suppresses the p53 signaling pathway through demethylation
Co-immunoprecipitation (Co-IP) assay was carried out
to ascertain whether LSD1 protein interacts with AR and p53
proteins in the LNCaP and LNCaP-AI cells. Western blot analysis
indicated that AR protein and p53 protein were detected in the
immunoprecipitated proteins by LSD1 antibody. LSD1 protein was
detected in the immunoprecipitated proteins by the AR antibody or
p53 antibody (Fig. 5A), which
confirmed that LSD1 protein interacted with the AR and p53
proteins.
When LSD1 was overexpressed in the LNCaP cells or
knocked down in the LNCaP-AI cells, the protein expression levels
of AR and p53 were not obviously changed, while the methylation of
histone H3-K9 and protein p53 K370 was distinctly altered (Fig. 5B). Mono- and di-methylation of H3-K9
and di-methylation of p53 K370 were decreased with LSD1
overexpression and were increased following LSD1 knockdown. p53
transactivates pro-apoptotic genes Bax and PUMA and
cyclin-dependent kinase inhibitor p21, and transcriptionally
represses anti-apoptotic genes Bcl-2 and Bcl-xL. Western blotting
revealed that Bax, PUMA and p21 were downregulated, while Bcl-2 and
Bcl-xL were upregu-lated, when dimethylation of p53 K370 was
decreased in the LNCaP-LSD1 cells vs. the LNCaP-NC cells.
Expectedly, opposite results were noted in the LNCaP-AI-sh-LSD1
cells vs. the LNCaP-AI-sh-NC cells (Fig. 5B).
Discussion
In the present study, we verified that the
expression of LSD1 was upregulated in human PCa specimens, which
implied the oncogenic function of LSD1. Reportedly, overexpression
of LSD1 is involved in tumorigenesis, progression, relapse and poor
prognosis in PCa (14,15,17,28).
Development of AI growth is a major obstacle to the
treatment of PCa. An ideal cell model pair is deeply desired to
better understand the molecular mechanisms of AI transition. The
LNCaP-AI cell line was generated by a 3-month culture of
androgen-dependent LNCaP cells in androgen-deprived medium
(29,30) and was defined by the ability of
proliferating under androgen-ablated conditions. The pair of cell
lines was perfectly suitable for our investigation. During the
androgen-deprived process, LNCaP cells showed an obvious
neuroendocrine-like morphology (31), which possibly rescued PCa cells from
androgen ablation (32–34).
We compared the biological behaviors of the parental
LNCaP and LNCaP-AI cells and it was shown that LNCaP-AI cells
proliferated well and restored the secretory capacity of PSA in the
absence of androgen. This phenomenon, in accordance with the PSA
levels in clinical patients with AIPC, validated the success of the
AIPC cell model establishment. Notably, LNCaP-AI and LNCaP cells
proliferated faster and produced a higher PSA concentration in the
androgen-containing environment, which showed that LNCaP-AI cells
remained androgen-responsive. Based on this finding, the priority
in clinical management of AIPC patients is to assure effective
androgen ablation. In addition, our study revealed that LNCaP-AI
cells had a lower apoptosis rate and G0/G1
arrest under androgen deprivation. Reduction in apoptosis and cell
cycle dysfunction play vital roles in cancer development and
progression, which might assist AI transition of PCa during
androgen ablation.
AR facilitates androgen-independent cellular
proliferation and cell cycle progression and contributes to the
development of AIPC (35,36). p53 affects apoptosis and cell cycle
progression, and its frequent disability may result in tumor
development and the failure of anti-neoplastic therapies (37,38).
Our data showed overexpression of LSD1 and AR and downregulation of
p53 in the LNCaP-AI cell line and in its established process.
Following overexpression of LSD1, LNCaP cells
manifested rapid proliferation, increased PSA production and less
apoptosis and G0/G1 arrest under androgen
ablation, along with downregulation of cleaved caspases and
cyclins/CDKs; knockdown of LSD1 inhibited growth and PSA secretion
in the LNCaP-AI cells and induced enhanced apoptosis and cell cycle
arrest at G0/G1 phase in the absence of
androgen, accompanied with upregulation of cleaved caspases and
cyclins/CDKs. Co-IP assay revealed that LSD1 protein interacted
with AR and p53 proteins in the LNCaP and LNCaP-AI cells, in
accordance with previous reports (4,6).
