Introduction
Over the past decade, the incidence rate of thyroid
cancer has greatly increased (1).
Thyroid cancer can be histologically classified into many subtypes,
including follicular, anaplastic, medullary and papillary thyroid
cancer (PTC), among which PTC is the most prevalent. PTCs usually
have a good survival and prognosis with a 5-year survival rate
higher than 95% (2); however, these
cancers occasionally become aggressive and deadly through
dedifferentiation into other subtypes, such as dedifferentiated
thyroid cancer. Current treatments include surgery, thyroid hormone
and radioactive iodine (RAI) therapy.
Identification of cancer-driving genes has been
consistently a hotspot in cancer genomic research, and to date, 547
cancer genes are annotated in the COSMIC database (3). The common approach to detect driver
searches for genes that are significantly mutated in a cohort of
cancer samples as compared to the background mutation rate, is
through MutSigCV (4) and MuSiC
(5). Application of MutSig to 496
paired tumor/normal samples has found many over-mutated cancer
drivers in thyroid cancer, such as BRAF, NRAS, HRAS, EIF1AX and
KRAS (6). However, new evidence
shows that many driver genes may occur at a low frequency; for
example, some cancer drivers are mutated in a small fraction (e.g.,
<1%) of tumors (7). Therefore,
current tools may overlook potential drivers that are mutated at a
low frequency in the cancer genome, and methods that could identify
these low-frequency mutated driver genes are urgently needed.
Methods such as OncodriveFM (8)
tend to detect genes that have bias toward the accumulation of
variants with high functional impact measured by SIFT (9), PolyPhen2 (10) and MutationAssessor (11). Another new method Dendrix based on
mutual exclusivity was developed to find sets of genes in which the
majority of cancer samples have at least one mutation, while
display a mutation in one of the genes (12). These prediction tools complementary
to existing methods provide new opportunities to identify cancer
genes that drive tumor formation and progression.
In the present study, we describe the analysis of
somatic mutations detected by whole exome sequencing of 446
normal/tumor pairs of thyroid cancer samples from the Cancer Genome
Atlas (TCGA) database, OncodriveFM and Dendrix were applied to
prioritize cancer driver genes and pathways. We identified 53
cancer driver candidates and 75 pathways with significant bias of
functional impact. In addition, we analyzed DNA methylation status,
copy number variation, expression levels and fusion genes among
these driver candidates. We found that two genes, FHOD3 and SRP72,
were hypomethylated, overexpressed and involved in major deletions,
suggesting that they play an oncogenic role in thyroid cancer. Our
study highlights the importance of identifying low-frequency
mutated cancer-driving genes in an integrated way.
Materials and methods
Prediction of the functional effect of
cancer mutations, genes and pathways
A total of 23,620 somatic mutations detected by
whole-exome sequencing of 446 tumor/normal pairs were obtained from
TCGA (http://cancergenome.nih.gov/,
download on July 14, 2015). Functional impact of somatic mutations
in the coding genome was classified with Ensembl Variant Effect
Predictor (VEP) (13). Cancer genes
and pathways were predicted by OncodriveFM (8) and Dendrix (12) programs and all the parameters were
set to default. Genes and pathways with Q-value <0.05 were
regarded as cancer gene and pathway candidates. GO enrichment
analysis was performed for all of the driver candidates from the
home page of the GOC website (14)
(http://geneontology.org/).
DNA methylation, RNA-seq data processing
and expression analyses
DNA methylation data for 389 thyroid cancer samples
were obtained from TCGA, and the undefined value was replaced with
an average β value. The average β value was computed for each gene
and cancer sample. RNA-seq data of 18 papillary thyroid carcinoma
biopsies and 4 normal thyroid tissues were obtained from the study
of Costa et al (GSE64912) (15). Read alignment with human genome 19
was conducted with TopHat2 (16),
and read count was computed with bedtools v2.22.1 (17) for each gene. Differentially
expressed genes were determined with DEseq2 (18) package in R between tumor and normal
tissues with cutoffs of a false discovery rate (FDR) ≤0.5 and
absolute fold-change ≥2.
Sources of copy number variation and
fusion gene data
We obtained copy number variations of 501 thyroid
cancer samples which were detected by SNP array and publically
available at the Broad Institute (6) (gdac.broadinstitute.org). RNA-seq data from the study
of Costa et al was aligned to the human genome with Tophat2,
fusion genes were detected with TopHat-Fusion (16), and all of the parameters were set by
default.
Statistical analysis
Data are presented as mean, and differences between
different groups were drawn with the Wilcoxon rank sum test in R.
P<0.05 was considered to indicate a statistically significant
difference and the null hypothesis was rejected.
Results
Catalogue of somatic mutations
A total of 23,620 somatic mutations detected by
whole-exome sequencing of 446 thyroid cancer specimens were
obtained from TCGA. Among these, 23,070 were single-nucleotide
variants (SNVs), 550 were small insertions or deletions.
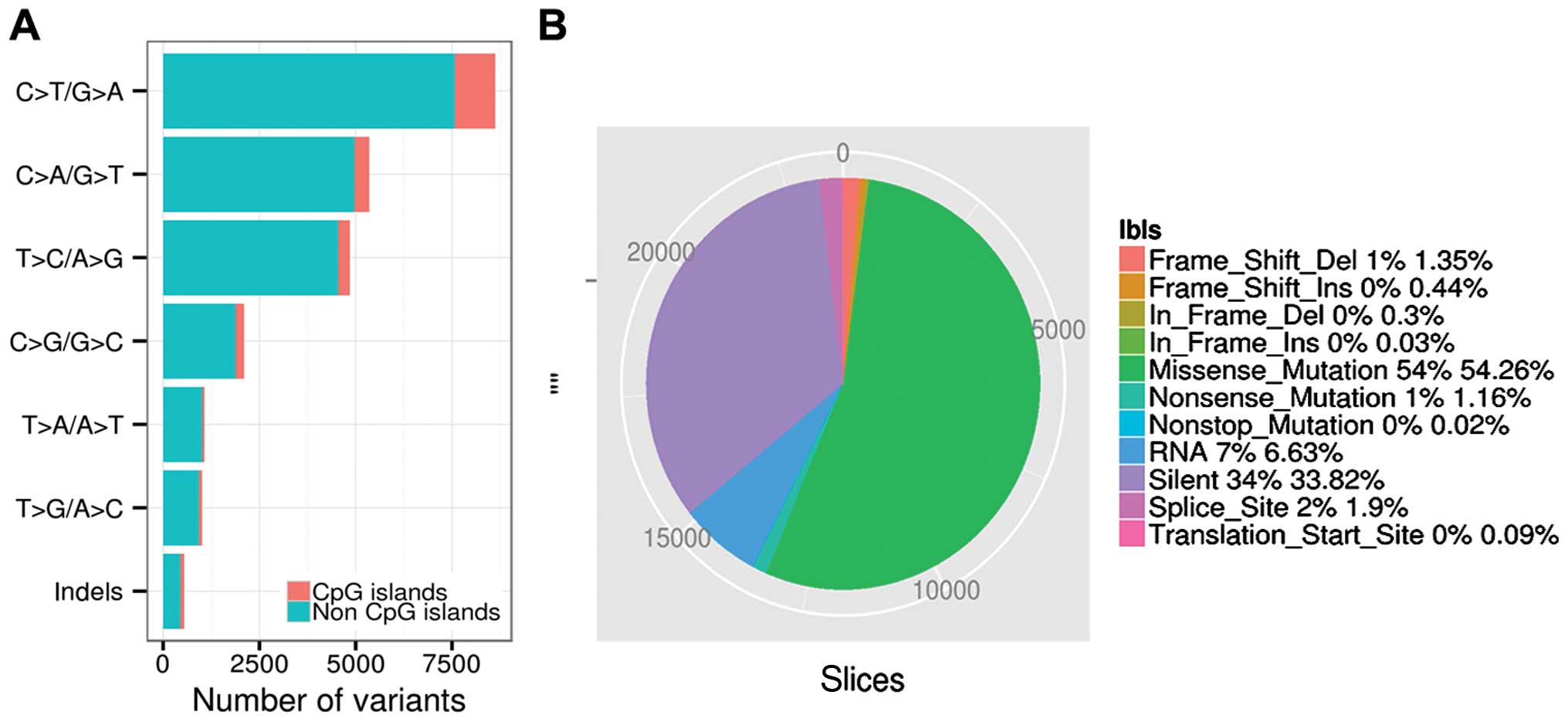
C>T/G>A, C>A/G>T and T>C/A>G accounted for 32.10,
21.04 and 19.21% of the variant types in the non-CpG sites, and
4.42, 1.65 and 1.35% of variant types in the CpG islands.
