Introduction
Human T-cell leukemia virus type 1 (HTLV-1) is the
etiologic agent of adult T cell leukemia (ATL), an aggressive
neoplasia of CD4+ T cells (1,2). The
HTLV-1 transcriptional transactivator protein Tax
transactivates the expression of many cellular genes in addition to
the viral LTR (3–5). The cellular genes activated by
Tax include several involved in cell growth, which suggests
that it is the expression of this protein that deregulates T cell
growth during HTLV-1 infection and, indeed, the constitutive
expression of Tax is correlated with immortalization in T
cells and the transformation of other cell types (6–9).
Tax corresponds to a 40-kDa transforming protein (10,11)
from the pathogenic retrovirus HTLV-1 that induces the expression
of various family members of the transcription factor AP-1, such as
c-Jun, JunD, c-Fos, and Fra-1, at the level of RNA expression in T
cells (12,13).
The Jun-N-terminal kinase (JNK) is the only member
of MAP kinases to phosphorylate c-Jun, the main component of AP-1
complexes, and also has ATF-2 and Elk-1 as substrates (14,15).
In mammalian cells, three MAPK families have been clearly
characterized: namely the classical MAPK (also known as ERK), C-Jun
N-terminal kinase/stress-activated protein kinase (JNK/SAPK) and
p38 kinase. The JNK group of mitogen-activated protein kinases
(MAPKs) is activated in response to the treatment of cells with
inflammatory cytokines and by exposure to environmental stress. JNK
activation is mediated by a protein kinase cascade composed of a
MAPK kinase and a MAPK kinase kinase (14–16).
JNK and p38 kinases were initially proposed to mediate apoptosis in
neuronal cells (17), and
phosphorylation of c-Jun is necessary for neuronal cell death
(18). The use of kinase inhibitors
and overexpression of dominant-negative mutant forms of MAPKs have
demonstrated a role of JNK and/or p38 kinase in apoptosis induced
in non-neuronal cells by various stimuli, including estrogen,
cisplatin, UV-B radiation, and singlet oxygen (19,20).
Recently, a link between EGR-1 and JNK has been demonstrated
(21), and JNK-1 was found to be
able to regulate the expression of EGR-1 (22). Although EGR-1 was cloned and
characterized as an 'immediate-early response' gene, several
studies have shown that EGR-1 plays a functional role in the
regulation of growth and suppression of the transformation of
several types of human cancer cells (23,24).
Moreover, the majority of primary human glioblastoma and
astrocytomas show decreased or deleted expression of EGR-1,
indicating a potential growth suppressive role in human
glioblastomas (25,26) and small cell lung carcinoma
(27). EGR-1 rapidly activates
signal transduction pathways involving the MAPKs that activate the
Egr-1 promoter. Inducible Egr-1 gene expression is mediated in part
by the extracellular signal-regulated kinase (ERK), c-Jun
NH2-terminal kinase (JNK) and p38 pathways.
Our results revealed that EGR-1 is somewhat
detectable in quiescent human Jurkat cell lines. This expression is
strongly enhanced by PMA treatment. Blocking of EGR-1 expression by
a specific siRNA did not affect the expression of JNK induced by
Tax in Jurkat cells transfected to express the Tax
protein. Finally, our data showed that EGR-1 is constitutively
activated in Jurkat leukemia T cells expressing the Tax
protein, suggesting that EGR-1 is important but not a determinant
for the activity for Tax-induced proliferation of Jurkat
cells.
Materials and methods
Reagents
The protease inhibitors phenylmethylsulfonyl
fluoride (PMSF), leupeptin, pepstatin, aprotinin, EDTA and bestatin
were purchased from Roche (USA); T4 polynucleotide kinase and
poly(dI-dC)2 were obtained from Amersham Pharmacia Biotech
(Piscataway, NJ, USA). Tris-borate-EDTA buffer and
acrylamide-bisacrylamide (29:1) were obtained from Bio-Rad
(Richmond, CA, USA). Luciferase assay reagent, lysis buffer and the
pGL-3 luciferase vector were obtained from Promega (Madison, WI,
USA). All-trans-retinoic acid (ATRA), dimethyl sulphoxide
(DMSO), Triton X-100, 1,4-dithiotreitol (DTT), paraformaldehyde,
protease inhibitor cocktail, phosphatase inhibitor cocktail,
Tween-20, Temed, 30% acrylamide, TPA and ionomycin were purchased
from Sigma (USA). Anti JNK-1, -JNKp, -EGR-1, -β-actin, -HTLV-1 Tax
(sc-34096) antibodies were purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Cell Death Detection ELISA was purchased
from Roche (Indianapolis IN, USA).
Cell culture
Jurkat T cells and Jurkat T cells transfected to
express Tax protein, were grown in RPMI-1640 medium
containing 10% heat-inactivated FBS, 200 mM glutamine,
non-essential amino acids, penicillin, and streptomycin sulfate.
Before treatment, the cells were grown overnight (16–20 h) in
medium containing 0.5% heat-inactivated FBS and subsequently
stimulated in the presence of the same medium with low
concentrations of serum. ATRA was diluted in culture medium
containing 0.5% FBS, keeping the DMSO concentration below 0.5%.
Appropriate controls containing the same amount of solvent were
included in each experiment. Intermittent passage in
G418-containing medium was performed to ensure retention of the
plasmid (28).
Plasmid construction and preparation of
nuclear extracts
The plasmid expressing the wild-type of the
Tax protein and the Tax-inducible cell line, were a
gift from Dr Warner Greene (Gladstone Institute of Virology and
Immunology, University of California, San Francisco, San Francisco,
CA, USA). The human IL-2 promoter-enhancer fragment (−500 to +60)
was subcloned from plasmid SV-IL-2-CAT into the luciferase vector
pGL-2 (Promega) (28). The
AP-1-luciferase reporter plasmid driven by the rat prolactin
minimal promoter (−36 to +37) under the control of four copies of
the human AP-1 site (28) was
kindly provided by M. Rincón and R.A. Flavell (Section of
Immunobiology, Howard Hughes Medical Institute, Yale University
School of Medicine, New Haven, CT, USA). Plasmids containing
multimers of the recognition sites for NF-κB and AP-1 were
constructed and linked to the pLuc-prolactin minimal promoter
plasmid (28). The orientation for
each element was confirmed by restriction enzyme cleavage. The
tandem sequences used to construct the different multimer plasmids
were as follows: i) four copies of the AP-1-responsive element (the
12-O-tetradecanoylphorbol-13-acetate [TPA]-responsive
element of the human collagenase promoter),
(5′-TCGATTGAGTCAGGGTAA-3′), and ii) two copies of the NF-κB-binding
site of the human Igκ light chain enhancer (5′-GGGACTTTCC-3′)
(28).
