Introduction
Hepatocellular carcinoma (HCC) is the third leading
cause of cancer-related death worldwide (1). It is estimated to cause approximately
half a million deaths annually. Several risk factors for HCC have
been reported, including infection with hepatitis B and hepatitis C
viruses, dietary intake of afratoxin, and alcohol consumption.
However, the molecular pathogenesis of HCC is not fully
understood.
Epigenetic changes are the mechanisms of
tumorigenesis as well as genetic changes such as chromosomal
alternations, gene amplifications, deletions, and mutations. DNA
methylation of CpG islands within the promoter regions of
tumor-suppressor genes is known to inhibit transcriptional
initiation, and thereby silence these genes. Tumor-suppressor genes
that are frequently methylated in HCC include APC (2), CDKN2A (3), RASSF1A (4), and GSTP1 (5). Aberrant DNA methylation of various
tumor-suppressor genes is suggested to be correlates with
biological features and clinical outcome of HCC (6,7).
In the present study, we aimed to identify novel
genes that are silenced by DNA hypermethylation in HCC. We screened
for genes with promoter DNA hypermethylation using a genome-wide
methylation microarray analysis in primary HCC tumors by comparison
with their non-tumor tissue counterparts. Further methylation
analyses, combined with expression analyses, revealed novel genes
that were downregulated by aberrant promoter hypermethylation in
HCC.
Materials and methods
Primary tumors and cell lines
Paired tumor and non-tumor tissues were obtained
from HCC patients who underwent surgery at the Hospital of Tokyo
Medical and Dental University. All specimens were immediately
frozen in liquid nitrogen and were stored at −80°C until required.
Table I summarizes the clinical
characteristics of a total of 47 patients (20 in the discovery set
and 27 in the validation set) in the present study. The protocol of
this study was approved by the ethics committees and conducted in
accordance with the Declaration of Helsinki. Informed consent was
obtained from each patient.
 | Table IPatient characteristics. |
Table I
Patient characteristics.
|
Characteristics | Discovery
set
(n=20) | Validation
set
(n=27) |
|---|
| Age (years) | 65.4±7.6 | 64.0±9.3 |
| Gender | | |
| Male | 15 | 22 |
| Female | 5 | 5 |
| Etiology | | |
| Hepatitis B | 3 | 5 |
| Hepatitis C | 11 | 14 |
| Other | 6 | 8 |
| No. of HCC
tumors | | |
| Single | 10 | 15 |
| Multiple | 10 | 12 |
| Maximum tumor size
(cm) | 5.9±3.0 | 7.0±4.8 |
| Child-Pugh class
(A/B/C) | 20/0/0 | 27/0/0 |
| Clinical stage | 0/7/8/5/0 | 0/8/10/8/1 |
|
(I/II/III/IVa/IVb)a | | |
| Background of the
liver tissue | | |
| Normal liver | 2 | 2 |
| Chronic
hepatitis | 6 | 11 |
| Liver
cirrhosis | 12 | 14 |
Three HCC cell lines, SNU449, Li7 and HLF, were
examined. All cell lines were maintained in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal calf serum.
DNA extraction and bisulfite
modification
Genomic DNA was isolated using the Puregene DNA
isolation kit (Gentra, Minneapolis, MN, USA). Bisulfite
modification of DNA was performed using an EZ DNA Methylation kit
(Zymo Research, Irvine, CA, USA).
Illumina HumanMethylation27 BeadChip
Genome-wide DNA methylation was analyzed by the
HumanMethylation27 BeadChip (Illumina, San Diego, CA, USA),
according to the instructions from the manufacturer. This Illumina
BeadChip interrogates 27,578 CpG sites, which were selected
predominantly from the promoter regions of an annotated 14,475
genes. Data were analyzed using Illumina GenomeStudio software.
Methylation values are expressed as a β-value (between 0 and 1) for
each CpG site, representing a continuous measurement from 0
(completely unmethylated) to 1 (completely methylated).
Differential methylation was assessed by comparing
the mean methylation level (β-value) of HCC tumor tissues with the
mean β-value of non-tumor liver tissues. Selection of significantly
differentially methylated loci was based on i) a β-value difference
[delta (Δ) β] of at least 0.15 between HCC tumor and non-tumor
samples and ii) a p-value of <0.01 as determined by paired
t-test with false-discovery rate (FDR) correction for multiple
comparisons, based on the Benjamini and Hochberg procedure
(8).