C>T/G>A, C>A/G>T and T>C/A>G were the three
predominant transitions in thyroid cancer (Fig. 1A). A total of 12,817, 274 and 5
single nucleotide variations were classified as missense, nonsense
and nonstop mutations, respectively by VEP. A total of 7,988 single
nucleotide variations were classified as silent. A total of 318 and
103 small deletions and insertions introduced translational
frameshifts, and 72 and 8 small deletions and insertions were in
frame mutations. A total of 448 and 22 mutations were located in
splicing sites and translation start sites, while non-synonymous
mutations accounted for >55% (13,091/23,620) of the total
variants (Fig. 1B). Thyroid cancer
showed a significantly lower non-synonymous mutation density (0.31
non-synonymous mutations per Mb per sample, on average) as compared
to other cancers, such as melanoma and lung cancer (4,19).
Cancer driver genes and pathways in
thyroid cancer
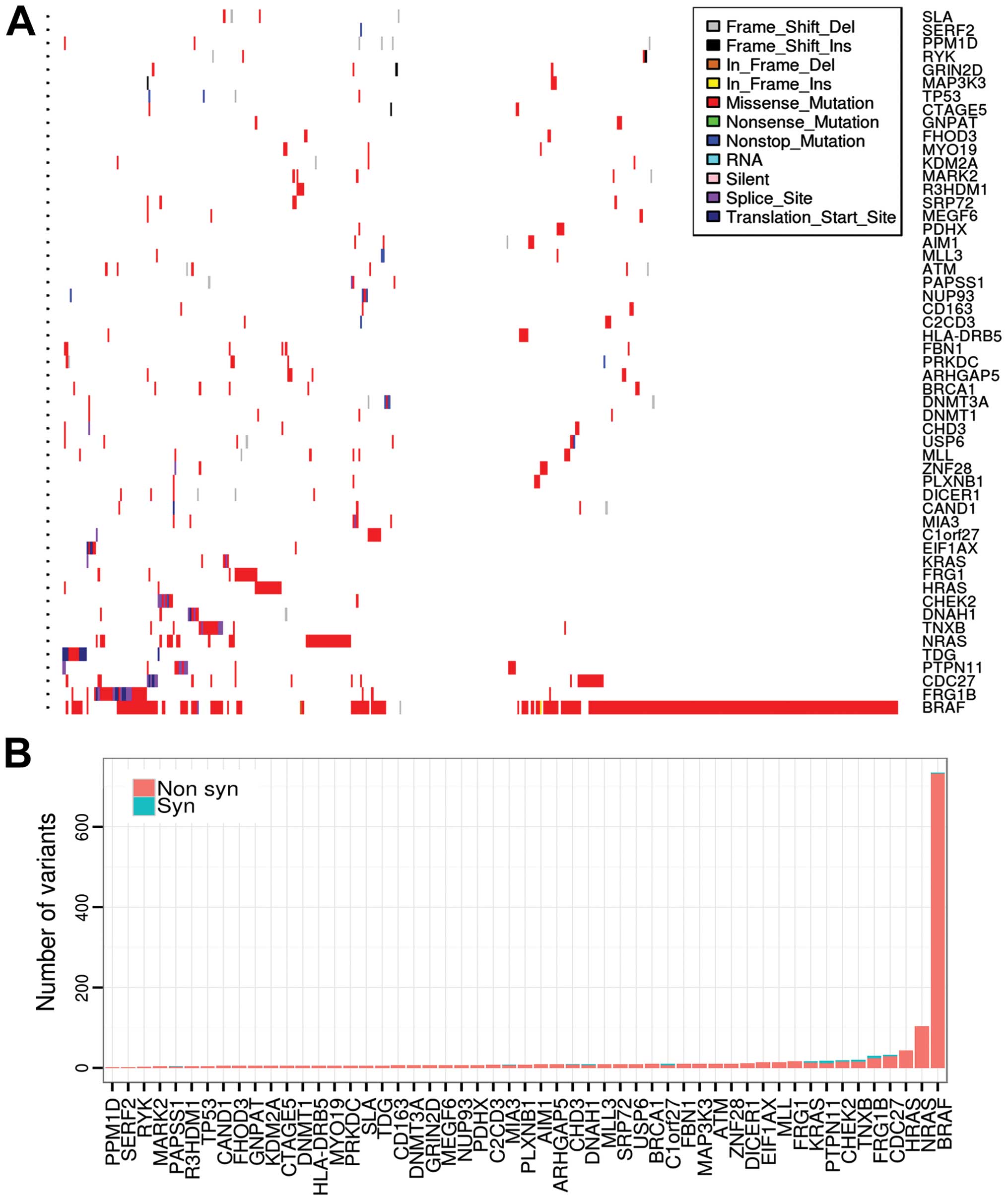
We applied OncodriveFM to identify driver genes in
thyroid cancer. In total, 53 genes were determined as driver
candidates by OncodriveFM. Among them, BRAF, NRAS, HRAS, KRAS,
PPM1D and EIF1AX are known recurrently mutated driver genes in
thyroid cancer, with mutation frequencies of 59.64, 8.52, 3.59,
1.12, 1.35 and 1.35% across all samples (6). However, most of the driver candidates
were not frequently mutated in thyroid cancer (Fig. 2A and B). The P53 signaling genes
were also determined as driver genes; TP53, ATM and CHEK2, showed
relatively low mutation frequencies (<2.5% in all cases),
suggesting the inactivation of the P53 signaling pathway in thyroid
cancer. Several known cancer genes of other cancer types were first
identified as drivers in thyroid cancer, such as BRCA1 in breast
cancer (20), MLL and MLL3 in
hepatocellular carcinoma (HCC) (21) and gastric cancer (22), ATM in glioma (23) and leukemia (24), PTPN11 in acute myeloid leukemia
(25) as well as DICER1 in
pleuropulmonary blastoma (26). In
addition, we also identified many new driver candidates, for
example, mitogen-activated protein kinase kinase kinase 3 (MEKK3)
and Transport and Golgi organisation protein 1 (TANGO). MEKK3 is
elevated in esophageal squamous cell carcinoma (ESCC), and
overexpression of MEKK3 indicates poor prognosis of ESCC (27). Breast and ovarian cancers show an
elevated MEKK3 protein level and increased NF-κB binding activity,
and overexpression of MEKK3 was found to activate NF-κB binding
activity and upregulate cell survival and anti-apoptotic genes such
as Bcl-2 and XIAP in U373 cells, which enhanced cellular resistance
to apoptosis induced by chemotherapeutic agents (28). Another driver candidate, MIA3, also
known as TANGO, is a member of the melanoma inhibitory activity
(MIA) gene family. It displays a tumor-suppressor function in
multiple cancer types, such as human colon and hepatocellular
carcinoma (29) and melanoma
(30).