siRNA preparation and transfection of
small interfering RNA
The siRNAs for Egr-1 and JNK-1 were obtained as
ready-annealed, purified duplex probes, and the scrambled control
siRNAs were purchased from Shanghai Genechem Co.: siRNA for Egr-1
(sense, 5′-CAGCAGCAGCAGCAGCAGCTT-3′ and antisense,
5′-AAGCTGCTGCGCTGCTGCTG-3′); siRNA oligonucleotides for JNK-1
(sense, 5′-AAGCCCAGTA ATATAGTAGTA-3′ and antisense,
5′-TACTACTATATTACTGGGCTT-3′). The cells were cultured in medium
without antibiotics and 24 h before transfection resulting in
confluency of the cell monolayer by 50–70%. Specific EGR-1 and
JNK-1 siRNAs or non-silencing siRNA (70 nmol) were mixed with
Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's
recommendation and added to the cells. After 6 h at 37°C, the
medium was changed, and the cells were cultivated in RPMI-1640
medium supplemented with 10% heat-inactivated FBS.
Cell proliferation assay
Cell survival was measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
cell proliferation assay. Cells (10,000/well) were seeded in
96-well plates in 100 μl of medium containing the
appropriate amount of serum. Jurkat and Jurkat-Tax cells were
transfected or not with siRNA-control or siRNA-Egr-1 and were then
treated with ATRA 8 μM or solvent (DMSO) for 1, 2, 4 or 6
days, when 10 μl/well of 5 mg/ml MTT was added. After 4 h of
incubation at 37°C, the tetrazolium crystals were solubilized
overnight at 37°C with 10% SDS and 10 mM HCl, and the optical
density (OD) at 595 nm was measured.
Transient transfection and luciferase
assays
Transfection of cells was conducted by
electroporation, using an Electro Cell Manipulator 600 (BTX, San
Diego, CA, USA) using 130 V/1,700 μF capacitance. Briefly,
8×106 cells were transfected with 10 μg of
luciferase reporter plasmid and 5 μg of each expression
plasmid, and the mixture was incubated for 24 h. Transfected cells
were cultured in complete medium for 24 h and stimulated with ATRA
for another 8 h. Cells were harvested 32 h post-transfection,
washed twice in PBS, and treated with lysis buffer (Luciferase
assay; Promega) for 5 to 10 min on ice. Lysates were spun down for
1 min, and the total supernatants were analyzed using luciferase
reagent (Promega) and measured as a duplicate in a luminometer
(MicroLumat LB 96 P; Berthold) for 5 sec. Background measurement
was subtracted from each duplicate, and experimental values are
expressed either as recorded light units of luciferase activity or
as relative activity compared with extracts from unstimulated cells
(29). Nuclear extracts were
prepared as previously described (29,30).
Western blot analysis
The Jurkat cell line (5×107) was seeded
onto 6-well plates. Forty-eight hours after transfection, the cells
were collected and washed twice by cold PBS, and each well was
treated with 50 ml lysis buffer (2 mmol/l Tris-HCl, pH 7.4, 50
mmol/l NaCl, 25 mmol/l EDTA, 50 mmol/l NaF, 1.5 mmol/l
Na3VO4, 1% Triton X-100, 0.1% SDS,
supplemented with protease inhibitors 1 mmol/l
phenylmethylsulfonylfluoride, 10 mg/l pepstatin, 10 mg/l aprotinin
and 5 mg/l leupeptin) (all from Sigma). Protein concentrations were
determined using the Bradford protein assay. Equal amounts of
protein (50 mg) were separated on a 15% SDS-polyacrylamide gel and
transferred to a nitrocellulose membrane (Hybond-C; Amersham,
Freiburg, Germany). The membranes were blocked in 5% non-fat dry
milk in TBS for 1 h at room temperature and probed with anti-HTLV-1
Tax (sc-34096), anti-JNK-1 (sc-1648) or with anti-EGR-1 (sc-110)
antibody (dilution 1:500; Santa Cruz Biotechnology) overnight at
4°C. After 3 washings with TBS containing 0.1% Tween-20, the
membranes were incubated with anti-rabbit IgG-horseradish
peroxidase (1:5,000; Santa Cruz Biotechnology), and developed by
luminal-mediated chemiluminescence (Appylgen Technologies, Inc.,
China). To confirm equal protein loading, the membranes were
reprobed with a 1:1,000 dilution of an anti-actin antibody (Santa
Cruz Biotechnology). Densitometric analyses were performed using
Scion Image software (31).
DNA fragmentation
DNA fragmentation was determined using a Cell Death
Detection ELISA (Roche). Jurkat and Jurkat-Tax cells were treated
with apoptotic compounds for the indicated periods of time. Cells
were harvested by centrifugation and lysed in 0.5 ml of the lysis
buffer provided with the kit. Two milliliters of the extract was
used for the ELISA, which was performed as instructed by the
manufacturer. The OD at 405 nm was measured and the fold induction
of apoptosis was calculated using untreated cells as control.
Labeling of apoptotic cells with Annexin
V
Jurkat cells (500,000) were treated with RA as
indicated. The cells were washed with PBS and stained with Annexin
V-FITC (Pharmingen) and PI in binding buffer (10 mM HEPES (pH 7.4,
140 mM NaCl 2.5 mM CaCl2) for 15 min at room temperature
in the dark. Cells were subsequently analyzed by flow cytometry
(FACSCalibur) for apoptosis (FITC) and viability (PI).
Results
We previously reported that application of ATRA
signaling causes reduction in Tax activity in Jurkat leukemia cells
transfected to express the Tax protein (31). However, the role of EGR-1 in
Tax-induced T cell proliferation has not been fully understood. To
address this issue, the effects of EGR-1 on the transcriptional
activity of Tax and JNK proteins in the presence of ATRA signaling
pathways were examined.
