Introduction
Cigarette smoking is not only one of the most severe
public health issues but is also one of the most significant
etiological factors that contribute to the development of lung
cancer, chronic obstructive pulmonary disease, interstitial lung
diseases and bronchial asthma (1).
Based on the reports of the World Health Organization, smoking will
cause roughly 10 million deaths per annum by the year 2030
(2). Approximately 25% of these
deaths will be exclusively from lung cancer. Cigarette smoking has
been established as the most significant risk factor for lung
cancer, and individuals who smoke usually have a 15- to 30-fold
increased risk of developing lung cancer (3). Current treatment options are
inadequate due to the poor understanding of the molecular basis of
cigarette-induced malignant transformation of bronchial epithelial
cells and the subsequent development of lung cancer.
The 'loss of functionʼ of tumor-suppressor genes
appears to be a critical step in the pathogenesis of human lung
cancer. The gene for RNA-binding motif protein 5 (RBM5), also known
as LUCA-15 or H37, is mapped to human chromosome 3p21.3. This
region of the human chromosome is deleted in a large number of
human cancers and is presumed to contain one or more
tumor-suppressor genes (4).
Previous studies have shown that RBM5 is downregulated in many
different types of cancers, including ras-transformed Rat-1
embryonic fibroblastic cells (5),
breast (1) and prostate cancer
(6), human vestibular schwannoma
(7) and primary lung cancer
(8). It was reported that
expression of RBM5 mRNA is marginally upregulated whereas the
protein was significantly upregulated in breast tumor tissues
compared to that in non-tumor tissues. These data revealed that
RBM5-related factors are differentially expressed in breast cancer
and suggest the involvement of complex inter-related regulatory
networks involving alternative splicing, oncogenic expression and
tissue-specific function (9–11).
RBM5, through pre-mRNA splicing of multiple target
genes, has the ability to modulate apoptosis and cell cycle arrest
in several types of malignancies, particularly non-small cell lung
cancer cells (4,5,8,12–18).
In addition, the loss of RBM5 protein expression in primary lung
tumors correlates with increased lymph node metastasis (13). RBM5 expression is also negatively
correlated with the smoking status in patients with lung cancer
(12). Cigarette smoking directly
affects human bronchial epithelial cells, which happen to be the
major component of the airway epithelium. However, the effect of
cigarette smoking on RBM5 expression in bronchial epithelial cells
is not clear; in addition, it would be important to determine
whether this cigarette smoke-induced cancerous transformation of
bronchial epithelial cells involves RBM5. Thus, by developing an
appropriate in vitro model, we attempted to address this
issue and to elucidate the role of RBM5 in the transformation of
bronchial epithelial cells.
Materials and methods
Cell culture
Human bronchial epithelial BEAS-2B cells were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). The cells are non-tumorigenic in nude mice.
BEAS-2B cells were maintained in RPMI-1640 medium (Gibco-BRL,
Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS), 100
U/ml penicillin and 0.1 mg/ml streptomycin [penicillin and
streptomycin solution (100X)] (both from Gibco, Waltham, MA, USA)
at 37°C in a humidified atmosphere containing 5%
CO2.
Preparation of cigarette smoke extract
(CSE)
The 100% CSE was generated as previously described
(19). Briefly, cigarette smoke was
prepared from research-grade cigarettes obtained from the Tobacco
Research and Development Center of the University of Kentucky, USA.
A syringe-driven apparatus device was designed and operated to
allow a stream of smoke to flow into a tube-shaped trap. The smoke
then entered a flask submerged in liquid nitrogen. The amount of
smoke obtained was calculated by the increase in the weight inside
the flask. The smoke particulates were collected in
dimethylsulfoxide (DMSO) at a concentration of 40 mg/ml and were
sterile-filtered through a 0.22-μm-pore filter (Millipore,
Watford, UK), aliquoted and then stored at −80°C, for use in
subsequent experiments.
Cell proliferation assay
Exponentially growing BEAS-2B cells were exposed to
varying concentrations (0, 25, 50 or 100 μg/ml) of CSE or
DMSO (CON) for 8 days, and fresh CSE was added each day. After
exposure, the cells were washed twice with phosphate-buffered
saline (PBS) and then allowed to recover for a period of 2 weeks.
In this way, we generated five cell populations for each
concentration of CSE. After exposure to CSE, the proliferative
changes of five cell populations were examined by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltet-razolium bromide (MTT)
assay. Briefly, the cells (5×103) were cultured in each
well of 96-well plates for 72 h. During the last 4 h of incubation,
the cells were exposed to 20 μl/well of 5 mg/ml MTT solution
(Sigma-Aldrich, St. Louis, MO, USA). The resulting formazan was
dissolved in 150 μl of DMSO, and the absorbance was measured
at 490 nm using a SpectraMax 190 microplate reader (Molecular
Devices, Sunnyvale, CA, USA). Similarly, the cell viability was
measured using the cell viability assay. In addition, the
morphology of each individual group of cells was examined under a
phase contrast microscope (CKX41; Olympus, Japan).
Cell invasion and migration assays
The invasion and migration of the cells were
determined by Transwell invasion and wound-healing assays. Briefly,
the cells (5×103) were cultured in FBS-free RPMI-1640
medium in the top chambers of 24-well Transwell plates (0.4
μm; Corning Costar, Cambridge, MA, USA) coated with Matrigel
(BD Biosciences). The lower chambers were filled with RPMI-1640
supplemented with 10% FBS as a chemoattractant for 72 h. The cells
that had invaded to the bottom of the top chamber were stained with
crystal violet stain dissolved in 2% ethanol, and at least 10
different fields were counted under a phase contrast microscope to
calculate the invasion efficiency.
For the wound-healing assay, the cells
(1×105) were first starved in 2% FBS medium in 24-well
plates overnight. The next day, a scratch was created in the cell
layer with the small tip of a ruler. The scratched areas were
washed with PBS to remove free cells, and the adherent cells were
cultured in complete medium for one more week. This procedure was
followed by longitudinal photo imaging under a phase contrast
microscope.
Western blot analysis
After treatment, the cultured cells were lysed using
RIPA lysis buffer. The protein concentration was determined using a
Bio-Rad protein assay kit (Bio-Rad), and protein samples (40
μg) were separated on a 10% sodium dodecyl
sulfate-polyacrylamide gel and transferred onto a polyvinylidene
difluoride membrane. The membranes were then blocked with 5%
non-fat dry milk in Tris-buffered saline for 2 h, followed by
incubation with primary antibodies, including anti-RBM5 (ab85504),
anti-cyclin A (ab80792), anti-cyclin D1 (ab7958),
anti-cyclin-dependent kinase 4 (CDK4) (EPR4513, ab108357),
anti-CDK6 (EPR4515, ab124821), anti-p53 (EPR17343, ab174977),
anti-p21 (ab18209), anti-matrix metalloproteinase (MMP)-2
(ab110168), anti-MMP-9 (ab5707), anti-hypoxia-inducible factor
(HIF)-1α (ab10363), anti-vascular endothelial growth factor (VEGF)
(ab46154), anti-K-ras (ab102007), anti-C-myc (ab39688), anti-Bax
(E63, ab32503), anti-bcl-2 (ab117115) and anti-β-actin (E247,
ab32572) from Abcam (Cambridge, MA, USA) as well as
anti-cleaved-caspase-3 (#9661) and anti-cleaved-caspase-9 (#9509)
from Cell Signaling Technology (Beverly, MA, USA), at 1:500
dilutions for 2 h at room temperature. The membranes were then
washed three times with 0.1% Tween-20/PBS prior to incubation with
the appropriate secondary antibody conjugated with peroxidase
(anti-rabbit or anti-mouse IgG) (Santa Cruz Biotechnology) for 1.5
h. Finally, the proteins were detected using the enhanced
chemiluminescence detection reagent (Millipore, Billerica, MA,
USA), and their levels were quantified by densitometry using
Quantity One software (Bio-Rad Laboratories).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted using TRIzol solution
(Takara Biotechnology Co., Dalian, China), according to the
manufacturer's instructions. cDNAs were reverse transcribed using a
reverse transcription kit (Takara Biotechnology), according to the
manufacturer's instructions. Semi-quantitative real-time PCR was
then performed using the following primers: RBM5 forward,
5′-ACACGATGGATGGAAGCCA-3′ and reverse, 5′-TCTGCTCTGCCTCTGACTT-3′;
glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) forward,
5′-GGGTGATGCTGGTGCTGAGTATGT-3′ and reverse,
5′-AAGAATGGGAGTTGCTGTTGAAGTC-3′. These oligonucleotides were
synthesized by Shanghai Promega Biological Products, Ltd.