Dendrix was developed to identify sets of genes
which are mutated in a large fraction of cancer samples and whose
mutations are mutually exclusive. We next analyzed somatic
mutations of thyroid cancer with Dendrix. In total, 10,839 genes
were reported as mutated in at least one patient. We performed
Dendrix for sets with sizes ranging from 2 to 5. When k=2, the pair
BRAF and NRAS was sampled 96.1% of the time. When k=3, the triplet
(BRAF, NRAS and KRAS) was sampled with a frequency of 22.2%. For
k=4, no gene set had sample frequency >1%. The pair (BRAF, NRAS)
and triplet (BRAF, NRAS and KRAS) were the most prevalent gene sets
in the mutual exclusivity test, further supporting the importance
of BRAF, NRAS and KRAS in the tumorigenesis of thyroid cancer.
We analyzed the enrichment of GO terms of the 53
cancer gene candidates, and 27 GO terms were determined with
significant statistical evidence (Table
I). The majority of GO terms were cancer-associated, such as
apoptotic signaling pathway, regulation of protein metabolic
process and cell cycle and DNA damage checkpoint. These findings
further support that the driver candidates identified by
OncodriveFM and Dendrix have critical functions in thyroid cancer.
OncodriveFM also revealed 75 pathways with high FM bias in thyroid
cancer, such as regulation of actin cytoskeleton, melanoma, thyroid
cancer, renal cell carcinoma, bladder cancer, non-small cell lung
cancer (NSCLC), alcoholism, endometrial cancer, prostate cancer,
glioma, chronic myeloid leukemia and acute myeloid leukemia
(Table II).
 | Table IEnrichment of GO terms for driver
candidates in thyroid cancer. |
Table I
Enrichment of GO terms for driver
candidates in thyroid cancer.
| GO terms | No. of genes | Fold-change of
enrichment | P-value |
|---|
| Replicative
senescence | 3 | >5 | 2.21e-02 |
| Intrinsic apoptotic
signaling pathway in response to DNA damage | 5 | >5 | 8.74e-03 |
| Signal transduction
in response to DNA damage | 5 | >5 | 4.46e-02 |
| Response to
ionizing radiation | 6 | >5 | 6.61e-03 |
| Double-strand break
repair | 6 | >5 | 6.90e-03 |
| DNA damage
checkpoint | 6 | >5 | 1.09e-02 |
| DNA integrity
checkpoint | 6 | >5 | 1.59e-02 |
| Mitotic cell cycle
checkpoint | 6 | >5 | 2.44e-02 |
| Intrinsic apoptotic
signaling pathway | 6 | >5 | 3.08e-02 |
| Cell cycle
checkpoint | 8 | >5 | 2.06e-03 |
| Response to
radiation | 9 | >5 | 1.18e-02 |
| Cellular response
to nitrogen compound | 12 | >5 | 3.06e-04 |
| Cellular response
to organonitrogen compound | 11 | >5 | 1.38e-03 |
| Cell cycle
process | 13 | >5 | 7.82e-03 |
| Response to
nitrogen compound | 12 | >5 | 2.42e-02 |
| Positive regulation
of protein modification process | 13 | 4.73 | 1.68e-02 |
| Cellular component
morphogenesis | 13 | 4.54 | 2.61e-02 |
| Cell cycle | 14 | 4.41 | 1.42e-02 |
| Regulation of
protein modification process | 16 | 3.97 | 8.52e-03 |
| Macromolecular
complex subunit organization | 17 | 3.41 | 2.82e-02 |
| Macromolecule
modification | 22 | 3.13 | 2.26e-03 |
| Regulation of
protein metabolic process | 19 | 3.13 | 2.20e-02 |
| Cellular protein
modification process | 20 | 2.99 | 2.10e-02 |
| Protein
modification process | 20 | 2.99 | 2.10e-02 |
| Cellular component
organization or biogenesis | 33 | 2.65 | 8.59e-06 |
| Cellular component
organization | 32 | 2.63 | 2.58e-05 |
| Response to
stress | 23 | 2.62 | 2.43e-02 |
| Table IITop 20 cancer-driving pathways
detected by OncodriveFM in thyroid cancer. |
Table II
Top 20 cancer-driving pathways
detected by OncodriveFM in thyroid cancer.