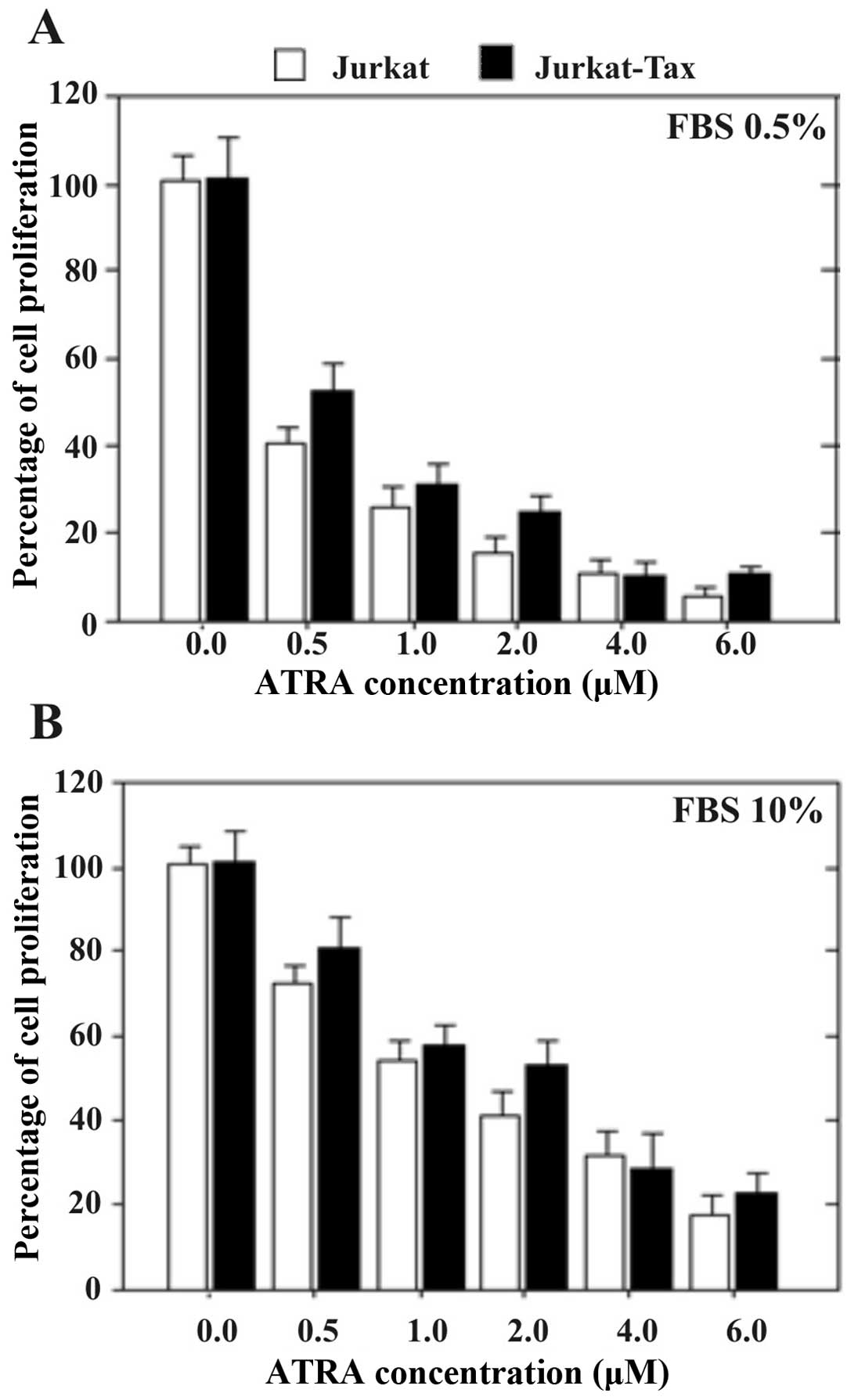
Jurkat cells expressing the Tax protein
exhibit significantly higher anti-proliferative activity when grown
in low concentrations of serum (0.5% FBS) in comparison to high
serum concentration (10% FBS)
Jurkat cells (wt) and Jurkat cells expressing the
Tax protein were grown with increasing concentrations of
ATRA for 1, 2, or 3 days in the presence of a low (0.5%) or high
(10%) concentration of serum. Cell growth was measured using the
MTT assay. Fig. 1A shows the
results obtained in Jurkat and Jurkat-Tax cells after 24 h of
incubation.
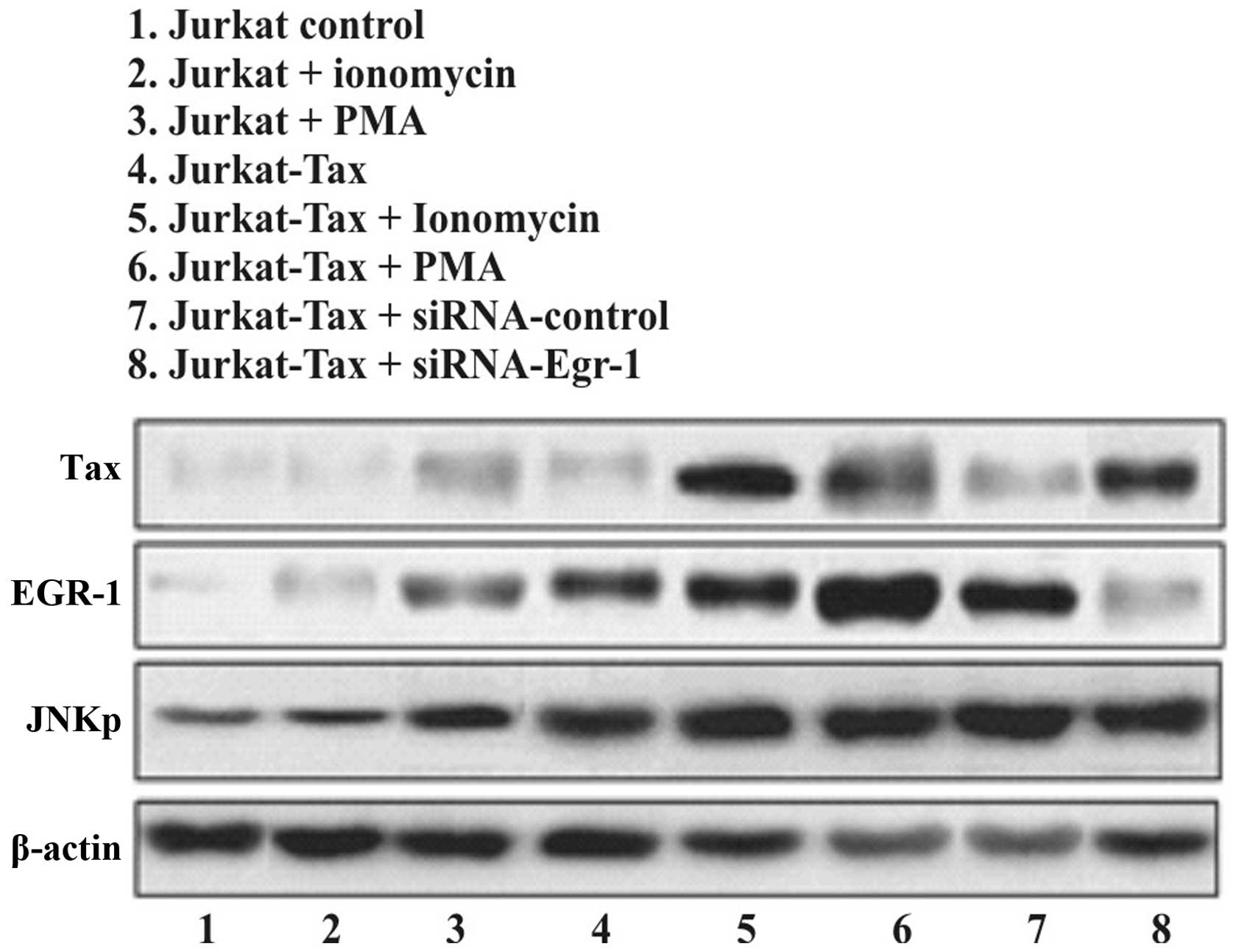
Small interfering RNA against EGR-1 did
not affect the induction of Tax protein in Jurkat and Jurkat cells
transfected to express the Tax protein
EGR-1 is not required for the expression of Tax
protein induced by certain stimuli. We investigated whether the
siRNA-EGR-1 induced or blocked Tax activity, compared with the
induction of EGR-1 and JNK-1 proteins. Cells were incubated for 16
h in medium containing 0.5% FBS prior to ionomycin or TPA
stimulation as positive control, or treated with siRNA-control and
siRNA against EGR-1 (Fig. 2). A
significant activation of Tax (top), EGR-1 (middle) and JNK-1
(bottom) was observed after 120 min of exposure to the stimuli.