However, LSD1 overexpression and knockdown did not obviously change
the protein expression levels of AR and p53, but the methylation of
histone H3-K9 and protein p53 K370. Overexpression of LSD1 reduced
mono- and di-methylation of H3-K9 and di-methylation of p53 K370
and knockdown of LSD1 increased these methylations. Demethylation
of mono- and di-methylated residues of H3-K9 is an important marker
of AR-activated gene expression (4). Overexpression of LSD1 increased PSA
secretion in the LNCaP cells, while knockdown of LSD1 reduced PSA
production in the LNCaP-AI cells, which implied that LSD1 regulated
the expression of the AR target gene through histone demethylation.
Demethylation of di-methylated residues of p53 K370 negatively
regulates the interaction of p53 and co-activator 53BP1 and
represses the transcriptional activity of p53 (6).
Apoptosis is an active cell suicide process and
maintains cellular homeostasis. However, cancer cells can override
apoptosis through upregulating anti-apoptotic machinery and/or
downregulating the pro-apoptotic program (39). Previous research has revealed that
tumor suppressor and transcription factor p53 regulates apoptosis
through transactivation of Bax and PUMA and transcriptional
repression of Bcl-2 and Bcl-xL (24,40,41).
Control of the cell cycle monitors cell growth and DNA integrity,
but uncontrolled cell cycle progression can contribute to genomic
instability and onco-genesis (42).
p53 transcriptionally controls the p21 gene, which acts as a
cyclin-CDK inhibitor and takes charge of negative regulation of the
cell cycle (43,44). Our data revealed that overexpression
of LSD1 in LNCaP cells resulted in decreased protein levels of Bax,
PUMA and p21 and increased protein levels of Bcl-2 and Bcl-xL,
while knockdown of LSD1 in LNCaP-AI cells increased the expression
levels of Bax, PUMA and p21 and reduced the expression levels of
Bcl-2 and Bcl-xL. Taken together, these findings indicate that LSD1
regulates the activity of p53 through demethylation, and thus
regulates apoptosis and the cell cycle.
In conclusion, LSD1 is upregulated in human prostate
cancer and plays an oncogenic role. During androgen ablation, LSD1
may contribute to AI transition of prostate cancer LNCaP cells
through activation of the AR signaling pathway and suppression of
the p53 signaling pathway. Our findings may elucidate another
mechanism involved in AIPC development.
Acknowledgments
The present study was funded by the National Natural
Science Foundation of China (nos. 30973008 and 81272847) and the
Program for New Century Excellent Talents in University (no.
NCET-13-0239 to Y.X.).
Abbreviations:
|
PCa
|
prostate cancer
|
|
AIPC
|
androgen-independent prostate
cancer
|
|
AI
|
androgen-independent
|
|
LSD1
|
lysine-specific demethylase 1
|
|
AR
|
androgen receptor
|
|
PSA
|
prostate-specific antigen
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ren SC, Chen R and Sun YH: Prostate cancer
research in China. Asian J Androl. 15:350–353. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Y, Lan F, Matson C, Mulligan P,
Whetstine JR, Cole PA, Casero RA and Shi Y: Histone demethylation
mediated by the nuclear amine oxidase homolog LSD1. Cell.
119:941–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Metzger E, Wissmann M, Yin N, Müller JM,
Schneider R, Peters AH, Günther T, Buettner R and Schüle R: LSD1
demeth-ylates repressive histone marks to promote androgen-receptor
dependent transcription. Nature. 437:436–439. 2005.PubMed/NCBI
|
|
5
|
Wissmann M, Yin N, Müller JM, Greschik H,
Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R,
et al: Cooperative demethylation by JMJD2C and LSD1 promotes
androgen receptor-dependent gene expression. Nat Cell Biol.
9:347–353. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang J, Sengupta R, Espejo AB, Lee MG,
Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT,
Jenuwein T, et al: p53 is regulated by the lysine demethylase LSD1.
Nature. 449:105–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scoumanne A and Chen X: The
lysine-specific demethylase 1 is required for cell proliferation in
both p53-dependent and -independent manners. J Biol Chem.