| Pathway name | Pathway ID | Gene number | P-value | Q-value |
|---|
| Regulation of actin
cytoskeleton | hsa04810 | 213 | 1.10e-182 | 1.39e-181 |
| Chemokine signaling
pathway | hsa04062 | 189 | 8.79e-178 | 1.06e-176 |
| Natural killer cell
mediated cytotoxicity | hsa04650 | 132 | 1.11e-174 | 1.27e-173 |
| Hepatitis C | hsa05160 | 131 | 9.96e-184 | 1.42e-182 |
| Melanoma | hsa05218 | 71 | 3.11e-213 | 2.37e-211 |
| Thyroid cancer | hsa05216 | 29 | 1.80e-213 | 2.05e-211 |
| Renal cell
carcinoma | hsa05211 | 70 | 7.99e-219 | 1.82e-216 |
| Long-term
potentiation | hsa04720 | 70 | 9.00e-190 | 1.71e-188 |
| ErbB signaling
pathway | hsa04012 | 88 | 1.50e-183 | 2.02e-182 |
| Neurotrophin
signaling pathway | hsa04722 | 119 | 8.30e-190 | 1.71e-188 |
| Serotonergic
synapse | hsa04726 | 112 | 1.40e-193 | 3.54e-192 |
| Alcoholism | hsa05034 | 177 | 1.02e-206 | 5.80e-205 |
| Glioma | hsa05214 | 65 | 1.67e-197 | 5.45e-196 |
| Non-small cell lung
cancer | hsa05223 | 54 | 1.56e-197 | 5.45e-196 |
| Bladder cancer | hsa05219 | 42 | 1.39e-204 | 6.34e-203 |
| Endometrial
cancer | hsa05213 | 52 | 1.89e-192 | 4.31e-191 |
| Prostate
cancer | hsa05215 | 88 | 1.94e-189 | 3.40e-188 |
| Chronic myeloid
leukemia | hsa05220 | 73 | 9.45e-196 | 2.69e-194 |
| Long-term
depression | hsa04730 | 61 | 9.04e-187 | 1.47e-185 |
| Acute myeloid
leukemia | hsa05221 | 57 | 1.69e-185 | 2.57e-184 |
DNA methylation in thyroid cancer
Epigenetic alterations such as methylation of
cytosine-guanine dinucleotides (CpG) play an important role in
human carcinogenesis. We obtained DNA methylation data from TCGA
and analyzed its association with driver genes in thyroid cancer.
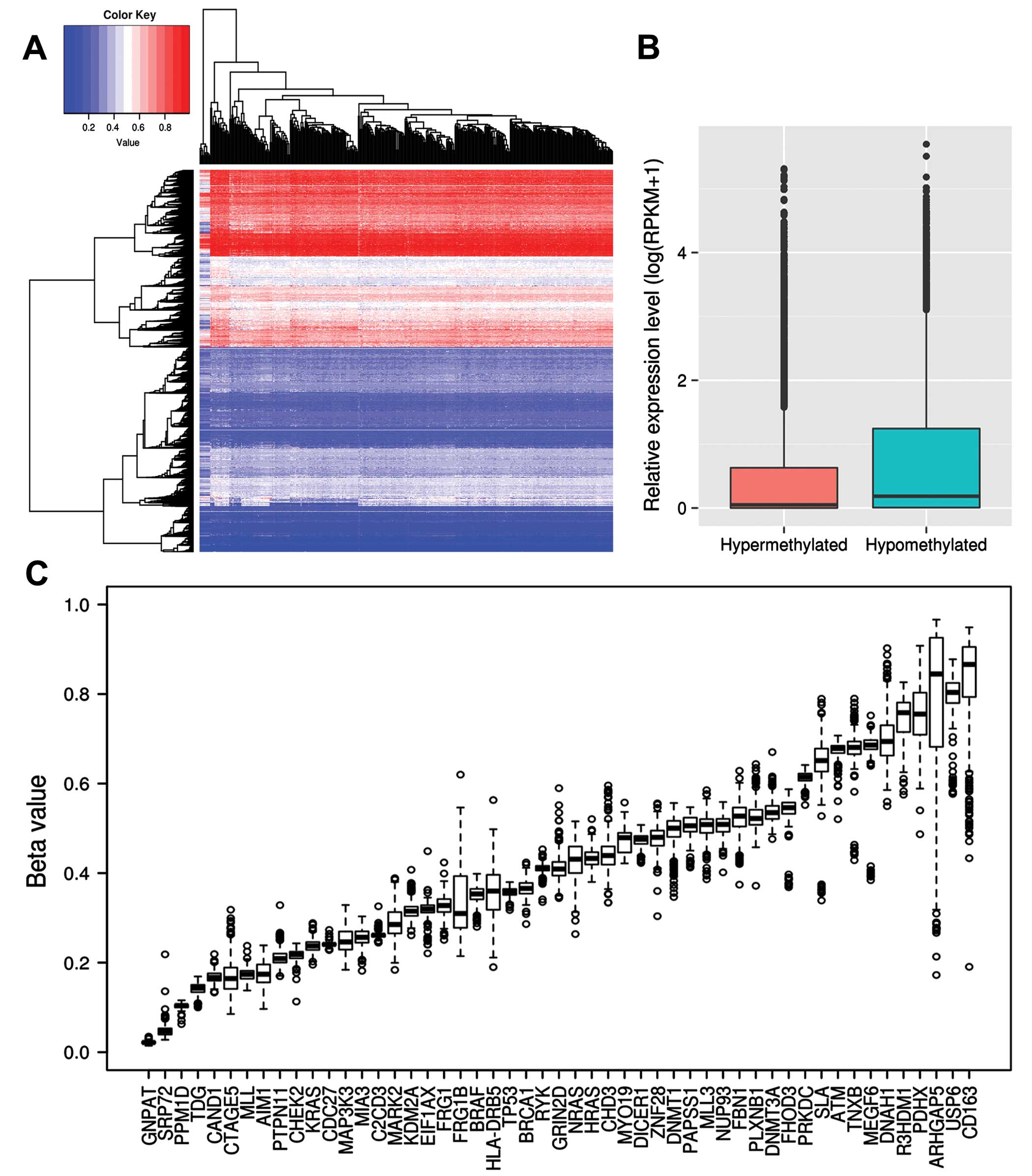
First of all, we used hierarchical clustering to analyze DNA
methylation profiling and found two clusters of hypermethylated and
hypomethylated genes (Fig. 3A).
This suggested that genes with hypermethylation or hypomethylation
on promoters are associated with tumor suppressors or oncogenes
(31). We selected the top 500
hypermethylated and hypomethylated genes which had the highest or
lowest mean β value, respectively, and analyzed functional
enrichment with GO terms.
Hypomethylated genes were significantly enriched in
80 GO terms. Among them, many were found to be associated with cell
cycle regulation, metabolic process, cell division and gene
expression, while, hypermethylated genes were enriched in only in 9
GO terms, such as detection of stimulus, G-protein coupled receptor
signaling pathway and sensory perception of smell. In addition, we
obtained RNA-seq data of 18 papillary thyroid carcinoma biopsies
and 4 normal thyroid tissues from the study of Costa et al
(15). We analyzed the expression
levels of hypermethylated and hypomethylated genes. The
hypermethylated genes were found to be significantly lower
expressed as compared to the hypomethylated ones (RPKM, 3.08 vs.