(Shanghai, China). The relative levels of target gene mRNA
transcripts to the control GAPDH were determined by quantitative
RT-PCR (qRT-PCR) on a Stratagene Mx3000P Real-Time PCR system
(Stratagene, La Jolla, CA, USA) using the FastStart Universal
SYBR-Green Master reagent (Roche, Mannheim, Germany) and specific
primers (Table I). The relative
levels of target gene mRNA transcripts to the control GAPDH were
calculated by the formula: 2−∆∆Ct.
 | Table IList of primer sequences used for qPCR
analyses. |
Table I
List of primer sequences used for qPCR
analyses.
| Gene | Forward primer | Reverse primer |
|---|
| GAPDH |
5′-AGAAGGCTGGGGCTCATTTG-3′ |
5′-AGGGGCCATCCACAGTCTTC-3′ |
| p53 |
5′-CCATCTACAAGCAGTCACAG-3′ |
5′-CAAATCTACAAGCAGTCACAG-3′ |
| p21 |
5′-ACTTCGACTTTGTCACCGAGA-3′ |
5′-GAGGCACAAGGGTACAAGACA-3′ |
| MMP-9 |
5′-CGAACTTCGACACTGACAAGAAGT-3′ |
5′-GCACGCTGGAATGATCTAAGC-3′ |
| MMP-2 |
5′-CTGGGTTTACCCCCTGATGTCC-3′ |
5′-AACCGGGGTCCATTTTCTTCTTT-3′ |
| Caspase-3 |
5′-GAAACCTCCGTGGATTCAAA-3′ |
5′-AGCCCATTTCAGGGTAATCC-3′ |
| Caspase-9 |
5′-TCCTGGTACATCGAGACCTTG-3′ |
5′-AAGTCCCTTTCGCAGAAACAG-3′ |
| Bax |
5′-ACCAAGAAGCTGAGCGAGTG-3′ |
5′-CCCAGTTGAAGTTGCCATCA-3′ |
| Bcl-2 |
5′-ACGACTTCTCCCGCCGCTAC-3′ |
5′-CCCAGCCTCCGTTATCCTG-3′ |
| Cyclin A |
5′-TGGACCTTCACCAGACCTAC-3′ |
5′-GGTTGAGGAGAGAAACACCA-3′ |
| Cyclin D1 |
5′-AGTTGCTGCAAATGGAACTG-3′ |
5′-AAAGGTCTGTGCATGTTTGC-3′ |
| CDK4 |
5′-TTTGATCTCATTGGATTGCC-3′ |
5′-AGGTCAGCATTTCCAGCAG-3′ |
| CDK6 |
5′-TGGAGTGTTGGCTGCATATT-3′ |
5′-ACAGGGCACTGTAGGCAGAT-3′ |
| HIF-1α |
5′-CCACAGGACAGTACAGGATG-3′ |
5′-TCAAGTCGTGCTGAATAATACC-3′ |
| VEGF |
5′-AAACCCTGAGGGAGGCTC-3′ |
5′-TACTTGCAGATGTGACAAGCCG-3′ |
| K-ras |
5′-GGGGAGGGCTTTCTTTGTGTA-3′ |
5′-GTCCTGAGCCTGTTTTGTGTC-3′ |
| C-myc |
5′-ACAGCTACGGAACTCTTGTGCGTA-3′ |
5′-GCCCAAAGTCCAATTTGAGGCAGT-3′ |
| RBM5 |
5′-ACACGATGGATGGAAGCCA-3′ |
5′-TCTGCTCTGCCTCTGACTT-3′ |
Lentiviral vector construction and
infection
Lentiviral vectors conjugated to green fluorescent
protein (GFP) were used to achieve high efficiency of infection and
subsequent stable expression of RBM5 protein in the target cells.
Recombinant pGC-LV-GV287-GFP vector with either the RBM5
(NM_005778) gene (LV-RBM5) or the scrambled control sequence
(LV-GV287) was constructed by GeneChem Co. (Shanghai, China). Next,
the cells (5×105) were seeded in a 6-well culture plate
and were incubated for 12 h to reach 30% confluency. Later, these
cells were infected with LV-RBM5 (RBM5 overexpression group) or
LV-GV287 (negative control group) lentiviral particles at a
multiplicity of infection (MOI) of 20 plaque-forming units/cell.
The plates were then incubated for 24 h prior to changing the
medium to fresh virus-free medium. Three days later, the GFP
expression was assessed in the target cells to evaluate the
efficiency of infection, and the cells were harvested for western
blotting and RT-PCR analyses.
Colony formation assay
The cells were digested with 0.25% trypsin
(Gibco-BRL, Grand Island, NY, USA) to obtain a single-cell
suspension with a density of 0.1×106 cells/ml. The cell
suspensions were then transferred into 6-well plates (300
cells/well) and incubated at 37°C in an atmosphere of 5%
CO2 for 21 days. The formed cell colonies were stained
with crystal violet dye in 50% methanol and 10% acetic acid, and
the stained colonies that had >50 cells were counted in a
blinded manner. The colony formation efficiency was calculated as
the ratio of the colony number to the plated cell number.
Flow cytometric analysis
Cells were trypsinized, washed twice with ice-cold
PBS, fixed in 70% ethanol and stained with propidium iodide (PI; 5
μg/ml PI in PBS containing 0.1% Triton X-100 and 0.2 mg/ml
RNase A) overnight at 4°C in the dark. The distribution of
different cell phases was analyzed by flow cytometry using ModFit
LT software.
Furthermore, the harvested cells were stained with
Annexin V-PE and 7-AAD. The percentages of apoptotic cells were
analyzed by flow cytometric analysis.
Tumorigenic and histological studies
BALB/c athymic nude male mice (nu/nu; 5 weeks) were
purchased from the Institute of Zoology, Chinese Academy of
Sciences, Beijing, China. The care and use of the animals was in
accordance with the Animal Care guidelines, and the protocol was
approved by Jilin University, Animal Care Committee. The cells were
washed and resuspended in PBS. A total of 5×106 cells
suspended in a 150-μl volume were subcutaneously injected
into the right flanks of each nude mouse. The size of the tumors
was measured using a caliper, starting on day 7 after cell
injection. The animals were palpated for tumor appearance once a
week and were sacrificed as soon as the tumor nodules reached 1.0
cm in size. Xenograft tissues were immediately harvested after
euthanasia, fixed in neutral buffered formalin, and embedded in
paraffin for routine histological evaluation by staining
5-μm-thick sections with hematoxylin and eosin (H&E)
stain.
Statistical analysis
All experiments were performed at least in
triplicate, and the data are presented as means ± standard
deviation (SD). Statistical significance was determined by analysis
of the t-test using SPSS version 17.0 (SPSS, Inc., Chicago, IL,
USA) software. A P-value of <0.05 was considered to indicate a
statistically significant result.
Results
CSE exposure promotes the cancerous
transformation of normal human bronchial epithelial cells
We evaluated the effect of CSE on non-cancerous
human bronchial epithelial BEAS-2B cells by exposure to varying
concentrations of CSE (0, 25, 50 and 100 μg/ml,
respectively) or DMSO (CON) for 8 days, followed by a recovery
period of 2 weeks. The results of the MTT assay showed that the
cell population, which survived the toxic effects of CSE for 8
days, acquired phenotypic changes after a recovery period of 2
weeks, including enhanced cell proliferation (Fig. 1A) and shorter doubling times during
the recovery period.
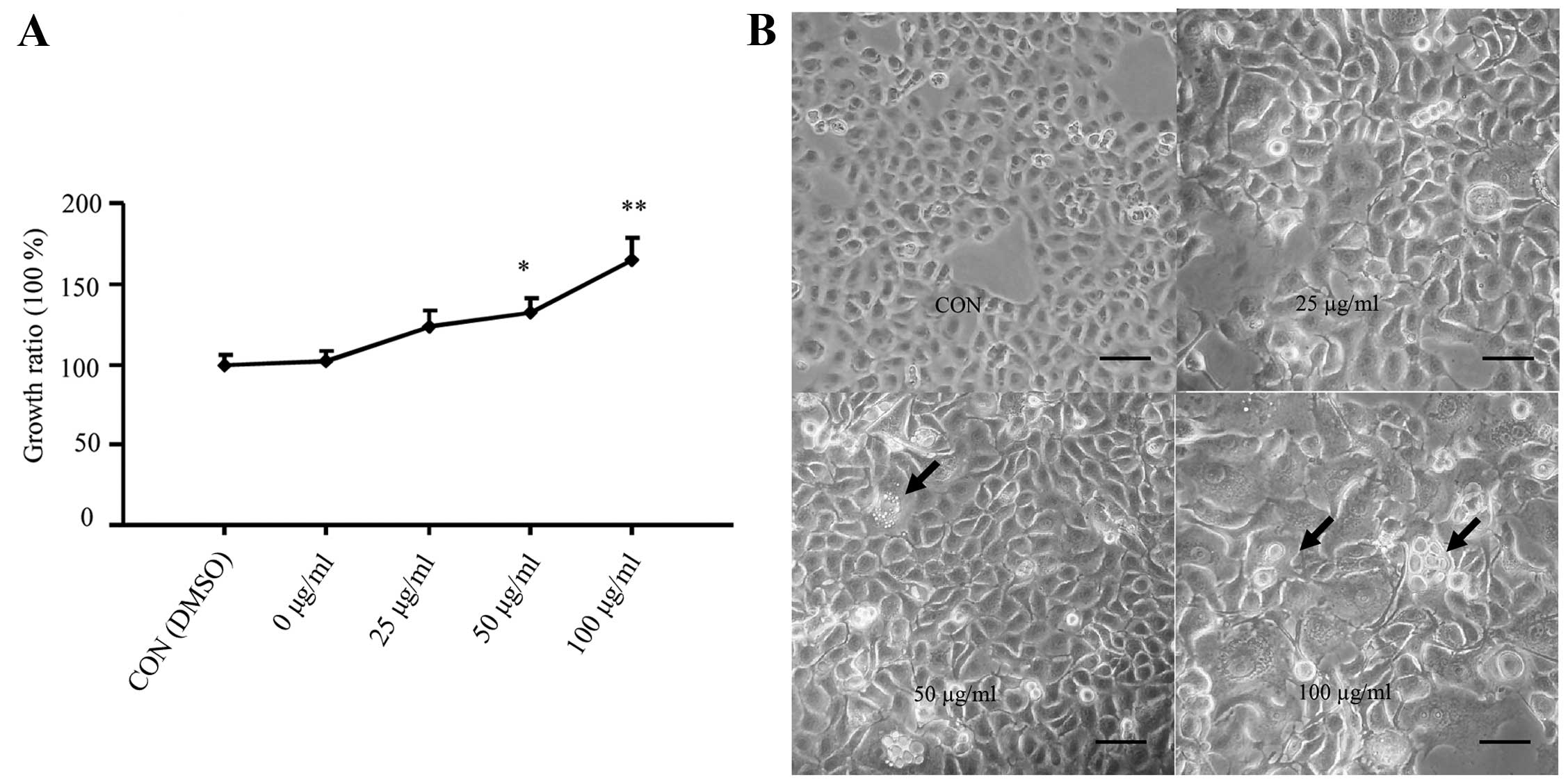 | Figure 1Analysis of the effect of CSE on
normal human bronchial epithelial cells. (A) The effects of CSE on
the growth rates of BEAS-2B cells as determined by the MTT assay.