282:15471–15475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nagasawa S, Sedukhina AS, Nakagawa Y,
Maeda I, Kubota M, Ohnuma S, Tsugawa K, Ohta T, Roche-Molina M,
Bernal JA, et al: LSD1 overexpression is associated with poor
prognosis in basal-like breast cancer, and sensitivity to PARP
inhibition. PLoS One. 10:e01180022015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim S, Janzer A, Becker A, Zimmer A,
Schüle R, Buettner R and Kirfel J: Lysine-specific demethylase 1
(LSD1) is highly expressed in ER-negative breast cancers and a
biomarker predicting aggressive biology. Carcinogenesis.
31:512–520. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X
and Song Y: Overexpression of LSD1 promotes proliferation,
migration and invasion in non-small cell lung cancer. PLoS One.
7:e350652012. View Article : Google Scholar
|
|
11
|
Hayami S, Kelly JD, Cho HS, Yoshimatsu M,
Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, et al:
Overexpression of LSD1 contributes to human carcinogenesis through
chromatin regulation in various cancers. Int J Cancer. 128:574–586.
2011. View Article : Google Scholar
|
|
12
|
Kauffman EC, Robinson BD, Downes MJ,
Powell LG, Lee MM, Scherr DS, Gudas LJ and Mongan NP: Role of
androgen receptor and associated lysine-demethylase coregulators,
LSD1 and JMJD2A, in localized and advanced human bladder cancer.
Mol Carcinog. 50:931–944. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Konovalov S and Garcia-Bassets I: Analysis
of the levels of lysine-specific demethylase 1 (LSD1) mRNA in human
ovarian tumors and the effects of chemical LSD1 inhibitors in
ovarian cancer cell lines. J Ovarian Res. 6:752013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Etani T, Suzuki T, Naiki T, Naiki-Ito A,
Ando R, Iida K, Kawai N, Tozawa K, Miyata N, Kohri K, et al: NCL1,
a highly selective lysine-specific demethylase 1 inhibitor,
suppresses prostate cancer without adverse effect. Oncotarget.
6:2865–2878. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kahl P, Gullotti L, Heukamp LC, Wolf S,
Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J,
Metzger E, et al: Androgen receptor coactivators lysine-specific
histone demethylase 1 and four and a half LIM domain protein 2
predict risk of prostate cancer recurrence. Cancer Res.
66:11341–11347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu CY, Hsieh CY, Huang KE, Chang C and
Kang HY: Cryptotanshinone down-regulates androgen receptor
signaling by modulating lysine-specific demethylase 1 function. Int
J Cancer. 131:1423–1434. 2012. View Article : Google Scholar
|
|
17
|
Willmann D, Lim S, Wetzel S, Metzger E,
Jandausch A, Wilk W, Jung M, Forne I, Imhof A, Janzer A, et al:
Impairment of prostate cancer cell growth by a selective and
reversible lysine-specific demethylase 1 inhibitor. Int J Cancer.
131:2704–2709. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kashyap V, Ahmad S, Nilsson EM, Helczynski
L, Kenna S, Persson JL, Gudas LJ and Mongan NP: The lysine specific
demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate
cancer. Mol Oncol. 7:555–566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ketscher A, Jilg CA, Willmann D, Hummel B,
Imhof A, Rüsseler V, Hölz S, Metzger E, Müller JM and Schüle R:
LSD1 controls metastasis of androgen-independent prostate cancer
cells through PXN and LPAR6. Oncogenesis. 3:e1202014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tamura K, Furihata M, Tsunoda T, Ashida S,
Takata R, Obara W, Yoshioka H, Daigo Y, Nasu Y, Kumon H, et al:
Molecular features of hormone-refractory prostate cancer cells by
genome-wide gene expression profiles. Cancer Res. 67:5117–5125.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Steinkamp MP, O'Mahony OA, Brogley M,
Rehman H, Lapensee EW, Dhanasekaran S, Hofer MD, Kuefer R,
Chinnaiyan A, Rubin MA, et al: Treatment-dependent androgen
receptor mutations in prostate cancer exploit multiple mechanisms
to evade therapy. Cancer Res. 69:4434–4442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu R, Dunn TA, Wei S, Isharwal S, Veltri
RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al:
Ligand-independent androgen receptor variants derived from splicing
of cryptic exons signify hormone-refractory prostate cancer. Cancer
Res. 69:16–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harris WP, Mostaghel EA, Nelson PS and
Montgomery B: Androgen deprivation therapy: Progress in
understanding mechanisms of resistance and optimizing androgen
depletion. Nat Clin Pract Urol. 6:76–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rozan LM and El-Deiry WS: p53 downstream
target genes and tumor suppression: A classical view in evolution.