4.72, P<2.2e-16; Wilcoxon rank sum test) (Fig. 3B). Among all the driver candidates,
CD163, USP6, R3HDM1, DNAH1, ARHGAP5 and R3HDM1 showed
hypermethylation, while PPM1D, SRP72, GNPAT and TDG exhibited
hypomethylation (Fig. 3C),
suggesting they may be involved in tumorigenesis of thyroid cancer
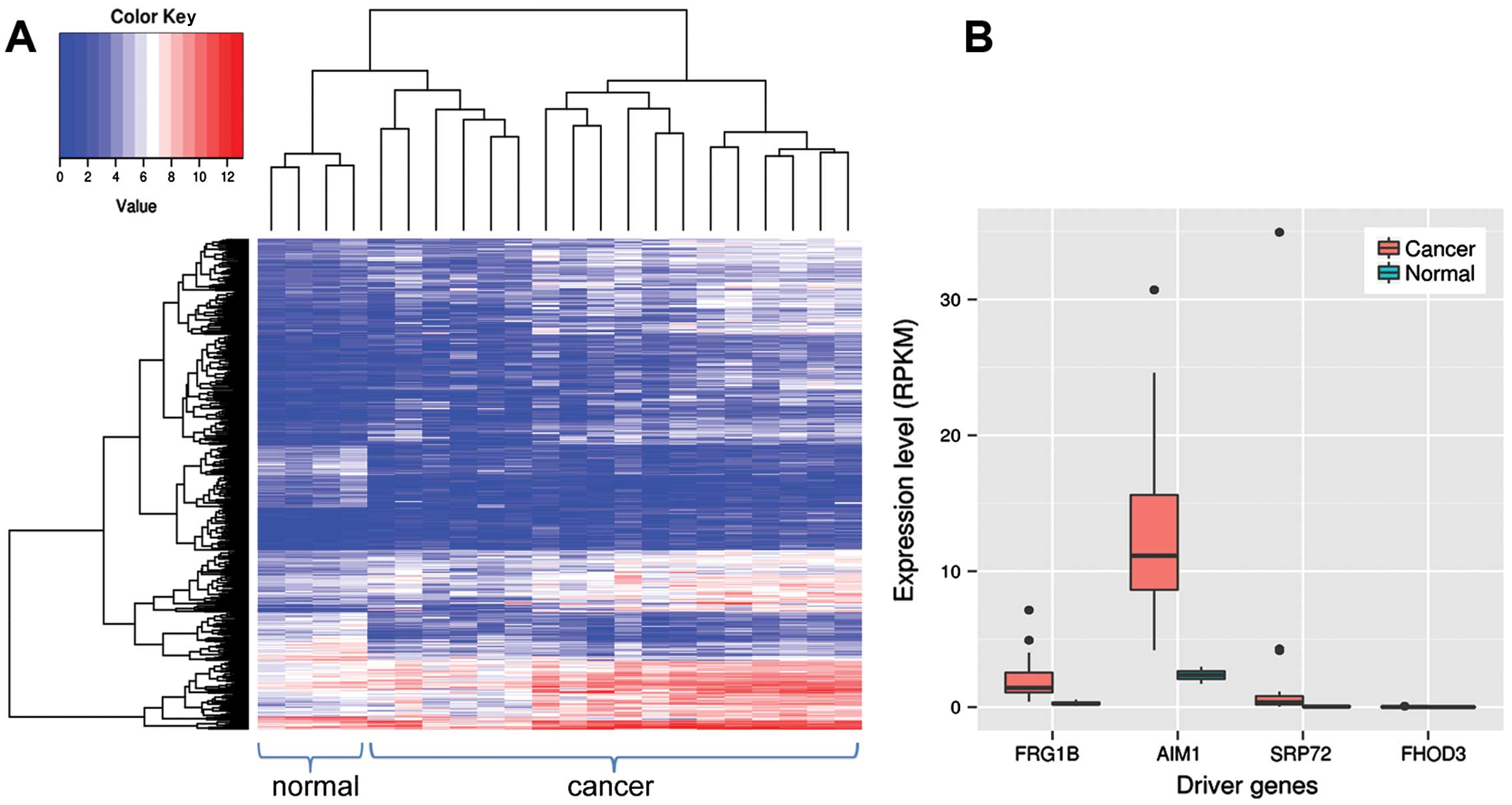
by altering the methylation status. We also identified 504
differentially expressed genes between thyroid cancer and normal
tissues (Fig. 4A). Among them, four
were driver gene candidates, including AIM1, FHOD3, SRP72 and FRG1B
(Fig. 4B). AIM1, SRP72 and FRG1B
are overexpressed and hypomethylated, which suggests they may have
oncogenic function in thyroid cancer.
Copy number variations in thyroid
cancer
We also obtained copy number variations of 501
thyroid cancer samples detected by the Broad Institute. Significant
focal gains and deletions (Q<0.25) were found in 247 samples
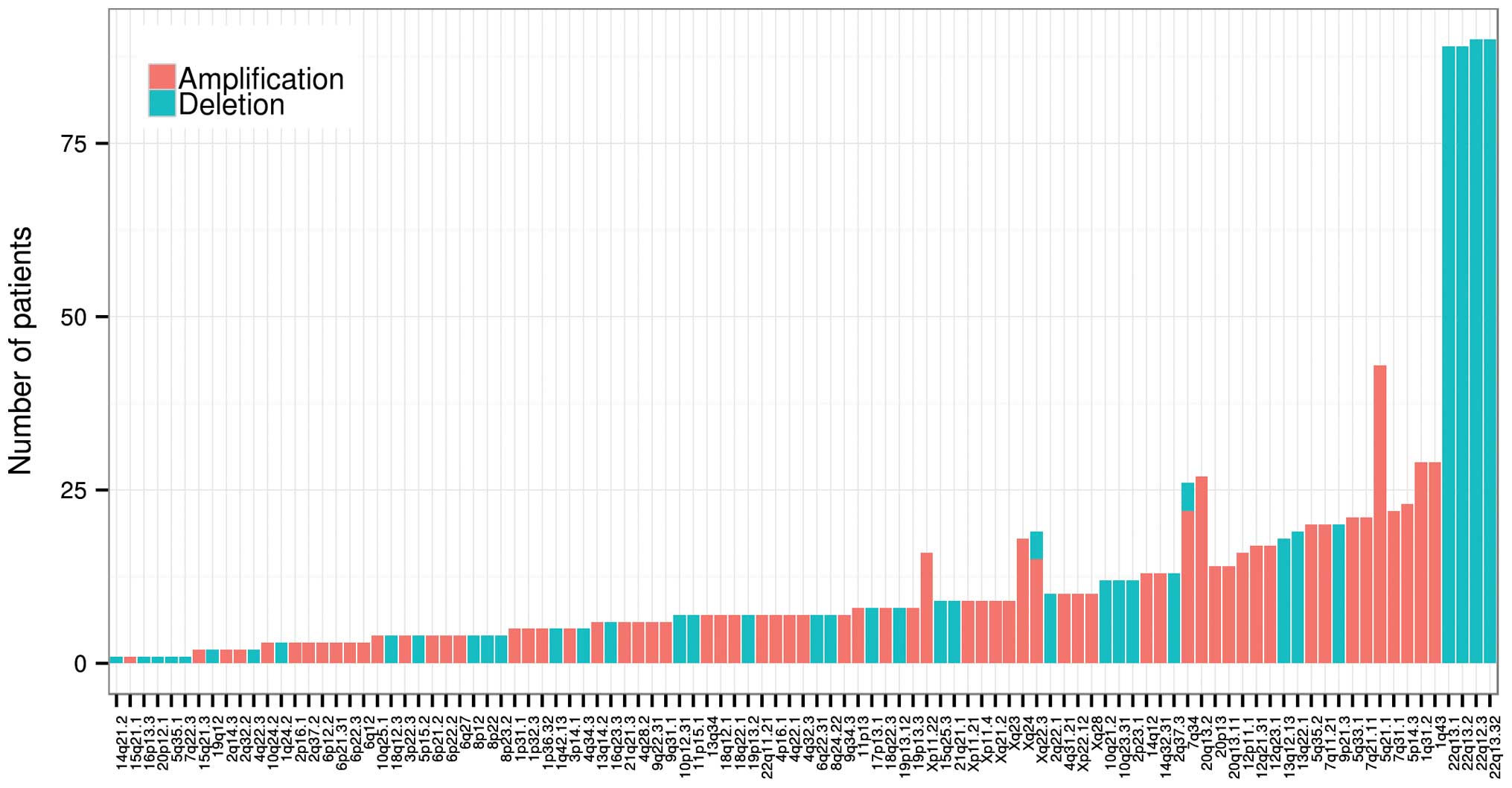
(247/501, 49.30%) at 107 loci (68 amplifications and 39 deletions).