The growth rates of control cells without CSE exposure were
designated as 0%. (B) Morphological changes in the BEAS-2B cells
following prolonged CSE exposure at varying concentrations. The
images were captured under a phase contrast light microscope. Scale
bar, 50 μm. (C and D) The invasive ability of the
CSE-transformed BEAS-2B cells as determined by the Transwell
invasion assay. Scale bar, 20 μm. *P<0.05,
**P<0.01 vs. the control. (E) The migration ability
of the CSE-transformed BEAS-2B cells exposed to varying
concentrations of CSE. The images were recorded after 0, 36 and 72
h of exposure. (F) The effects of CSE exposure on the expression of
oncogenes, K-ras, c-myc, cyclin A and cyclin D1. The expression of
β-actin was used as a loading control. (G) The normalized
expression of these oncogenes. *P<0.05,
**P<0.01 vs. the control. (H) Relative mRNA
expression of the oncogenes by RT-PCR. (I) Xenograft tumor growth
at 2 weeks after inoculation with CSE-transformed BEAS-2B cells
exposed to different CSE concentrations. The arrows indicate a
large tumor mass and a section of a representative tumor stained
with H&E. Scale bar, 50 μm. The data in all panels were
collected from three individual experiments. *P<0.05,
**P<0.01 vs. the control. |
In addition, we analyzed whether prolonged exposure
to CSE led to the cancerous transformation of non-tumorous
epithelial BEAS-2B cells, as they reflected an enhanced cell
proliferation. Therefore, to determine whether this transformation
occurred, we characterized the morphological changes in BEAS-2B
cells, longitudinally. During the recovery period after CSE
exposure, the cells treated with 100 μg/ml CSE showed
aberrant and condensed nuclei in addition to abnormal nuclear
cytoplasmic ratios, compared to the control cells (Fig. 1B). Similar changes were also
observed for the cells treated with 25 and 50 μg/ml CSE, but
the effects were not as obvious as with the higher dose (100
μg/ml) of CSE. Moreover, treatment with 100 μg/ml CSE
also revealed multiple layers of cell growth, indicative of the
loss of contact inhibition. These data thus indicate that exposure
to CSE promoted the cancerous transformation of BEAS-2B cells.
CSE-transformed BEAS-2B cells exhibit
enhanced invasion and migration ability
The invasion and migration of the CSE-transformed
BEAS-2B cells were determined by Transwell invasion and
wound-healing assays. As shown in Fig.
1C and D, CSE exposure (25, 50 and 100 μg/ml) or DMSO
increased the ability of these cells to invade the extracellular
matrix, compared to the control treatment. This effect was most
significant (P<0.01) following exposure to 100 μg/ml CSE.
Similarly, the wound-healing assay results also showed that CSE
exposure enhanced the migration of CSE-transformed cells compared
to the control group (Fig. 1E).
Prolonged exposure of CSE induces the
expression of oncogenes and xenograft growth
Next, we examined the protein and mRNA levels of
various oncogenes, including K-ras, c-myc and cyclin A and cyclin
D1, in the CSE-transformed BEAS-2B cells. Western blot analysis
showed that the expression of these proteins was significantly
enhanced in the CSE-transformed BEAS-2B cells treated with varying
concentrations of CSE (P<0.01) (Fig.
1F and G). In addition, the qPCR results demonstrated a
similarly significant (P<0.05) increase in the expression of
K-ras, c-myc, cyclin A and cyclin D1 mRNA levels in the
CSE-transformed BEAS-2B cells, compared with the control group
(Fig. 1H). These results indicate
that CSE regulated the expression of these oncogenes at the
transcriptional level in the CSE-transformed BEAS-2B cells.
Subsequently, we analyzed the ability of the
CSE-transformed BEAS-2B cells to induce xenograft growth. BEAS-2B
cells were treated with varying concentrations (0, 25, 50 or 100
μg/ml) of CSE or DMSO (CON) for 8 days, and then allowed to
recover for a period of 2 weeks. Next, we implanted BEAS-2B cells,
BEAS-2B cells treated with DMSO (CON cells) or CSE-transformed
BEAS-2B cells into immunodeficient mice (5 nude mice/cell line) and
observed that the BEAS-2B and CON cells did not form tumors, even
after 3 months of injection, while all mice (n=5) implanted with
the transformed BEAS-2B cells exposed to 100 μg/ml CSE
developed tumors with a latency period of ~2 weeks, indicating a
high efficiency of murine tumorigenicity (Fig. 1I). However, the transformed cells
exposed to 25 or 50 μg/ml CSE did not form tumors. The
histological examination further confirmed that the cells exposed
to 100 μg/ml CSE formed carcinomas, revealing that the
tumors were composed of variably sized nests of polyhedral cells
that were connected by thin stromal strands. These tumor cells were
moderately pleiomorphic, had shapes from round to elongated, and
had prominent multiple nuclei (Fig.
1I). We designated this CSE-induced cancerous transformation of
BEAS-2B cells as 'T3 cellsʼ.
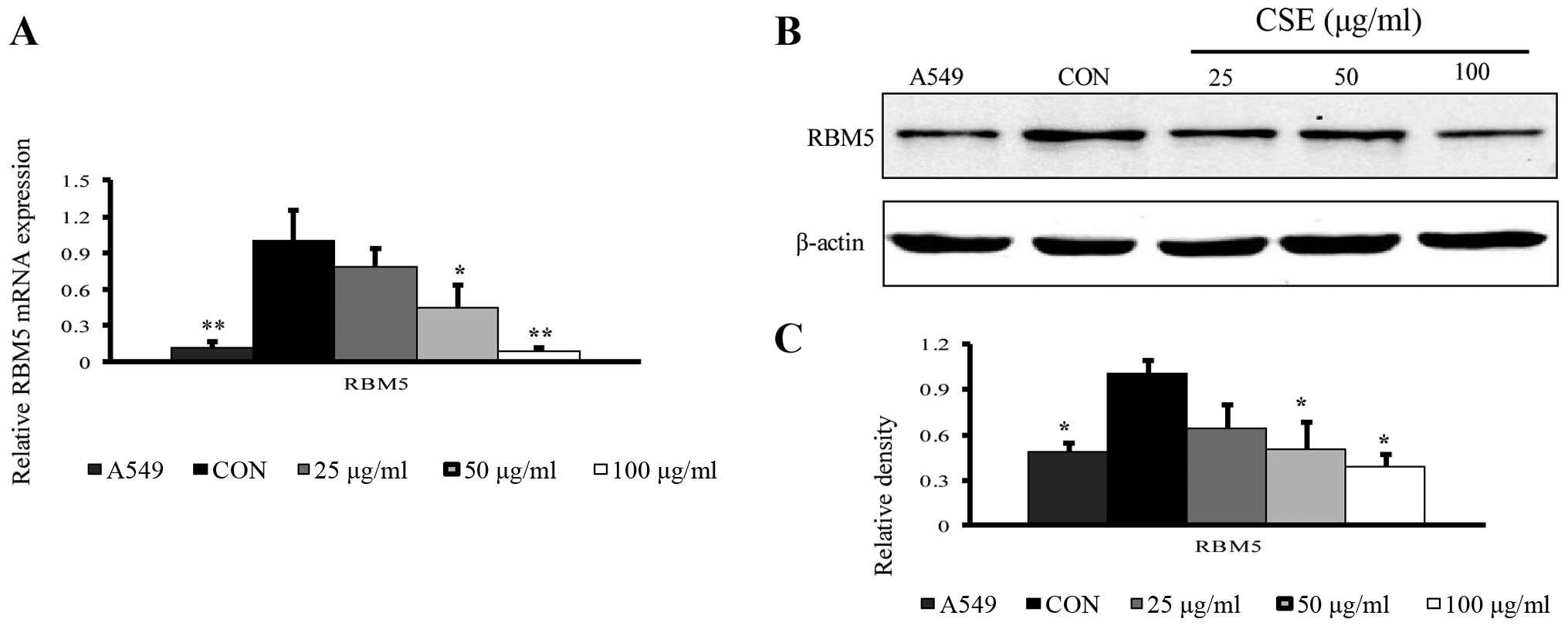
CSE-transformed BEAS-2B cells have
reduced expression of RBM5
To analyze the role of RBM5 in the cancerous
transformation of normal human bronchial epithelial cells, we first
determined the expression of RBM5 in these cells exposed to CSE at
varying concentrations. We observed that both at the mRNA (qRT-PCR)
and protein levels (western blotting), the RBM5 expression was
reduced with an increasing CSE concentration, compared to the
control group. The fold-change in the gene expression was
significantly reduced in the T3 cells (cells treated with 100
μg/ml CSE) (P<0.01) (Fig.
2A–C). It is important to mention that we also analyzed the
expression of RBM5 in A549 cells, which are inherently cancerous
cells, as a control and observed that RBM5 expression in these
cells was also less than that in the control cells.