Cell Death Differ. 14:3–9. 2007. View Article : Google Scholar
|
|
25
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qian J, Hirasawa K, Bostwick DG,
Bergstralh EJ, Slezak JM, Anderl KL, Borell TJ, Lieber MM and
Jenkins RB: Loss of p53 and c-myc overrepresentation in stage
T(2–3)N(1–3)M(0) prostate cancer are potential markers for cancer
progression. Mod Pathol. 15:35–44. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zegarra-Moro OL, Schmidt LJ, Huang H and
Tindall DJ: Disruption of androgen receptor function inhibits
proliferation of androgen-refractory prostate cancer cells. Cancer
Res. 62:1008–1013. 2002.PubMed/NCBI
|
|
28
|
Metzger E, Wissmann M and Schüle R:
Histone demethylation and androgen-dependent transcription. Curr
Opin Genet Dev. 16:513–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu S, Tsai SY and Tsai MJ: Molecular
mechanisms of androgen-independent growth of human prostate cancer
LNCaP-AI cells. Endocrinology. 140:5054–5059. 1999.PubMed/NCBI
|
|
30
|
Berthois Y, Katzenellenbogen JA and
Katzenellenbogen BS: Phenol red in tissue culture media is a weak
estrogen: Implications concerning the study of estrogen-responsive
cells in culture. Proc Natl Acad Sci USA. 83:2496–2500. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen R, Dorai T, Szaboles M, Katz AE,
Olsson CA and Buttyan R: Transdifferentiation of cultured human
prostate cancer cells to a neuroendocrine cell phenotype in a
hormone-depleted medium. Urol Oncol. 3:67–75. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burchardt T, Burchardt M, Chen MW, Cao Y,
de la Taille A, Shabsigh A, Hayek O, Dorai T and Buttyan R:
Transdifferentiation of prostate cancer cells to a neuroendocrine
cell phenotype in vitro and in vivo. J Urol. 162:1800–1805. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Deeble PD, Murphy DJ, Parsons SJ and Cox
ME: Interleukin-6- and cyclic AMP-mediated signaling potentiates
neuroendocrine differentiation of LNCaP prostate tumor cells. Mol
Cell Biol. 21:8471–8482. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vashchenko N and Abrahamsson PA:
Neuroendocrine differentiation in prostate cancer: Implications for
new treatment modalities. Eur Urol. 47:147–155. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Balk SP and Knudsen KE: AR, the cell
cycle, and prostate cancer. Nucl Recept Signal.
6:e0012008.PubMed/NCBI
|
|
36
|
Ramamurthy VP, Ramalingam S, Gediya L,
Kwegyir-Afful AK and Njar VC: Simultaneous targeting of androgen
receptor (AR) and MAPK-interacting kinases (MNKs) by novel
retinamides inhibits growth of human prostate cancer cell lines.
Oncotarget. 6:3195–3210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kastan MB, Canman CE and Leonard CJ: P53,
cell cycle control and apoptosis: Implications for cancer. Cancer
Metastasis Rev. 14:3–15. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Enoch T and Norbury C: Cellular responses
to DNA damage: Cell-cycle checkpoints, apoptosis and the roles of
p53 and ATM. Trends Biochem Sci. 20:426–430. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fernald K and Kurokawa M: Evading
apoptosis in cancer. Trends Cell Biol. 23:620–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu J and Zhang L: The transcriptional
targets of p53 in apoptosis control. Biochem Biophys Res Commun.
331:851–858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Galluzzi L, Morselli E, Kepp O, Tajeddine
N and Kroemer G: Targeting p53 to mitochondria for cancer therapy.
Cell Cycle. 7:1949–1955. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Agarwal ML, Taylor WR, Chernov MV,
Chernova OB and Stark GR: The p53 network. J Biol Chem. 273:1–4.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|