Among them, deletions at 2q13.32, 22q13.2, 22q13.1 and 22q12.3 were
the most frequent copy number variations in thyroid cancer, with an
occurrence rate of 17.96 (90/501), 17.76 (89/501), 17.76 (89/501)
and 17.96% (90/501), respectively (Fig.
5). Several known tumor suppressors and oncogenes were involved
in copy number variation, including BRAF (amplification and
deletion, 7q34), TP53 and BRCA1 (deletion, 17p13.1), EIF1AX
(amplification and deletion, Xp22.12 and Xq23). Many driver
candidates were also found to be implicated in the CNVs, including
FRG1, PAPSS1 and SRP72 (deletion, 4q22.3), CDC27, CDH3, PPM1D,
USP6, MYO19, (17p13.1), DNMT3A and R3HDM1 (2q14.3) and FHOD3
(18q12.3), NUP93(16p13.3) DNMT1 (19p13.2).
Fusion genes in thyroid cancer
We obtained RNA-seq data of 18 papillary thyroid
carcinoma samples from the study of Costa et al (15), and applied Tophat-Fusion to detect
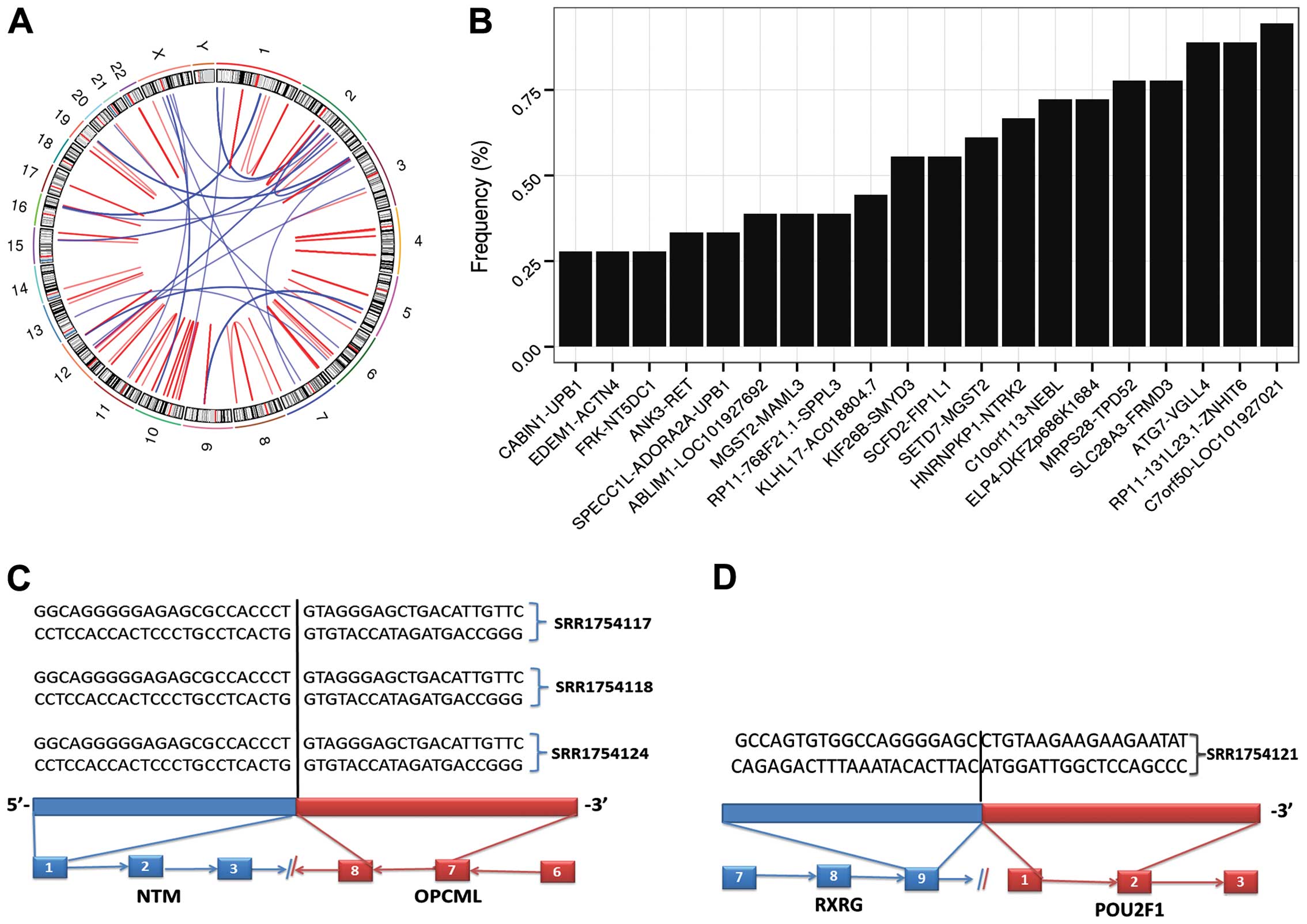
potential fusion genes in thyroid cancer. In total, we found 91
pairs of fusion genes in 18 cancer samples (Fig. 6A). C7orf50-LOC101927021, ATG7-VGLL4,
RP11-131L23.1-ZNHIT6, MRPS28-TPD52 and SLC28A3-FRMD3 were among the
most frequent fusion pairs in thyorid cancer, with a frequency of
94.44, 88.89, 88.89, 77.78 and 77.78% in the cancer samples
(Fig. 6B). We compared our list of
fusion gene pairs with known fusion genes of thyroid cancer from
TCGA Fusion Gene Data Portal (http://54.84.12.177/PanCanFusV2/). There were only two
known fusion gene pairs, ANK3-RET (33.33%) and PPARG-PAX8 (5.56%),
while the majority of pairs of fusion genes were newly identified.