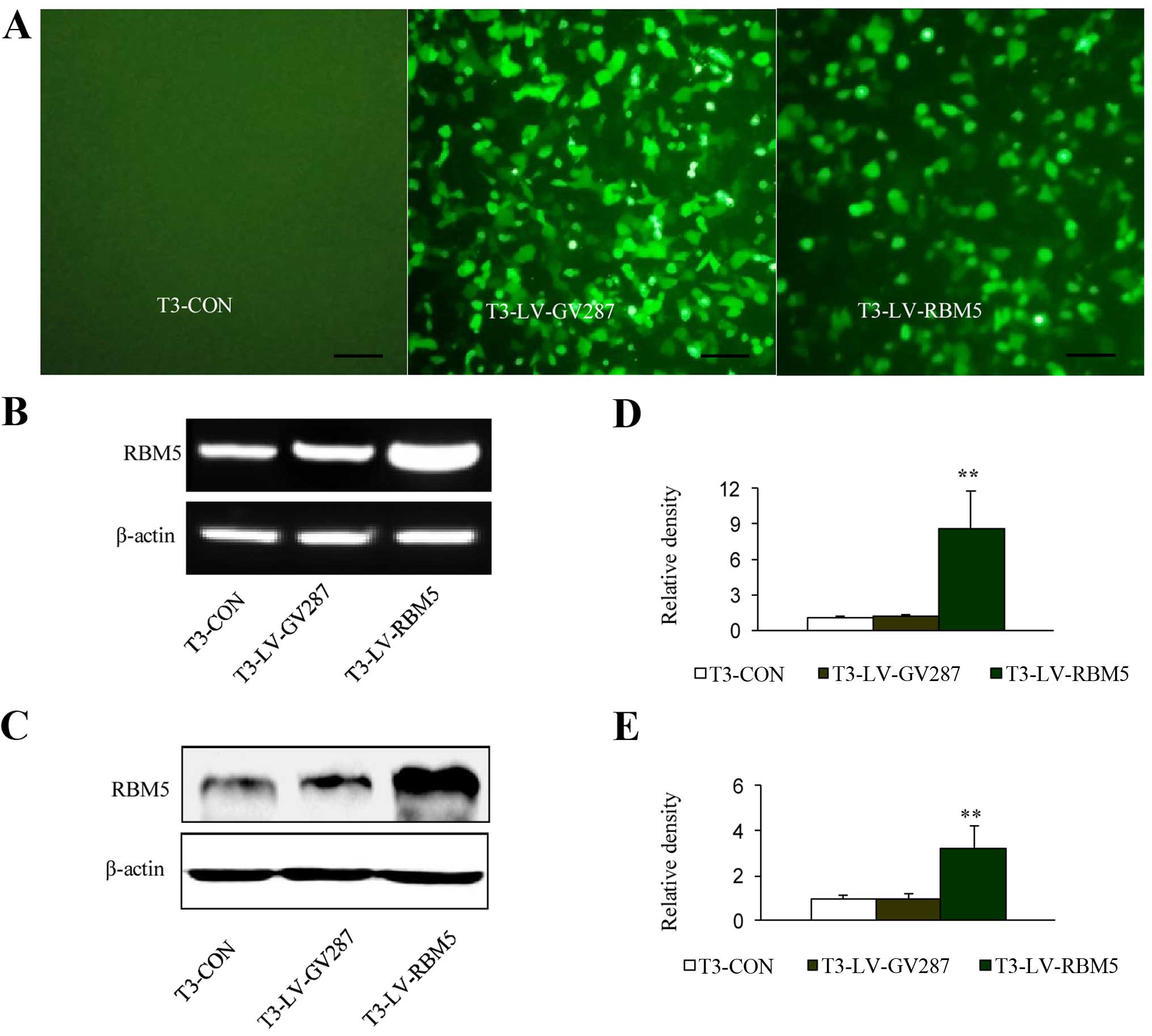
Analysis of RBM5 overexpression in the
CSE-transformed BEAS-2B cells infected with lentiviral vectors
The T3 cells were successfully infected with
recombinant lentiviral vectors expressing GFP, LV-RBM5 or vector
alone (LV-GV287). Using fluorescence microscopy, the GFP expression
was analyzed 3 days after the infection, and it was observed that
the efficiency of the infection was ~80% at an MOI of 20 (Fig. 3A). Therefore, an MOI of 20 was used
for all other infection experiments in the present study. The
RT-PCR analysis demonstrated a significant (P<0.01) increase in
the expression level of RBM5 mRNA in the T3-LV-RBM5 group, compared
with the negative control group (T3-LV-GV287 cells) and the
non-transfected control group (T3-CON cells) (Fig. 3B and D). Similarly, western blot
analysis also confirmed the overexpression of RBM5 (Fig. 3C and E).
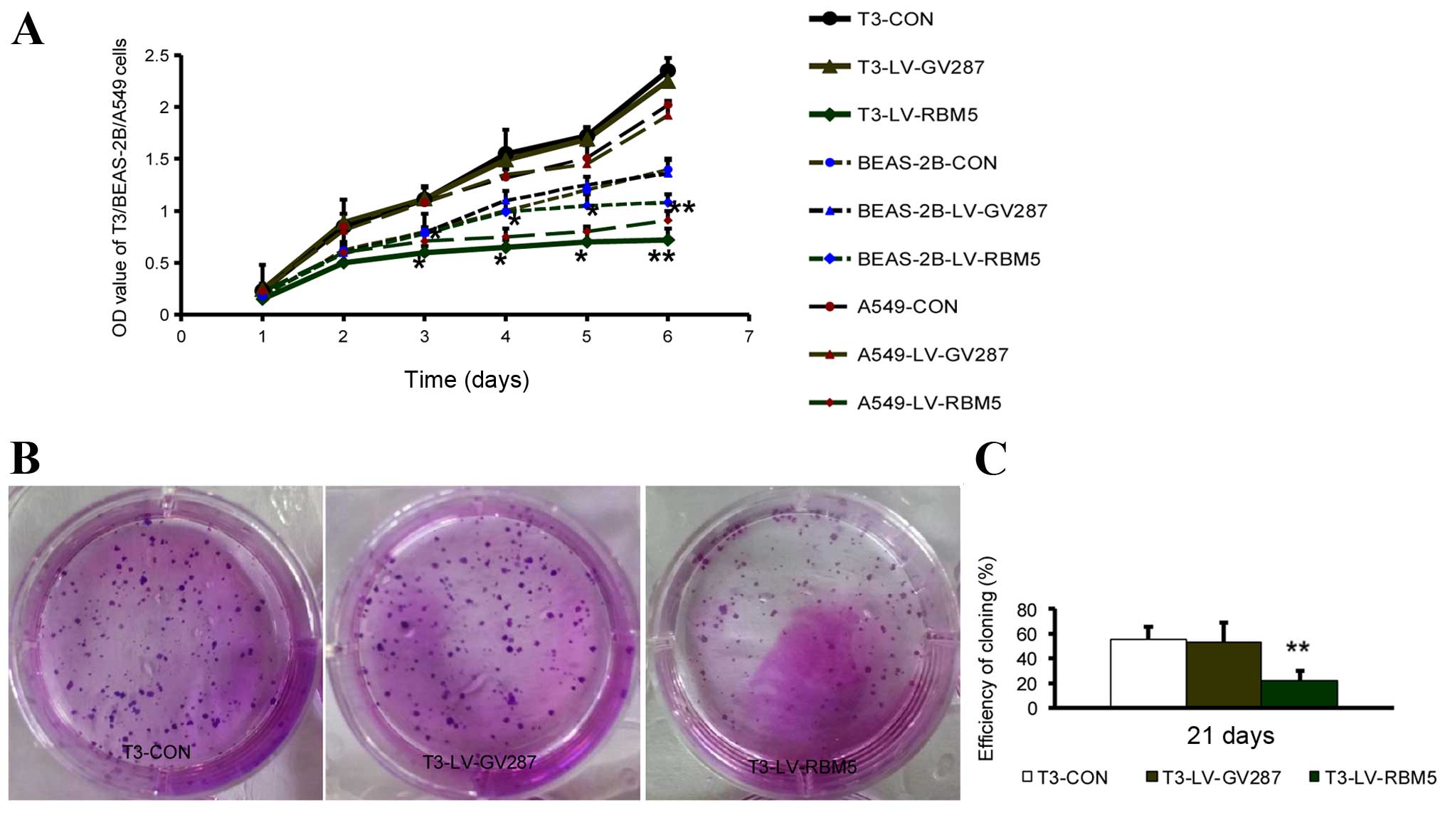
RBM5 overexpression inhibits the
proliferation and colony formation of CSE-transformed BEAS-2B
cells
Next, we analyzed whether an elevated RBM5
expression has any effect on the cell proliferation and colony
formation of CSE-transformed BEAS-2B cells. As shown in Fig. 4A, RBM5 overexpression markedly
inhibited the cell growth of the T3 cells overexpressing RBM5,
compared to the T3 cells transfected with the empty vector
(P<0.01). Simultaneously, RBM5 overexpression markedly inhibited
the cell growth of the A549 cells, compared to the A549 cells
transfected with the empty vector, while the growth of BEAS-2B
cells was not significantly changed after RBM5 overexpression for 3
days, compared to BEAS-2B cells transfected with the empty vector.
Moreover, the ability of these different groups of cells to form
colonies was assessed after culturing for an additional 21 days,
and images under a microscope were recorded. Quantitative analysis
of individual clones with >50 cells revealed a significantly
(P<0.01) reduced colony size and fewer numbers of cells in the
T3-LV-RBM5 group, compared to the T3-CON and T3-LV-GV287 groups
(Fig. 4B and C). This set of data
indicated that overexpression of RBM5 inhibited the proliferation
and the colony formation potential of the CSE-transformed BEAS-2B
cells.