However, the treatment of cells with siRNA against EGR-1 did not
affect the expression of either Tax or JNK-1 proteins (Fig. 2 top and bottom, respectively).
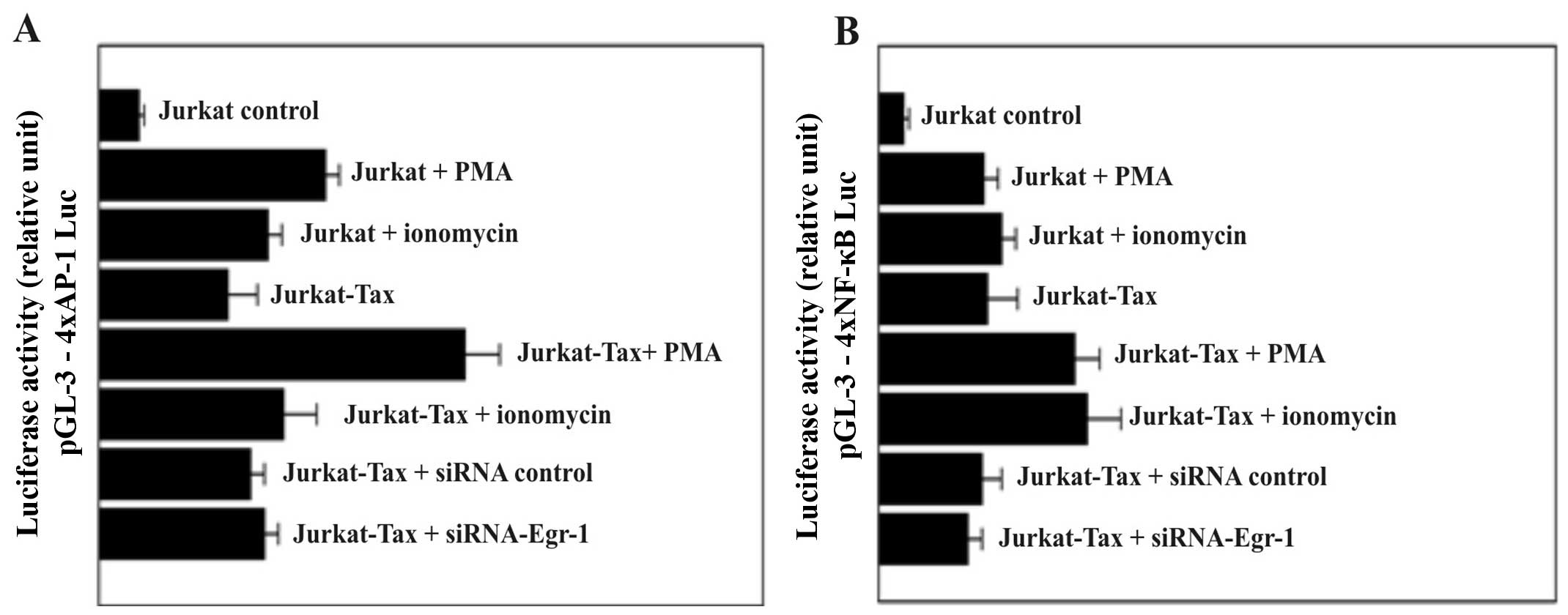
Firefly luciferase activity levels in
Jurkat-Tax cells transfected with reporter plasmids containing
multiple consensus sequences of NF-κB and AP-1 response elements
were not affected by inhibition of the EGR-1 expression
We transfected cells with a reporter plasmid
containing multiple consensus sequences of 4xAP-1 (Fig. 3A) and/or 4xNF-κB (Fig. 3B) response elements linked to a
minimal promoter controlling transcription of the firefly
luciferase cDNA. Both nuclear factors, AP-1 and NF-κB, play
important roles in inflammation, immune responses, cell growth and
apoptosis. PMA and ionomycin are potent activators of AP-1 and/or
NF-κB responsive genes. In the present study, we demonstrated that
transiently transfected cells with the 4xAP-1 (Fig. 3A) exhibited stronger and consistent
luciferase expression in response to PMA and ionomycin stimulation
compared with cells transfected with the luciferase vector carrying
the 4xNF-κB (Fig. 3B) response
element. At the same time, the expression of luciferase activity in
Jurkat-Tax cells was not affected by treatment of the cells with
either the siRNA-control or siRNA-Egr-1 (Fig. 3).