In addition, we found that three fusion genes were differentially
expressed between the thyroid cancer and normal tissues, including
NTM, SLC7A7 and RXRG. The three genes were all overexpressed in
thyroid cancer, with a fusion frequency: NTM-OPCML (16.67%),
RXRG-POU2F1 (5.56%) and SLC7A7-TRAJ24 (5.56%) (Fig. 6C and D). Finally, no driver
candidate was found to be implicated in fusion genes, indicating
that gene fusion may not be the primary mechanism for the
involvement of driver genes in thyroid cancer.
Discussion
In the present study, we carried out a full analysis
on the somatic mutations generated by whole exome sequencing of
thyroid cancer samples. We found 53 cancer gene candidates and 75
cancer pathways. Among them, BRAF, NRAS, HRAS, EIF1AX, CHEK2 and
PPM1D are recurrently mutated (6).
BRAF as a proto-oncogene regulates the MAP kinase/ERK signaling
pathway, which affects cell division, differentiation and
secretion. Mutations within BRAF contribute to carcinogenesis of a
variety of cancer types, such as thyroid cancer (32) and melanoma (33). KRAS, HRAS and NRAS are the most
common oncogenes from the RAS gene family. They encode proteins of
the GTPase superfamily which plays a great role in signal
transduction, protein biosynthesis, cell division, translocation of
proteins and transport of vesicles. Mutations in RAS proteins are
associated with ~30% of all human cancers (34). PPM1D is a member of the PP2C family
which is known to be a negative regulator of cell stress response
pathways. It has been reported to be involved in multiple human
tumors, such as lung cancer (35),
breast cancer and ovarian cancer (36). However, the majority of driver
candidates were new cancer genes with low mutation frequency. A
great advantage of OncodriveFM and Dendrix is that these two tools
identify genes and pathways which accumulate variants of high
function impact or mutationally exclusive, independent of the
cancer mutation frequency. Therefore, application of these tools in
cancer research enables us to better explore cancer-driving genes
and pathways in the cancer genome. In addition, we also identified
many drivers that were differentially expressed, hypermethylated,
hypomethylated and CNV-associated, such as SRP72 and FRG1B,
suggesting that these genes may contribute to the formation and
progression of thyroid cancer in various ways.
We found 91 pairs of fusion genes using RNA-seq
data. Three of them, NTM, SLC7A7 and RXRG fusion genes, were
overexpressed in thyroid cancer; however, their fusion partners
were not affected. These fusion genes are actively implicated in
various cancer types, for instance, RXRG is a member of the
Retinoid X receptors. It shows tumor-suppressor function in NSCLC
(37) and colon cancer (38). RXRG upregulation is associated with
dedifferentiation, advanced tumor stage and metastasis (39) and increased apoptosis (40) in thyroid cancer. POU class 2
homeobox 1 (POU2F1), also known as OCT1, is a ubiquitous member of
the POU transcription factor family. POU2F1 shows pro-proliferative
and pro-apoptotic activity in bladder carcinoma (41). Neurotrimin (NTM) and OPCML are
members of the IgLON family of immunoglobulin (Ig)
domain-containing glycosylphosphati-dylinositol (GPI)-anchored cell
adhesion molecules. OPCML as a tumor-suppressor gene is commonly
inactivated by either allele loss or DNA methylation in epithelial
ovarian cancer (42). SLC7A7 is
overexpressed in glioblastoma (GBM), and its overexpression
indicates a poor outcome of GBM (43). Genetic variants in SLC7A7 are
associated with the risk of glioma (44). Therefore, pairs of these fusion
genes may play an important role in the carcinogenesis of thyroid
cancer.
In conclusion, taken together, we successfully found
a set of cancer-related genes, pathways and fusion genes in thyroid
cancer. The findings provide new insight into the pathogenesis of
thyroid cancer, therefore paving a potential avenue by which to
cure thyroid cancer, based on the disruption of driver genes,
pathways and fusion gene pairs.
References
|
1
|
Girardi FM, Barra MB and Zettler CG:
Analysis of pattern of occurrence of thyroid carcinoma between 2001
and 2010. Braz J Otorhinolaryngol. 81:541–548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hay ID, Thompson GB, Grant CS, Bergstralh
EJ, Dvorak CE, Gorman CA, Maurer MS, McIver B, Mullan BP, Oberg AL,
et al: Papillary thyroid carcinoma managed at the Mayo Clinic
during six decades (1940–1999): Temporal trends in initial therapy
and long-term outcome in 2444 consecutively treated patients. World
J Surg. 26:879–885. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the catalogue of somatic
mutations in cancer. Nucleic Acids Res. 39(Database): D945–D950.
2011. View Article : Google Scholar :
|
|
4
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dees ND, Zhang Q, Kandoth C, Wendl MC,
Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis
ER, et al: MuSiC: Identifying mutational significance in cancer
genomes. Genome Res. 22:1589–1598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer T and Atlas G; Cancer Genome Atlas
Research Network: Integrated genomic characterization of papillary
thyroid carcinoma. Cell. 159:676–690. 2014. View Article : Google Scholar
|
|
7
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gonzalez-Perez A and Lopez-Bigas N:
Functional impact bias reveals cancer drivers. Nucleic Acids Res.
40:e1692012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reva B, Antipin Y and Sander C: Predicting
the functional impact of protein mutations: Application to cancer
genomics. Nucleic Acids Res. 39:e1182011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vandin F, Upfal E and Raphael BJ: De novo
discovery of mutated driver pathways in cancer. Genome Res.
22:375–385. 2012. View Article : Google Scholar :
|
|
13
|
McLaren W, Pritchard B, Rios D, Chen Y,
Flicek P and Cunningham F: Deriving the consequences of genomic
variants with the Ensembl API and SNP effect predictor.