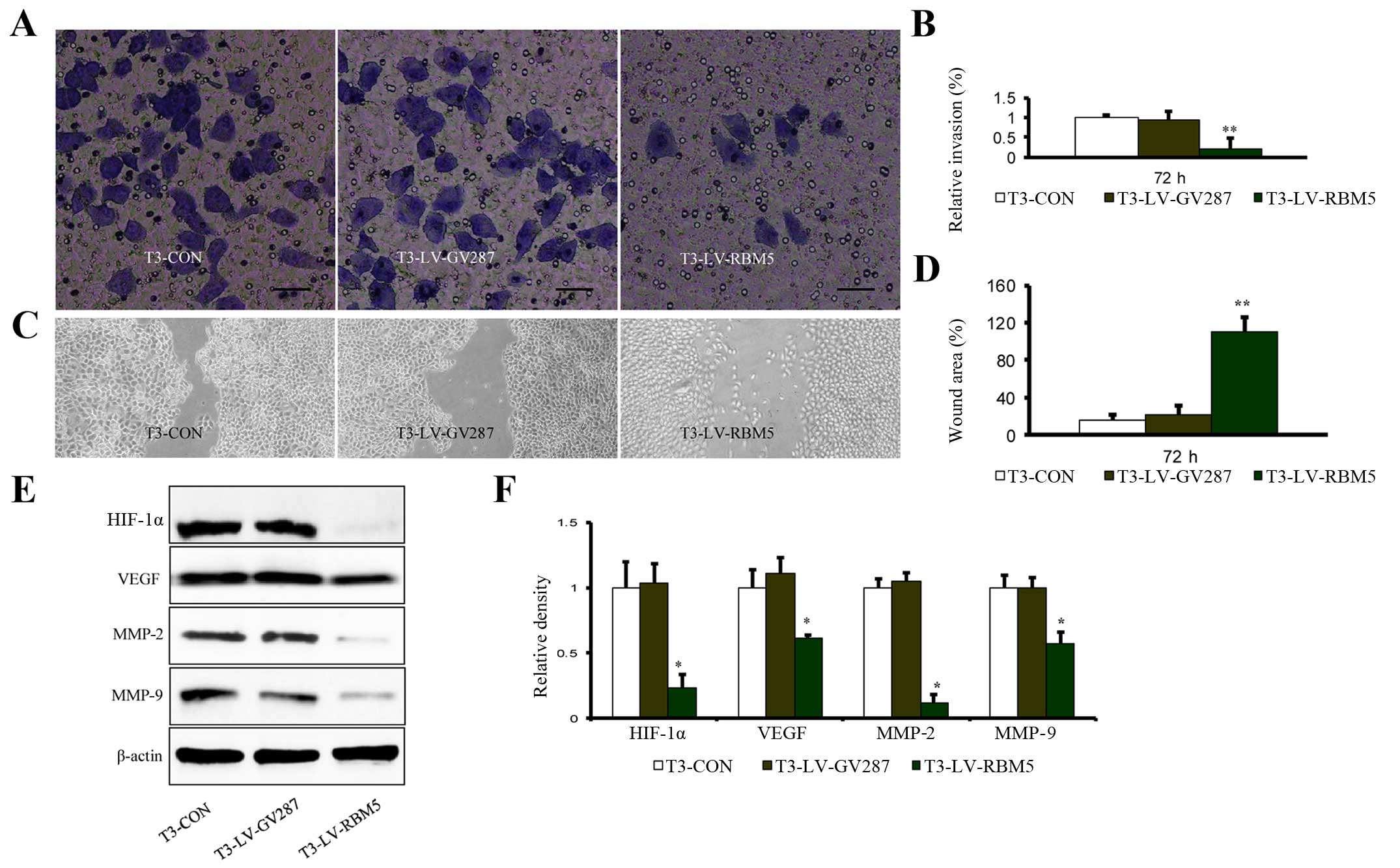
RBM5 overexpression inhibits the invasion
and migration of CSE-transformed BEAS-2B cells
We further determined the impact of RBM5 on the
invasion and migration ability of CSE-transformed BEAS-2B cells. As
shown in Fig. 5A and B, the ability
of T3-LV-RBM5 cells to invade through the extracellular matrix was
significantly reduced, compared with the T3-CON and T3-LV-GV287
cells. Similarly, the results of the wound-healing assay also
showed that compared with the control groups, the migration of the
T3-LV-RBM5 cells was markedly decreased (Fig. 5C and D). Furthermore, expression
analysis of the genes commonly implicated in cell invasion and
migration revealed that RBM5 overexpression decreased the relative
levels of HIF-1α, VEGF, MMP-2 and MMP-9 (Fig. 5E and F). These data demonstrated
that RBM5 inhibited the invasion and migration of CSE-transformed
BEAS-2B cells, most probably by regulating the expression of these
pathway-related genes.
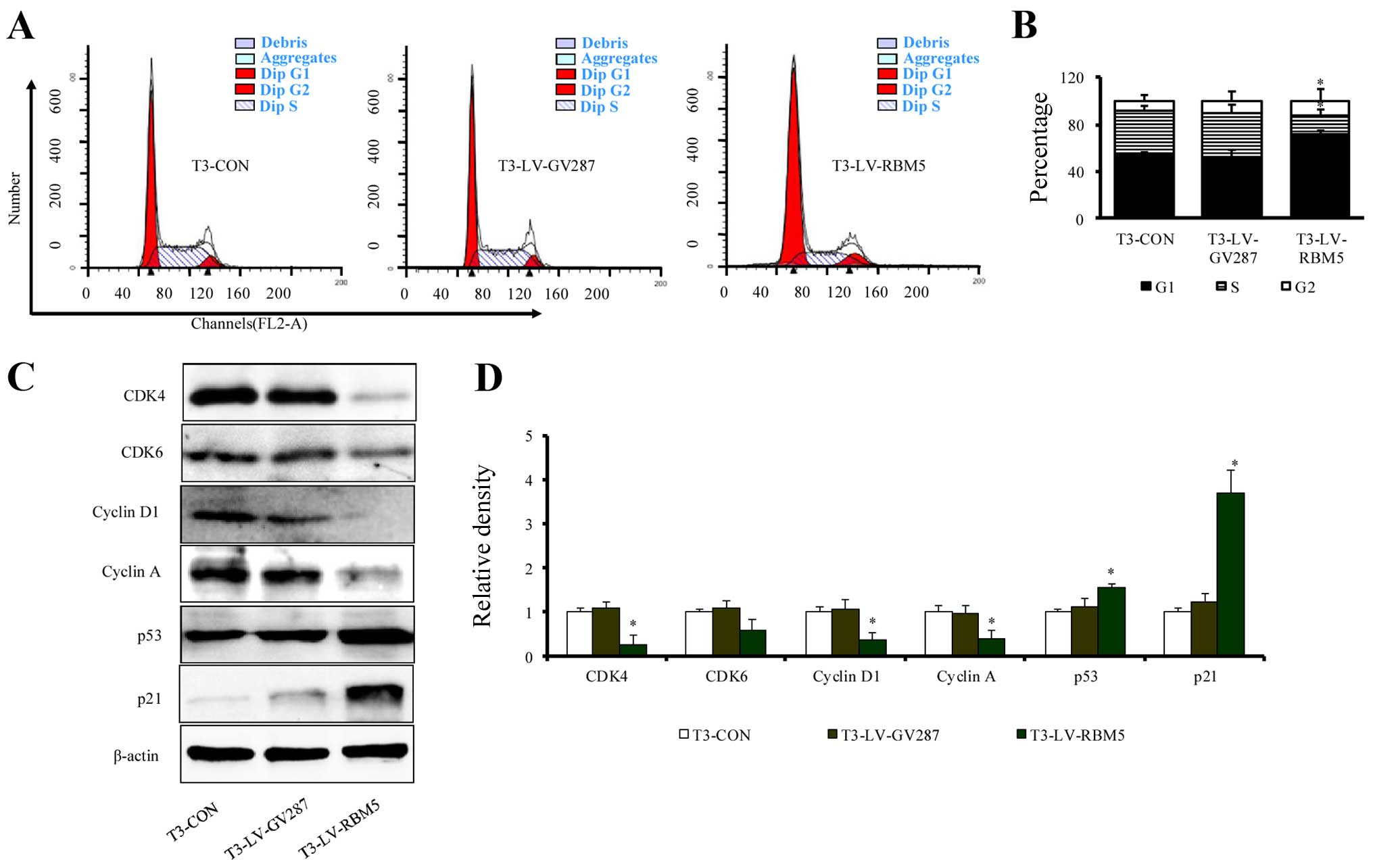
RBM5 overexpression induces cell cycle
arrest of CSE-transformed BEAS-2B cells at the G1/S phase
In an effort to understand the mechanism of RBM5
overexpression on inhibition of cell proliferation, we first
analyzed the effects of RBM5 on the cell cycle distribution in
CSE-transformed BEAS-2B cells using flow cytometry. The results
revealed that RBM5-overexpressing T3 cells (T3-LV-RBM5) had a
significantly increased fraction of cells in the G1 phase and a
decreased percentage in the S phase (Fig. 6A). The overall distribution of cells
in each phase of the cell cycle among all the groups is shown in
Fig. 6B. Further analysis of the
expression of cell cycle regulators indicated that the T3-LV-RBM5
cells had significantly decreased relative levels of CDK4, CDK6,
cyclin D1 and cyclin A, but increased levels of p53 and p21
(Fig. 6C and D). These data
indicate that RBM5 induced cell cycle arrest at the G1/S phase in
the CSE-transformed BEAS-2B cells, probably through modulating the
expression of cell cycle regulators.