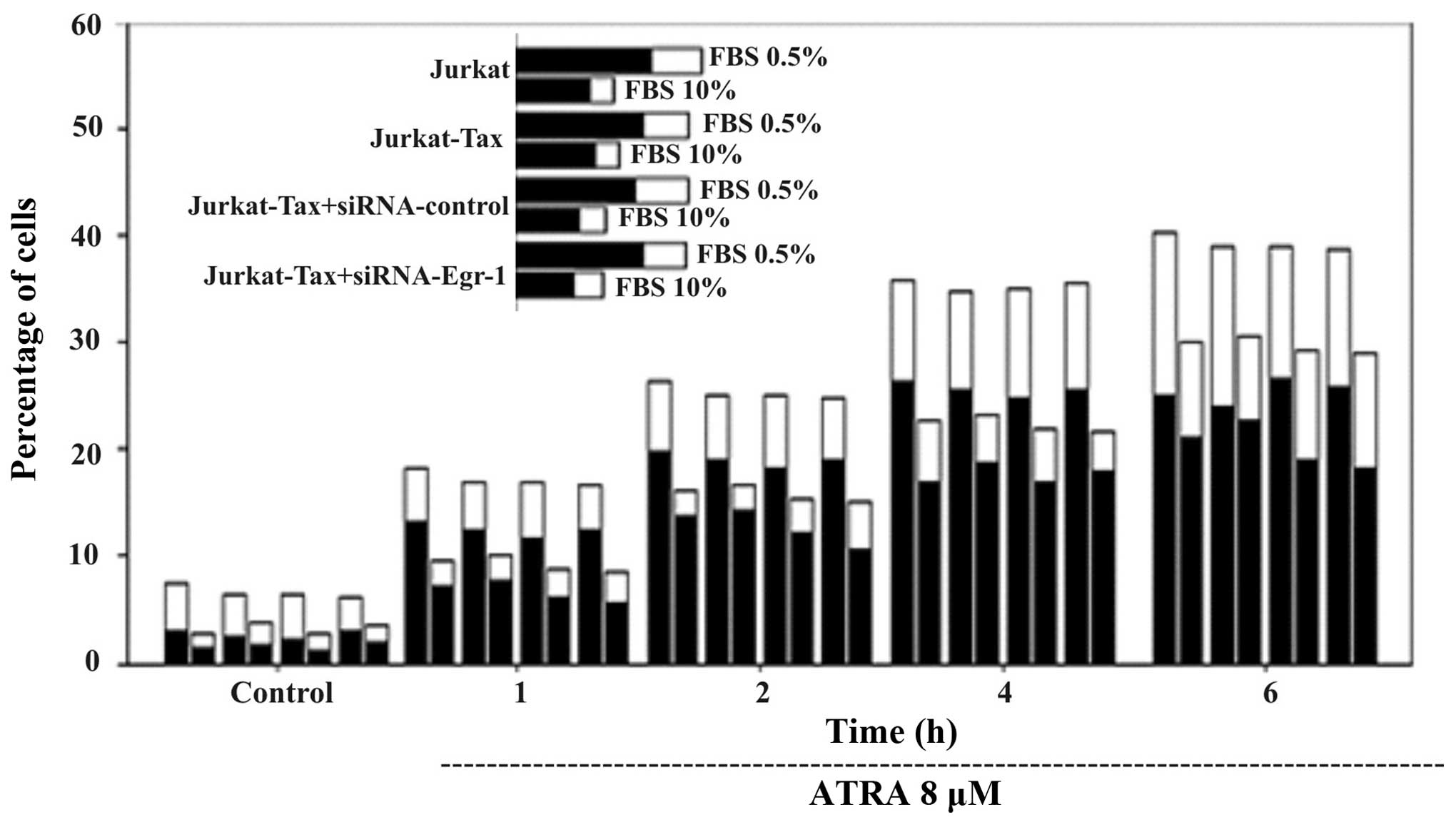
Silencing of Egr-1 expression does not
block ATRA-induced Jurkat-Tax cell apoptosis but is clearly
dependent on the concentration of FBS concentration (0.5 or
10%)
To determine whether the silencing of Egr-1
expression is correlated with the increase or reduction in
ATRA-induced Jurkat-Tax cell apoptosis, RNA interference was used.
As shown in Fig. 4, transient
transfection of a specific siRNA against Egr-1 (siRNA-Egr-1)
slightly attenuated ATRA-induced apoptosis in the Jurkat and
Jurkat-Tax-cells. However, these results were clearly dependent on
the concentration of FBS and on the time of incubation at a given
ATRA concentration. As shown in Fig.
4, Jurkat and Jurkat-Tax cells were incubated with either PBS
0.5% (left columns) or with PBS 10% (right columns) for the
indicated periods of time, and apoptosis was determined by double
staining with Annexin V-FITC and PI followed by cytometric
analysis. The percentages of Annexin V-positive/PI-negative cells
(indicative of early apoptosis) (black columns) and double-positive
cells (late apoptotic and/or necrotic) (white columns) are shown.
Cells incubated with vehicle for 1 h were used as control. The
induction of DEVDase activity was higher when a low concentration
of serum was present in the culture medium. The appearance of
Annexin V-positive (apoptotic) cells was evident after only 1 h of
incubation with ATRA, and reached a maximum after 4–6 h (Fig. 4). We showed that the percentage of
apoptotic cells was higher in the presence of a low serum
concentration (0.5% FBS) compared with a high serum concentration
(10% FBS).