Bioinformatics. 26:2069–2070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al The Gene Ontology Consortium: Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Costa V, Esposito R, Ziviello C, Sepe R,
Bim LV, Cacciola NA, Decaussin-Petrucci M, Pallante P, Fusco A and
Ciccodicola A: New somatic mutations and WNK1-B4GALNT3 gene fusion
in papillary thyroid carcinoma. Oncotarget. 6:11242–11251. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lawrence MS, Stojanov P, Mermel CH,
Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander
ES and Getz G: Discovery and saturation analysis of cancer genes
across 21 tumour types. Nature. 505:495–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhong Q, Peng H-L, Zhao X, Zhang L and
Hwang W-T: Effects of BRCA1/2 on ovarian and breast cancer
survival-response. Clin Cancer Res. 21:3807. 2015. View Article : Google Scholar
|
|
21
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zang ZJ, Cutcutache I, Poon SL, Zhang SL,
McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al:
Exome sequencing of gastric adenocarcinoma identifies recurrent
somatic mutations in cell adhesion and chromatin remodeling genes.
Nat Genet. 44:570–574. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Golding SE, Rosenberg E, Adams BR,
Wignarajah S, Beckta JM, O'Connor MJ and Valerie K: Dynamic
inhibition of ATM kinase provides a strategy for glioblastoma
multiforme radiosensiti-zation and growth control. Cell Cycle.
11:1167–1173. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Negi SS and Brown P: rRNA synthesis
inhibitor, CX-5461, activates ATM/ATR pathway in acute
lymphoblastic leukemia, arrests cells in G2 phase and induces
apoptosis. Oncotarget. 6:18094–18104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bentires-Alj M, Paez JG, David FS,
Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A,
Sugarbaker DJ, et al: Activating mutations of the noonan
syndrome-associated SHP2/PTPN11 gene in human solid tumors and
adult acute myelogenous leukemia. Cancer Res. 64:8816–8820. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Seki M, Yoshida K, Shiraishi Y, Shimamura
T, Sato Y, Nishimura R, Okuno Y, Chiba K, Tanaka H, Kato K, et al:
Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma.
Cancer Res. 74:2742–2749. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hasan R, Sharma R, Saraya A, Chattopadhyay
TK, DattaGupta S, Walfish PG, Chauhan SS and Ralhan R: Mitogen
activated protein kinase kinase kinase 3 (MAP3K3/MEKK3)
overexpression is an early event in esophageal tumorigenesis and is
a predictor of poor disease prognosis. BMC Cancer. 14:22014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samanta AK, Huang HJ, Bast RC Jr and Liao
WS-L: Overexpression of MEKK3 confers resistance to apoptosis
through activation of NFkappaB. J Biol Chem. 279:7576–7583. 2004.
View Article : Google Scholar
|
|
29
|
Arndt S and Bosserhoff AK: Reduced
expression of TANGO in colon and hepatocellular carcinomas. Oncol
Rep. 18:885–891. 2007.PubMed/NCBI
|
|
30
|
Arndt S and Bosserhoff AK: TANGO is a
tumor suppressor of malignant melanoma. Int J Cancer.
119:2812–2820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gonzalo S: Epigenetic alterations in
aging. J Appl Physiol. 1985(109): 586–597. 2010. View Article : Google Scholar
|
|
32
|
Xing M, Alzahrani AS, Carson KA, Shong YK,
Kim TY, Viola D, Elisei R, Bendlová B, Yip L, Mian C, et al:
Association between BRAF V600E mutation and recurrence of papillary
thyroid cancer. J Clin Oncol. 33:42–50. 2015. View Article : Google Scholar
|
|
33
|
Inumaru JS, Gordo KI, Fraga Junior AC,
Silva AM, Leal CB, Ayres FM, Wastowski IJ, Borges NF and Saddi VA:
Analysis of the BRAF V600E mutation in primary cutaneous melanoma.
Genet Mol Res. 13:2840–2848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Parikh C, Subrahmanyam R and Ren R:
Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic
potentials in mice. Cancer Res. 67:7139–7146. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang C, Chen Y, Wang M, Chen X, Li Y,
Song E, Liu X, Kim S and Peng H: PPM1D silencing by RNA
interference inhibits the proliferation of lung cancer cells. World
J Surg Oncol. 12:2582014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruark E, Snape K, Humburg P, Loveday C,
Bajrami I, Brough R, Rodrigues DN, Renwick A, Seal S, Ramsay E, et
al Breast and Ovarian Cancer Susceptibility Collaboration: Wellcome
Trust Case Control Consortium: Mosaic PPM1D mutations are
associated with predisposition to breast and ovarian cancer.
Nature. 93:406–410. 2013.
|
|
37
|
Brabender J, Danenberg KD, Metzger R,
Schneider PM, Lord RV, Groshen S, Tsao-Wei DD, Park J, Salonga D,
Hölscher AH, et al: The role of retinoid X receptor messenger RNA
expression in curatively resected non-small cell lung cancer. Clin
Cancer Res. 8:438–443. 2002.PubMed/NCBI
|
|
38
|
Papi A, Rocchi P, Ferreri AM and Orlandi
M: RXRgamma and PPARgamma ligands in combination to inhibit
proliferation and invasiveness in colon cancer cells. Cancer Lett.
297:65–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Z, Zhou G, Nakamura M, Bai Y, Li Y,
Ozaki T, Mori I, Miyauchi A and Kakudo K: Retinoid X receptor γ
up-regulation is correlated with dedifferentiation of tumor cells
and lymph node metastasis in papillary thyroid carcinoma. Pathol
Int. 61:109–115. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Klopper JP, Hays WR, Sharma V, Baumbusch
MA, Hershman JM and Haugen BR: Retinoid X receptor-gamma and
peroxisome proliferator-activated receptor-gamma expression
predicts thyroid carcinoma cell response to retinoid and
thiazolidinedione treatment. Mol Cancer Ther. 3:1011–1020.
2004.PubMed/NCBI
|
|
41
|
Szekeres K, Koul R, Mauro J, Lloyd M,
Johnson J and Blanck G: An Oct-1-based, feed-forward mechanism of
apoptosis inhibited by co-culture with Raji B-cells: Towards a
model of the cancer cell/B-cell microenvironment. Exp Mol Pathol.
97:585–589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sellar GC, Watt KP, Rabiasz GJ, Stronach
EA, Li L, Miller EP, Massie CE, Miller J, Contreras-Moreira B,
Scott D, et al: OPCML at 11q25 is epigenetically inactivated and
has tumor-suppressor function in epithelial ovarian cancer. Nat
Genet. 34:337–343. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fan S, Meng D, Xu T, Chen Y, Wang J, Li X,
Chen H, Lu D, Chen J and Lan Q: Overexpression of SLC7A7 predicts
poor progression-free and overall survival in patients with
glio-blastoma. Med Oncol. 30:3842013. View Article : Google Scholar
|
|
44
|
Fan S, Zhao Y, Li X, Du Y, Wang J, Song X,
Zhou F, Chen H, Chen G, Zhao Y, et al: Genetic variants in SLC7A7
are associated with risk of glioma in a Chinese population. Exp
Biol Med (Maywood). 238:1075–1081. 2013. View Article : Google Scholar
|