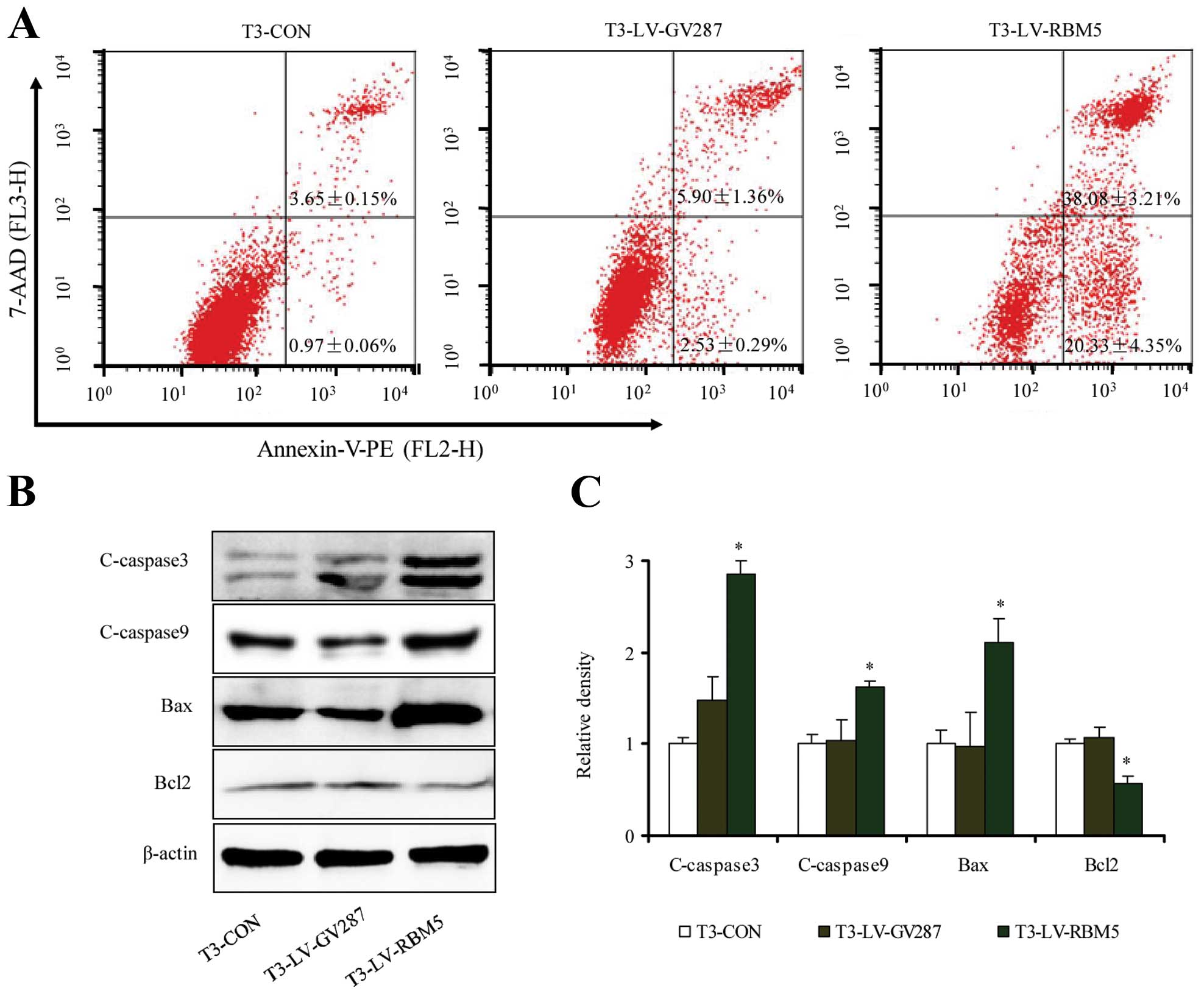
RBM5 overexpression induces apoptosis of
CSE-transformed BEAS-2B cells
To further understand the mechanism of RBM5 in cell
proliferation, we also analyzed the effect of RBM5 overexpression
on apoptosis by labeling the cells with FITC-Annexin V and PI,
followed by flow cytometric analysis. As shown in Fig. 7A, the percentage of apoptotic cells
in the T3-LV-RBM5 group was significantly higher than that in the
T3-CON and T3-LV-GV287 groups (P<0.05). Furthermore, RBM5
overexpression significantly increased the levels of cleaved
caspases-3 and -9, along with Bax expression. In contrast, it
inhibited the expression of the anti-apoptotic protein Bcl-2
(Fig. 7B and C). Collectively, RBM5
overexpression triggered the apoptosis of CSE-transformed BEAS-2B
cells.
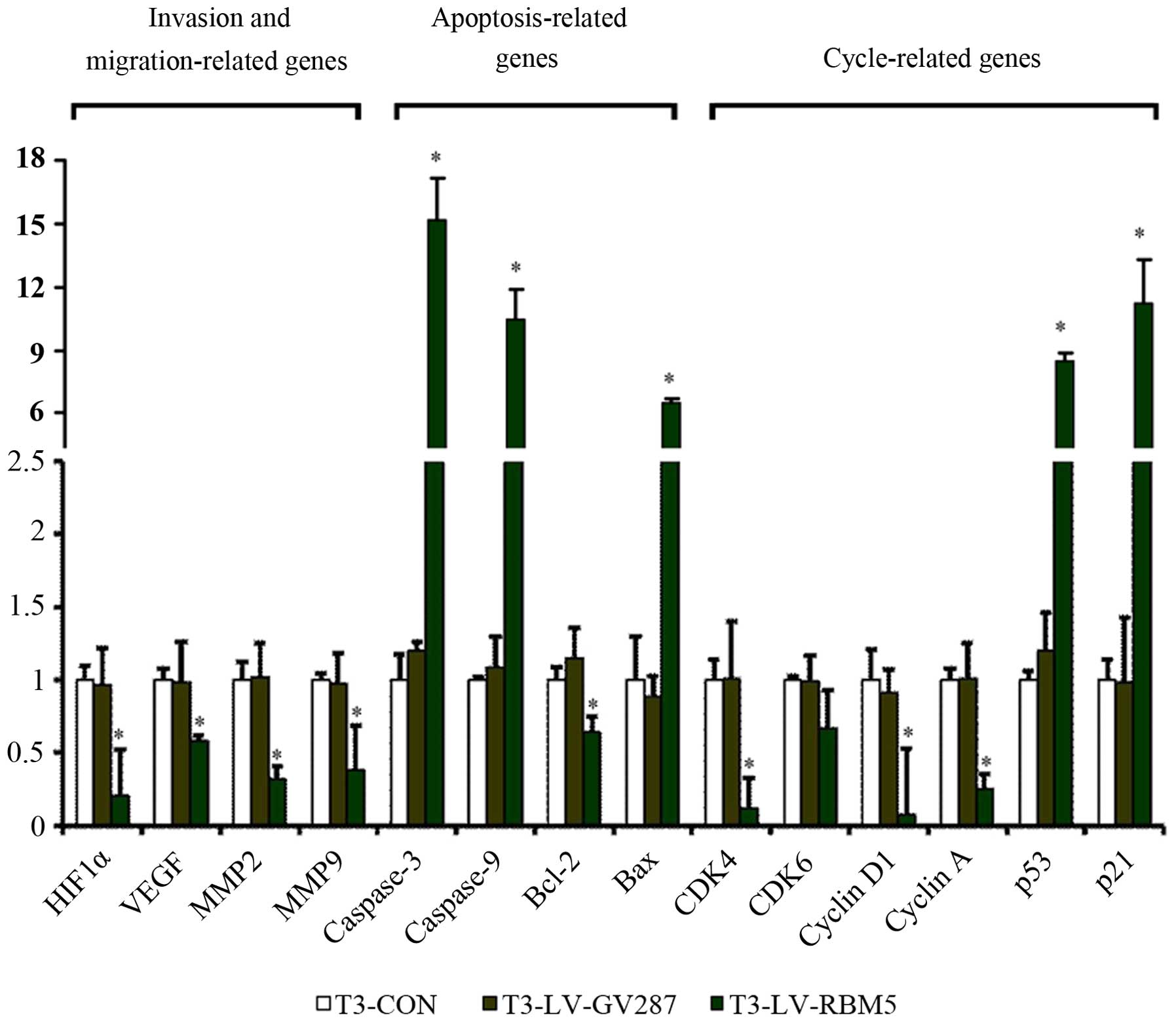
RBM5 modulates the relative mRNA levels
of many genes implicated in cell invasion, apoptosis and cell cycle
regulation
We established that RBM5 overexpression regulated
many genes implicated in cell invasion, migration, cell cycle
regulation and apoptosis at the protein level. To further identify
whether RBM5 overexpression has any effects on these genes at the
transcriptional (mRNA) level, we analyzed their relative expression
by qRT-PCR, compared against the housekeeping gene GAPDH. The
results demonstrated that T3 cells infected with LV-RBM5 had
upregulated relative levels of caspase-3, caspase-9 and Bax mRNA
transcripts (Fig. 8). In contrast,
T3-LV-RBM5 cells had significantly decreased mRNA transcript levels
of the anti-apoptotic gene Bcl-2 (P<0.05). Furthermore, RBM5
overexpression significantly reduced the relative levels of CDK4,
CDK6, cyclin D1 and cyclin A, but elevated the levels of p53 and
p21 mRNA transcripts (P<0.05), indicating that RBM5 induced cell
cycle arrest in the CSE-transformed BEAS-2B cells. In addition,
RBM5 overexpression significantly decreased the relative levels of
HIF-1α, VEGF, MMP-2 and MMP-9 expression (P<0.05), explaining
its effect on the inhibition of invasion and migration of the
CSE-transformed BEAS-2B cells.