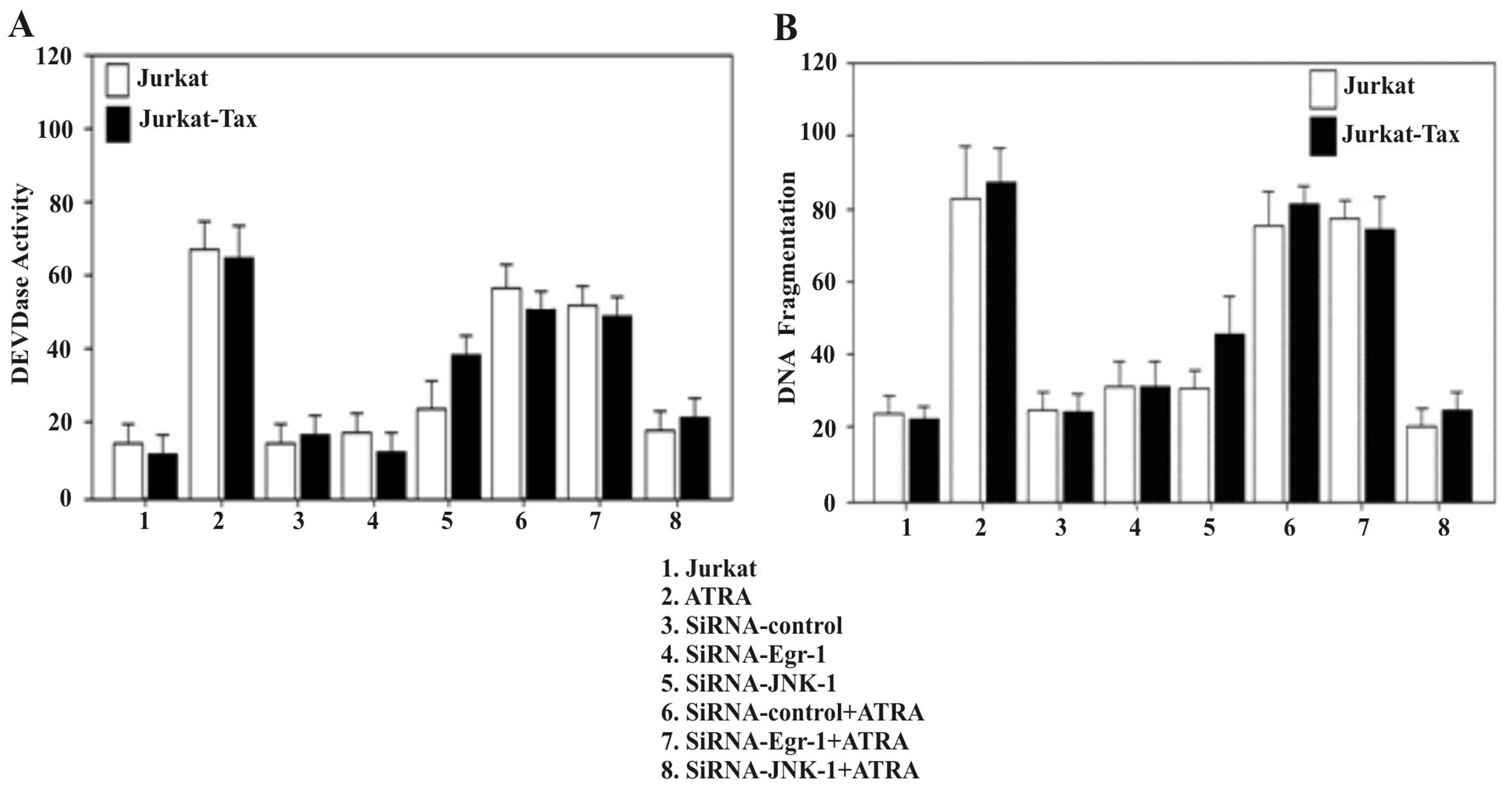
Induction of apoptosis by ATRA is not
altered by inhibition of the EGR-1 pathway
Several studies have demonstrated the inhibitory
effect of ATRA on the proliferation of Jurkat cells (31). We examined the ability of ATRA to
induce apoptosis in Jurkat and Jurkat-Tax cells by analysis of
DEVDase activity (Fig. 5A) and DNA
fragmentation (Fig. 5B). To examine
whether EGR-1 and/or JNK-1 activities are required for the
induction of apoptosis by ATRA in Jurkat-Tax cells, we examined the
effect of small interfering RNA inhibitors against Egr-1 and JNK-1
on the induction of apoptosis by ATRA. Jurkat cells were treated
with siRNA-Egr-1 or siRNA-JNK-1 or non-silencing siRNA as the
control, prior to treatment with 8 μM ATRA for 2 h. Fig. 5A shows that the specific siRNA-Egr-1
had no effect on the apoptosis induced by ATRA, as determined by
DEVDase activity (Fig. 5A) and by
DNA fragmentation (Fig. 5B). In
contrast, inhibition of JNK-1 by a specific siRNA reduced
ATRA-induced apoptosis (Fig. 5).
The results demonstrated that the induction of apoptosis by ATRA in
Jurkat-Tax cells was clearly dependent on the presence of JNK
kinase activity (Fig. 5).
Discussion
The HTLV-1 transcriptional activator protein
Tax transactivates the expression of many cellular genes in
addition to the viral LTR (1,2,10,11,32).
Previous studies have demonstrated that the Tax response of the
IL-2R α chain promoter is mediated by the activation of NF-κB
(33,34). Tax increases the expression of NF-κB
proteins (35) and prolongs its
localization to the nucleus where it is transcriptionally active
(35,36). Tax also interacts specifically with
IκB proteins and induces its degradation (34,37,38),
and in vitro studies have shown that several retinoids are
strong inducers of apoptosis in a wide variety of cancer cell lines
(39–41).
Here, we showed that activation of Tax protein by
JNK-1 kinase was not dependent on the presence or not of the early
growth response factor-1 gene product. Luciferase assay showed that
AP-1 consensus elements were required for strong luciferase
activity, while the NF-κB response element remained low, suggesting
that activation of JNK is required for ATRA-induce apoptosis in
Jurkat-Tax cells which is necessary for the transcriptional
activation of c-jun. There is a strong and sustained activation of
JNK-1 and EGR-1 before and after ATRA treatment of Jurkat-Tax
cells. JNK activity correlates with the induction of apoptosis, as
determined by measurement of DEVDase activity, DNA fragmentation
and labelling of cells with Annexin V. Our data demonstrated that
the activation or blocking of EGR-1 does not have any effect on the
activation and expression of JNK-1 kinase in Jurkat-Tax leukemia
cells. Importantly, JNK activation was sufficient for the induction
of apoptosis by ATRA, since inhibition of EGR-1 by a specific siRNA
had no effect on ATRA-induce apoptosis. As observed, the strong
activation of JNK-1 and EGR-1 induced by PMA and ionomycin was
further increase in both Jurkat and Jurkat-Tax cells. However,
EGR-1 did not appear to be essential for Tax activation, suggesting
that it may not be necessary for JNK mediated-Jurkat-Tax cell
proliferation. More probably the role of EGR-1 in Tax-induce
proliferation of Jurkat cells is independent of JNK-1 kinase
activity. Activation of JNK-1 kinase in Jurkat-Tax cells led to the
phosphorylation of c-Jun a member of the transcription factor AP-1
(14–18). The results suggest that activation
of JNK-1 kinase in Jurkat-Tax cells may lead to the phosphorylation
of certain anti-apoptotic proteins promoting cell proliferation. On
the other hand, in the presence of ATRA, the JNK kinase activity
may lead to the activation of pro-apoptotic protein favoring the
induction of apoptosis. The JNK signaling pathway is involved in a
variety of cellular responses, and the outcomes of cellular
response are varied and complicated. Similar to the JNK pathway,
the involvement of EGR-1 in apoptosis is also diverse (22,23,42,43).
It has been shown that both EGR-1 and JNK signaling promotes cell
death (21,44), whereas it has also been shown that
both cascades enhance survival (42,45),
cell growth (46), and
differentiation (47). These data
therefore indicate that the JNK or EGR-1 pathway is required for
apoptosis and survival depending on cell types and conditions.
Although it is known that JNK kinase participates in the induction
of apoptosis and/or cell survival by various chemical drugs, the
real contribution of EGR-1 in Tax-transfected Jurkat cells
remains to be examined.
Acknowledgments
We gratefully acknowledge Dr Warner Greene
(Gladstone Institute of Virology and Immunology, University of
California and San Francisco, CA, USA) for providing the plasmid
expressing the wild-type of the Tax protein and the Tax-inducible
cell line. The present study was supported by the Biomedical
Experimental Laboratory and Intramural Regular Research Grant,
UTA-6710-14, University of Tarapacá, Arica-Chile.
References
|
1
|
Araya N, Sato T, Yagishita N, Ando H,
Utsunomiya A, Jacobson S and Yamano Y: Human T-lymphotropic virus
type 1 (HTLV-1) and regulatory T cells in HTLV-1-associated
neuroinflammatory disease. Viruses. 3:1532–1548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hinuma Y, Nagata K, Hanaoka M, Nakai M,
Matsumoto T, Kinoshita KI, Shirakawa S and Miyoshi I: Adult T-cell
leukemia: Antigen in an ATL cell line and detection of antibodies
to the antigen in human sera. Proc Natl Acad Sci USA. 78:6476–6480.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cann AJ, Rosenblatt JD, Wachsman W, Shah
NP and Chen IS: Identification of the gene responsible for human T-
cell leukaemia virus transcriptional regulation. Nature.
318:571–574. 1985. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Felber BK, Paskalis H, Kleinman-Ewing C,
Wong-Staal F and Pavlakis GN: The pX protein of HTLV-I is a
transcriptional activator of its long terminal repeats. Science.
229:675–679. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sodroski J, Rosen C, Goh WC and Haseltine
W: A transcriptional activator protein encoded by the x-lor region
of the human T-cell leukemia virus. Science. 228:1430–1434. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grassmann R, Berchtold S, Radant I, Alt M,
Fleckenstein B, Sodroski JG, Haseltine WA and Ramstedt U: Role of
human T-cell leukemia virus type 1 X region proteins in
immortalization of primary human lymphocytes in culture. J Virol.
66:4570–4575. 1992.PubMed/NCBI
|
|
7
|
Siekevitz M, Feinberg MB, Holbrook N,
Wong-Staal F and Greene WC: Activation of interleukin 2 and
interleukin 2 receptor (Tac) promoter expression by the trans
activator (tat) gene product of human T-cell leukemia virus, type
I. Proc Natl Acad Sci USA. 84:5389–5393. 1987. View Article : Google Scholar
|
|
8
|
Ballard DW, Bohnlein E, Lowenthal JW, Wano
Y, Franza BR and Greene WC: HTLV-I tax induces cellular proteins
that activate the kappa B element in the IL-2 receptor alpha gene.
Science. 241:1652–1655. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leung KY and Nabel GJ: HTLV-1
transactivator induces interleukin-2 receptor expression through an
NF-kappa B-like factor. Nature. 333:776–778. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Franchini G: Molecular mechanisms of human
T-cell leukemia/ lymphotropic virus type I infection. Blood.
86:3619–3639. 1995.PubMed/NCBI
|
|
11
|
Franklin AA and Nyborg JK: Mechanisms of
Tax regulation of human T cell leukemia virus type I gene
expression. J Biomed Sci. 2:17–29. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bergers G, Graninger P, Braselmann S,
Wrighton C and Busslinger M: Transcriptional activation of the
fra-1 gene by AP-1 is mediated by regulatory sequences in the first
intron. Mol Cell Biol. 15:3748–3758. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Angel P and Karin M: The role of Jun, Fos
and the AP-1 complex in cell-proliferation and transformation.
Biochim Biophys Acta. 1072:129–157. 1991.PubMed/NCBI
|
|
14
|
Fuchs SY, Xie B, Adler V, Fried VA, Davis
RJ and Ronai Z: c-Jun NH2-terminal kinases target the
ubiquitination of their associated transcription factors. J Biol
Chem. 272:32163–32168. 1997. View Article : Google Scholar
|
|
15
|
Yao M, Nguyen TV and Pike CJ:
Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal
kinase-dependent down-regulation of Bcl-w. J Neurosci.
25:1149–1158. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: Conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999.PubMed/NCBI
|
|
17
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Behrens A, Sibilia M and Wagner EF:
Amino-terminal phosphorylation of c-Jun regulates stress-induced
apoptosis and cellular proliferation. Nat Genet. 21:326–329. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang CC and Shapiro DJ: Activation of the
p38 mitogen-activated protein kinase pathway by estrogen or by
4-hydroxytamoxifen is coupled to estrogen receptor-induced
apoptosis. J Biol Chem. 275:479–486. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu R, Mandlekar S, Tan TH and Kong AN:
Activation of p38 and c-Jun N-terminal kinase pathways and
induction of apoptosis by chelerythrine do not require inhibition
of protein kinase C. J Biol Chem. 275:9612–9619. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hoffmann E, Ashouri J, Wolter S, Doerrie
A, Dittrich-Breiholz O, Schneider H, Wagner EF, Troppmair J,
Mackman N and Kracht M: Transcriptional regulation of EGR-1 by the
interleukin-1-JNK-MKK7-c-Jun pathway. J Biol Chem. 283:12120–12128.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parra E, Ferreira J and Ortega A:
Overexpression of EGR-1 modulates the activity of NF-κB and AP-1 in
prostate carcinoma PC-3 and LNCaP cell lines. Int J Oncol.