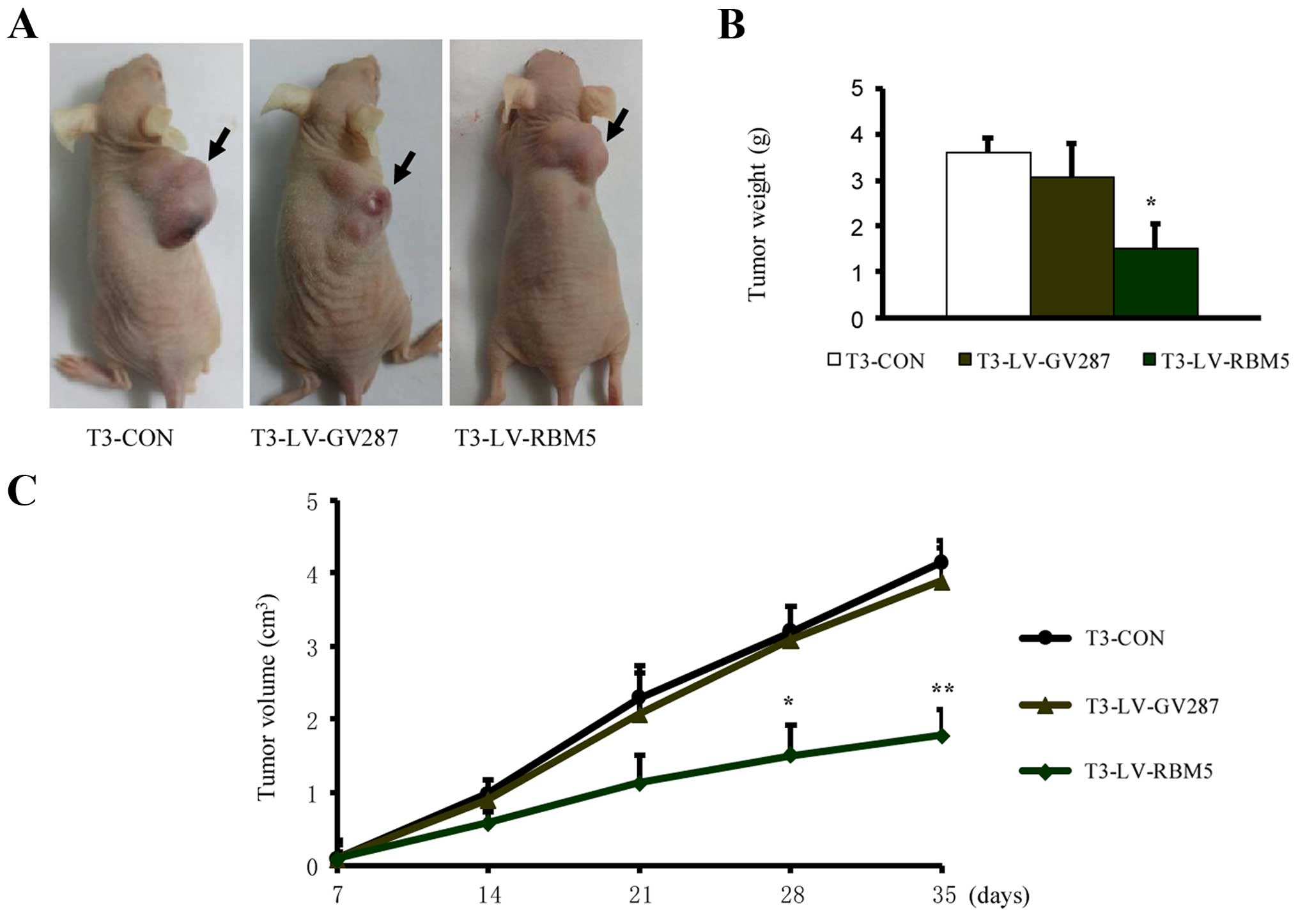
RBM5 overexpression inhibits the
xenograft growth of CSE-transformed BEAS-2B cells
Finally, we determined the effect of RBM5
overexpression on the in vivo growth of CSE-transformed
BEAS-2B cells. The T3 cells overexpressing LV-RBM5 resulted in a 55
and 50% reduction in tumor weight and volume, respectively,
measured after 4 weeks, compared to the control groups (Fig. 9). Thus, these data indicate that
RBM5 overexpression significantly inhibited xenograft growth.
Discussion
In the present study, we established a cellular
model of continual CSE exposure of human non-tumorous bronchial
epithelial cells and showed that CSE can damage bronchial
epithelial cells by inducing hyperplasic growth, malignant cell
transformation and tumorigenesis. We observed that BEAS-2B cells
surviving 8 days of repeated CSE exposure, followed by a recovery
period of 2 weeks, were endowed with phenotypic changes that were
characteristic of oncogenic transformation, including increased
cell proliferation, enhanced invasion and migration activity, and
more importantly, the ability to promote xenograft growth.
Similarly, at the cellular level, we observed upregulation of the
expression of certain oncogenes, including c-myc, K-ras, cyclin A
and cyclin D1 in the transformed BEAS-2B cells. Our results
revealing enhancement of the neoplastic transformation of BEAS-2B
cells by prolonged treatment with CSE were consistent with
previously published data (20).
Meanwhile, analysis of RBM5 expression in these transformed cells
revealed that it was downregulated at both the mRNA and protein
levels and was equivalent to the expression in another tumor cell
line (A549), which was analyzed as a control. This observation led
us to believe that RBM5 expression may be negatively correlated
with smoking-related lung cancer.
To further understand the correlation between RBM5
overexpression and cigarette smoke-induced cancerous transformation
of bronchial epithelial cells, we overexpressed wild-type RBM5
using the recombinant lentiviral vector LV-RBM5 into the
transformed BEAS-2B cells. Further analysis of these transformed
cells revealed that RBM5 overexpression significantly inhibited
their cell proliferation and ability to invade and migrate along
with their colony-forming potential. It also induced G1/S arrest
and apoptosis. In addition, the xenograft growth of transformed
BEAS-2B cells was inhibited as a result of RBM5 overexpression. The
RBM5 overexpression resulted in inhibition of cell cycle regulatory
genes, including CDK4, CDK6, cyclin A and cyclin D1, while the
expression of p53 and p21 was increased. Similarly, the analysis of
apoptosis-related genes (caspase-3 and caspase-9, Bax and Bcl-2)
revealed that RBM5 overexpression significantly altered their
expression. The levels of cleaved caspase-3 and caspase-9 as well
as Bax expression were increased, whereas the expression level of
the anti-apoptotic gene Bcl-2 was decreased. Moreover, we analyzed
the expression of regulatory genes (HIF-1α, VEGF, MMP-2 and MMP-9)
associated with cell migration and invasion and observed that their
expression was significantly downregulated by RBM5 overexpression.
Thus, our study demonstrated that RBM5 overexpression inhibited the
proliferation of CSE-transformed BEAS-2B cells through regulation
of cell cycle arrest and apoptosis by modulating the expression of
key genes involved in both of these pathways.
RBM5 is a nuclear RNA-binding protein that is widely
distributed in mammalian tissues (21). Accumulating evidence suggests that
RBM5 functions as a tumor-suppressor protein by inhibiting tumor
transformation and progression (22). The expression of RBM5 mRNA
transcript and protein has been observed to be decreased in 70–80%
of lung cancers (8). Despite the
increasing evidence suggesting that RBM5 down-regulation plays an
important role in lung cancer initiation, progression, metastasis
and drug resistance (4,8,12,23),
its functional mechanism remains unclear. Our studies on the
antitumor mechanisms of RBM5 were mostly focused on its role in the
cell cycle and apoptosis. Another previous study in leukemic cells
showed the involvement of RBM5 overexpression in inhibiting cell
proliferation by extending the G1 phase of the cell cycle (24). RBM5 has also been suggested to
inhibit both in vitro and in vivo tumor growth of
lung cancer cells, with antitumor mechanisms involving G1 cell
cycle arrest (5). The normal cell
progression from the G1-S-G2-M-G0 phases during the cell division
cycle is tightly controlled by the protein kinase activity of a
class of serine-threonine kinases (CDKs). CDK4 and CDK6, called the
cyclin-dependent kinases, as well as cyclin A and cyclin D1 are
important factors during normal cell progression from G1-S-G2-M-G0
(25–27). The G1/S phase transition is
negatively regulated by p21 and p27 through the degradation of
cyclin/CDK complexes (28,29). In addition, p53 is an important
regulator of p21 expression (30).
Thus, based on our data we speculated that overexpression of RBM5
would upregulate p53 expression, which in turn would elevate the
expression of p21. This p21 expression would then subsequently
inhibit the cyclin/CDK complex, eventually leading to cell cycle
arrest at the G1/S phase in CSE-transformed BEAS-2B cells.
It has been previously suggested that RBM5 modulates
apoptosis by regulating the alternative splicing of
apoptosis-associated pre-mRNAs, such as CASP2 and FAS/CD95
(12,31). Increased levels of RBM5 promote
apoptosis through several pathways, including Fas, TNF-α, TRAIL and
p53-mediated apoptosis (32,33).
Induction of apoptosis by RBM5 has also been correlated with
altered levels of Bcl-2, Bcl-x, and Bax as well as activation of
cleaved caspases-9 and -3 (4,16,34).
In addition, it has been suggested that upregulation or
downregulation of RBM5 causes changes in the transcription levels
of ~35 genes known to control cell proliferation and apoptosis
(17). Our results were consistent
with these previous findings in lung cancer and further support the
notion that RBM5 may function as a tumor suppressor by promoting
the induction of apoptosis in CSE-transformed BEAS-2B cells.
The progression of cancer to different sites largely
involves the ability of cancer cells to invade and migrate. The
inhibition of these pathways is essentially one of the strategies
to combat cancer (35). The
migration and invasion of cancer cells are regulated by many
factors, such as chemokines, their receptors, the
epithelial-mesenchymal transition process, matrix-degrading and
antioxidant enzymes, such as MMPs, which mediate the degradation of
extracellular matrix proteins (36,37).
Among these proteases, MMP-2 and MMP-9 are the most prominent.
Furthermore, the migration and invasion can also be modulated by
hypoxic conditions and angiogenesis regulators, including HIF-1α
and VEGF (38,39). In the present study, we identified
that RBM5 overexpression significantly inhibited the migration and
invasion of CSE-transformed BEAS-2B cells by significantly
attenuating the expression of HIF-1α, VEGF, MMP-2 and MMP-9
proteins.
In conclusion, we demonstrated that RBM5 expression
was inhibited in CSE-transformed BEAS-2B cells and that subsequent
overexpression of RBM5 in these cells significantly inhibited the
proliferation of cigarette smoke-induced transformed BEAS-2B cells
through induction of cell cycle arrest and apoptosis. Furthermore,
RBM5 overexpression also reduced the xenograft growth of
CSE-transformed BEAS-2B cells. Therefore, we hypothesized that RBM5
may be a promising candidate for intervention of smoking-related
lung cancer, and our findings may provide new insights into the
understanding of the precise mechanism underlying the antitumor
activity of RBM5.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81472169 and
81241069).