39:345–352. 2011.PubMed/NCBI
|
|
23
|
Parra E and Ferreira J: The effect of
siRNA-Egr-1 and camptothecin on growth and chemosensitivity of
breast cancer cell lines. Oncol Rep. 23:1159–1165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parra E, Ortega A and Saenz L:
Down-regulation of Egr-1 by siRNA inhibits growth of human prostate
carcinoma cell line PC-3. Oncol Rep. 22:1513–1518. 2009.PubMed/NCBI
|
|
25
|
Liu C, Yao J, Mercola D and Adamson E: The
transcription factor EGR-1 directly transactivates the fibronectin
gene and enhances attachment of human glioblastoma cell line U251.
J Biol Chem. 275:20315–20323. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Calogero A, Arcella A, De Gregorio G,
Porcellini A, Mercola D, Liu C, Lombari V, Zani M, Giannini G,
Gagliardi FM, et al: The early growth response gene EGR-1 behaves
as a suppressor gene that is down-regulated independent of ARF/Mdm2
but not p53 alterations in fresh human gliomas. Clin Cancer Res.
7:2788–2796. 2001.PubMed/NCBI
|
|
27
|
Zhang H, Chen X, Wang J, Guang W, Han W,
Zhang H, Tan X and Gu Y: EGR1 decreases the malignancy of human
non-small cell lung carcinoma by regulating KRT18 expression. Sci
Rep. 4:54162014.PubMed/NCBI
|
|
28
|
Parra E, Varga M, Hedlund G, Kalland T and
Dohlsten M: Costimulation by B7-1 and LFA-3 targets distinct
nuclear factors that bind to the interleukin-2 promoter: B7-1
negatively regulates LFA-3-induced NF-AT DNA binding. Mol Cell
Biol. 17:1314–1323. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parra E, Varga M, Sigvardsson M, Hedlund
G, Kalland T, Leanderson T, Sjogren H and Dohlsten M: Costimulation
of human CD4+ T cells with LFA-3 and B7 induce distinct
effects on AP-1 and NF-kappa B transcription factors. J Immunol.
155:1132–1140. 1995.PubMed/NCBI
|
|
30
|
Parra E: Activation of MAP kinase family
members triggered by TPA or ionomycin occurs via the protein
phosphatase 4 pathway in Jurkat leukemia T cells. Mol Med Rep.
5:773–778. 2012.
|
|
31
|
Parra E and Gutiérrez L: Growth inhibition
of Tax-activated human Jurkat leukemia T cells by all-trans
retinoic acid requires JNK-1 inhibition. Oncol Rep. 29:387–393.
2013.
|
|
32
|
Azran I, Schavinsky-Khrapunsky Y and Aboud
M: Role of Tax protein in human T-cell leukemia virus type-I
leukemogenicity. Retrovirology. 1:202004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Good L, Maggirwar SB and Sun SC:
Activation of the IL-2 gene promoter by HTLV-I tax involves
induction of NF-AT complexes bound to the CD28-responsive element.
EMBO J. 15:3744–3750. 1996.PubMed/NCBI
|
|
34
|
Harhaj EW and Harhaj NS: Mechanisms of
persistent NF-kappaB activation by HTLV-I tax. IUBMB Life.
57:83–91. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li XH, Murphy KM, Palka KT, Surabhi RM and
Gaynor RB: The human T-cell leukemia virus type-1 Tax protein
regulates the activity of the IkappaB kinase complex. J Biol Chem.
274:34417–34424. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xiao G and Sun SC: Activation of IKKalpha
and IKKbeta through their fusion with HTLV-I tax protein. Oncogene.
19:5198–5203. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Petropoulos L, Lin R and Hiscott J: Human
T cell leukemia virus type 1 tax protein increases NF-kappa B dimer
formation and antagonizes the inhibitory activity of the I kappa B
alpha regulatory protein. Virology. 225:52–64. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Verma IM: Nuclear factor (NF)-kappaB
proteins: Therapeutic targets. Ann Rheum Dis. 63(Suppl 2):
ii57–ii61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Holmes WF, Soprano DR and Soprano KJ:
Synthetic retinoids as inducers of apoptosis in ovarian carcinoma
cell lines. J Cell Physiol. 199:317–329. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang XK: Vitamin A and apoptosis in
prostate cancer. Endocr Relat Cancer. 9:87–102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang D, Holmes WF, Wu S, Soprano DR and
Soprano KJ: Retinoids and ovarian cancer. J Cell Physiol. 185:1–20.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu C, Rangnekar VM, Adamson E and Mercola
D: Suppression of growth and transformation and induction of
apoptosis by EGR-1. Cancer Gene Ther. 5:3–28. 1998.PubMed/NCBI
|
|
43
|
Virolle T, Adamson ED, Baron V, Birle D,
Mercola D, Mustelin T and de Belle I: The Egr-1 transcription
factor directly activates PTEN during irradiation-induced
signalling. Nat Cell Biol. 3:1124–1128. 2001. View Article : Google Scholar
|
|
44
|
Chen L, Wang S, Zhou Y, Wu X, Entin I,
Epstein J, Yaccoby S, Xiong W, Barlogie B, Shaughnessy JD Jr, et
al: Identification of early growth response protein 1 (EGR-1) as a
novel target for JUN-induced apoptosis in multiple myeloma. Blood.
115:61–70. 2010. View Article : Google Scholar :
|
|
45
|
Park JM, Greten FR, Li ZW and Karin M:
Macrophage apoptosis by anthrax lethal factor through p38 MAP
kinase inhibition. Science. 297:2048–2051. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Juretic N, Santibanez JF, Hurtado C and
Martinez J: ERK 1,2 and p38 pathways are involved in the
proliferative stimuli mediated by urokinase in osteoblastic SaOS-2
cell line. J Cell Biochem. 83:92–98. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Halawani D, Mondeh R, Stanton LA and Beier
F: p38 MAP kinase signaling is necessary for rat chondrosarcoma
cell proliferation. Oncogene. 23:3726–3731. 2004. View Article : Google Scholar : PubMed/NCBI
|