References
|
1
|
Chen M, Yang T, Meng X and Sun T:
Azithromycin attenuates cigarette smoke extract-induced oxidative
stress injury in human alveolar epithelial cells. Mol Med Rep.
11:3414–3422. 2015.
|
|
2
|
Lemjabbar-Alaoui H, Dasari V, Sidhu SS,
Mengistab A, Finkbeiner W, Gallup M and Basbaum C: Wnt and Hedgehog
are critical mediators of cigarette smoke-induced lung cancer. PLoS
One. 1:e932006.
|
|
3
|
Sasco AJ, Secretan MB and Straif K:
Tobacco smoking and cancer: A brief review of recent
epidemiological evidence. Lung Cancer. 45(Suppl 2): S3–S9.
2004.
|
|
4
|
Oh JJ, Razfar A, Delgado I, Reed RA,
Malkina A, Boctor B and Slamon DJ: 3p21.3 tumor suppressor gene
H37/Luca15/RBM5 inhibits growth of human lung cancer cells
through cell cycle arrest and apoptosis. Cancer Res. 66:3419–3427.
2006.
|
|
5
|
Mourtada-Maarabouni M, Sutherland LC,
Meredith JM and Williams GT: Simultaneous acceleration of the cell
cycle and suppression of apoptosis by splice variant delta-6 of the
candidate tumour suppressor LUCA-15/RBM5. Genes Cells. 8:109–119.
2003.
|
|
6
|
Zhao L, Li R, Shao C, Li P, Liu J and Wang
K: 3p21.3 tumor suppressor gene RBM5 inhibits growth of human
prostate cancer PC-3 cells through apoptosis. World J Surg Oncol.
10:2472012.
|
|
7
|
Welling DB, Lasak JM, Akhmametyeva E,
Ghaheri B and Chang LS: cDNA microarray analysis of vestibular
schwannomas. Otol Neurotol. 23:736–748. 2002.
|
|
8
|
Oh JJ, West AR, Fishbein MC and Slamon DJ:
A candidate tumor suppressor gene, H37, from the human lung
cancer tumor suppressor locus 3p21.3. Cancer Res. 62:3207–3213.
2002.
|
|
9
|
Rintala-Maki ND, Goard CA, Langdon CE,
Wall VE, Traulsen KE, Morin CD, Bonin M and Sutherland LC:
Expression of RBM5-related factors in primary breast tissue. J Cell
Biochem. 100:1440–1458. 2007.
|
|
10
|
Oh JJ, Grosshans DR, Wong SG and Slamon
DJ: Identification of differentially expressed genes associated
with HER-2/neu over-expression in human breast cancer cells.
Nucleic Acids Res. 27:4008–4017. 1999.
|
|
11
|
Wang W, Cassidy J, O'Brien V, Ryan KM and
Collie-Duguid E: Mechanistic and predictive profiling of
5-fluorouracil resistance in human cancer cells. Cancer Res.
64:8167–8176. 2004.
|
|
12
|
Sutherland LC, Wang K and Robinson AG:
RBM5 as a putative tumor suppressor gene for lung cancer. J Thorac
Oncol. 5:294–298. 2010.
|
|
13
|
Oh JJ, Taschereau EO, Koegel AK, Ginther
CL, Rotow JK, Isfahani KZ and Slamon DJ: RBM5/H37 tumor suppressor,
located at the lung cancer hot spot 3p21.3, alters expression of
genes involved in metastasis. Lung Cancer. 70:253–262. 2010.
|
|
14
|
Akhtar MJ, Ahamed M, Khan MA, Alrokayan
SA, Ahmad I and Kumar S: Cytotoxicity and apoptosis induction by
nanoscale talc particles from two different geographical regions in
human lung epithelial cells. Environ Toxicol. 29:394–406. 2014.
|
|
15
|
Niu Z, Jin W, Zhang L and Li X: Tumor
suppressor RBM5 directly interacts with the DExD/H-box protein
DHX15 and stimulates its helicase activity. FEBS Lett. 586:977–983.
2012.
|
|
16
|
Mourtada-Maarabouni M, Sutherland LC and
Williams GT: Candidate tumour suppressor LUCA-15 can regulate
multiple apoptotic pathways. Apoptosis. 7:421–432. 2002.
|
|
17
|
Mourtada-Maarabouni M, Keen J, Clark J,
Cooper CS and Williams GT: Candidate tumor suppressor
LUCA-15/RBM5/H37 modulates expression of apoptosis and cell cycle
genes. Exp Cell Res. 312:1745–1752. 2006.
|
|
18
|
Bechara EG, Sebestyén E, Bernardis I,
Eyras E and Valcárcel J: RBM5, 6, and 10 differentially regulate
NUMB alternative splicing to control cancer cell proliferation. Mol
Cell. 52:720–733. 2013.
|
|
19
|
Schamberger AC, Mise N, Jia J, Genoyer E,
Yildirim AÖ, Meiners S and Eickelberg O: Cigarette smoke-induced
disruption of bronchial epithelial tight junctions is prevented by
transforming growth factor-β. Am J Respir Cell Mol Biol.
50:1040–1052. 2014.
|
|
20
|
Du H, Sun J, Chen Z, Nie J, Tong J and Li
J: Cigarette smoke-induced failure of apoptosis resulting in
enhanced neoplastic transformation in human bronchial epithelial
cells. J Toxicol Environ Health A. 75:707–720. 2012.
|
|
21
|
Drabkin HA, West JD, Hotfilder M, Heng YM,
Erickson P, Calvo R, Dalmau J, Gemmill RM and Sablitzky F: DEF-3
(g16/NY-LU-12), an RNA binding protein from the 3p21.3 homozygous
deletion region in SCLC. Oncogene. 18:2589–2597. 1999.
|
|
22
|
Maarabouni MM and Williams GT: The
antiapoptotic RBM5/LUCA-15/H37 gene and its role in apoptosis and
human cancer: Research update. ScientificWorldJournal. 6:1705–1712.
2006.
|
|
23
|
Liang H, Zhang J, Shao C, Zhao L, Xu W,
Sutherland LC and Wang K: Differential expression of RBM5, EGFR and
KRAS mRNA and protein in non-small cell lung cancer tissues. J Exp
Clin Cancer Res. 31:362012.
|
|
24
|
Kobayashi T, Ishida J, Musashi M, Ota S,
Yoshida T, Shimizu Y, Chuma M, Kawakami H, Asaka M, Tanaka J, et
al: p53 transactivation is involved in the antiproliferative
activity of the putative tumor suppressor RBM5. Int J Cancer.
128:304–318. 2011.
|
|
25
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: Several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009.
|
|
26
|
Hochegger H, Takeda S and Hunt T:
Cyclin-dependent kinases and cell-cycle transitions: Does one fit
all? Nat Rev Mol Cell Biol. 9:910–916. 2008.
|
|
27
|
Fung TK and Poon RY: A roller coaster ride
with the mitotic cyclins. Semin Cell Dev Biol. 16:335–342.
2005.
|
|
28
|
Hedberg Y, Ljungberg B, Roos G and
Landberg G: Retinoblastoma protein in human renal cell carcinoma in
relation to alterations in G1/S regulatory proteins. Int J Cancer.
109:189–193. 2004.
|
|
29
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149.
2003.
|
|
30
|
Lv XJ, Zhao LJ, Hao YQ, Su ZZ, Li JY, Du
YW and Zhang J: Schisandrin B inhibits the proliferation of human
lung adenocarcinoma A549 cells by inducing cycle arrest and
apoptosis. Int J Clin Exp Med. 8:6926–6936. 2015.
|
|
31
|
Fushimi K, Ray P, Kar A, Wang L,
Sutherland LC and Wu JY: Up-regulation of the proapoptotic caspase
2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl
Acad Sci USA. 105:15708–15713. 2008.
|
|
32
|
Rintala-Maki ND and Sutherland LC:
LUCA-15/RBM5, a putative tumour suppressor, enhances multiple
receptor-initiated death signals. Apoptosis. 9:475–484. 2004.
|
|
33
|
Rintala-Maki ND, Abrasonis V, Burd M and
Sutherland LC: Genetic instability of RBM5/LUCA-15/H37 in MCF-7
breast carcinoma sublines may affect susceptibility to apoptosis.
Cell Biochem Funct. 22:307–313. 2004.
|
|
34
|
Sutherland LC, Lerman M, Williams GT and
Miller BA: LUCA-15 suppresses CD95-mediated apoptosis in Jurkat T
cells. Oncogene. 20:2713–2719. 2001.
|
|
35
|
Sleeman J and Steeg PS: Cancer metastasis
as a therapeutic target. Eur J Cancer. 46:1177–1180. 2010.
|
|
36
|
Verma S, Kesh K, Ganguly N, Jana S and
Swarnakar S: Matrix metalloproteinases and gastrointestinal
cancers: Impacts of dietary antioxidants. World J Biol Chem.
5:355–376. 2014.
|
|
37
|
Gencer S, Cebeci A and Irmak-Yazicioglu
MB: Matrix metalloproteinase gene expressions might be oxidative
stress targets in gastric cancer cell lines. Chin J Cancer Res.
25:322–333. 2013.
|
|
38
|
Ramakrishnan S, Subramanian IV, Yokoyama Y
and Geller M: Angiogenesis in normal and neoplastic ovaries.
Angiogenesis. 8:169–182. 2005.
|
|
39
|
Yamakuchi M, Lotterman CD, Bao C, Hruban
RH, Karim B, Mendell JT, Huso D and Lowenstein CJ: P53-induced
microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad
Sci USA. 107:6334–6339. 2010.
|