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was obtained using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA). qRT-PCR experiments were performed
with the LightCycler system using FastStart DNA Master Plus SYBR
Green I (Roche Diagnostics, Penzberg, Germany), as previously
described (9). The primers used are
listed in Table II. The endogenous
control for mRNA was ACTB.
| Table IISequences of the PCR primers used in
the study. |
Table II
Sequences of the PCR primers used in
the study.
| Purpose | Gene | Forward primer | Reverse primer |
|---|
| Quantitative
RT-PCR | ZNF154 |
5′-GCCTGTACCGTGATGTGATG-3′ |
5′-TTTTTCTCCAAGGTGCTGCT-3′ |
| MAP9 |
5′-GGTAGGTGTTACCGGCTTCA-3′ |
5′-CTCAACTCAGGCACACTCCA-3′ |
| SPINT2 |
5′-GGAAGGGAGGGGAGACTATG-3′ |
5′-AGAAATCGATCAGCGAGGAA-3′ |
| AKR1B1 |
5′-ACCTCCCACAAGGATTACCC-3′ |
5′-GGCAAAGCAAACTGGAAGAG-3′ |
| RSPH9 |
5′-TTAAGCGCGACTACCGCTAT-3′ |
5′-TCCACTCTGTGCAGTTCAGG-3′ |
| GRASP |
5′-TCGGCTTTGAGATCCAGACT-3′ |
5′-TCTGAGAACATTGCCTGACG-3′ |
| STEAP4 |
5′-CAGAACACACGCTCCTTCAA-3′ |
5′-CCAGCCTGGATGGTACCTAA-3′ |
| NXPE3 |
5′-TCTGCAGCTCAGAAAAGCAA-3′ |
5′-CTGTCGATGAAAGTGGCTGA-3′ |
| ACTB |
5′-GTCCACCTTCCAGCAGATGT-3′ |
5′-TGTTTTCTGCGCAAGTTAGG-3′ |
|
Methylation-specific PCR | ZNF154
M |
5′-ATGTTTTGCGTTGAACGTTAC-3′ |
5′-CTAAAATAACCGCCACGAAA-3′ |
| ZNF154
U |
5′-AGAATGTTTTGTGTTGAATGTTAT-3′ |
5′-AAACTAAAATAACCACCACAAAA-3′ |
| MAP9 M |
5′-GGTGGTTGTTTTAGCGATAC-3′ |
5′-TCCTAAACCGAACGAAAA-3′ |
| MAP9 U |
5′-TTGGGTGGTTGTTTTAGTGATAT-3′ |
5′-CAATCCTAAACCAAACAAAAA-3′ |
| SPINT2
M |
5′-TTTAGGTGCGTTTAGGGTC-3′ |
5′-ACCAATAACGAACGCCTATT-3′ |
| SPINT2
U |
5′-GGGTTTAGGTGTGTTTAGGGTT-3′ |
5′-ACCAATAACAAACACCTATTAAA-3′ |
| AKR1B1
M |
5′-GGGTCGGTTTTGTAGAGATC-3′ |
5′-CGCTAAAACCCAAAATACG-3′ |
| AKR1B1
U |
5′-TGGGGTTGGTTTTGTAGAGATT-3′ |
5′-CACTAAAACCCAAAATACAAA-3′ |
| RSPH9 M |
5′-GGGTTTTAGTTCGGATCGTC-3′ |
5′-ATAATCGACGACGAAACCAA-3′ |
| RSPH9 U |
5′-GTAGGGTTTTAGTTTGGATTGTT-3′ |
5′-ATAATCAACAACAAAACCAAAAA-3′ |
| GRASP M |
5′-TTATAAAGGGAGGCGATTC-3′ |
5′-CGACGAAAAATCATAACTCC-3′ |
| GRASP U |
5′-AGTTTATAAAGGGAGGTGATTT-3′ |
5′-CAACAAAAAATCATAACTCCAAC-3′ |
| STEAP4
M |
5′-GTATCGTTGGCGTTGGAC-3′ |
5′-GCGACGAAAAATTTACAAACA-3′ |
| STEAP4
U |
5′-GTATTGTTGGTGTTGGAT-3′ |
5′-ACAACAAAAAATTTACAAACA-3′ |
| NXPE3 M |
5′-GCGATAGTTGTAGTGTCGC-3′ |
5′-ACCCCCGACTACGATTAATA-3′ |
| NXPE3 U |
5′-GGGGTGATAGTTGTAGTGTTGT-3′ |
5′-CAACCCCCAACTACAATTAATAA-3′ |
| COBRA | STEAP4 |
5′-GGGATTTTTAGTTTGAATTTTT-3′ |
5′-ATTTACAAACACCTATTCTTCAAT-3′ |
Methylation-specific PCR (MSP)
MSP was performed, as previously described (10). Briefly, genomic DNA was treated with
sodium bisulfite and subjected to PCR using specific primer sets
(Table II).
Combined bisulfite and restriction
analysis (COBRA)
COBRA was performed, as previously described
(10). Briefly, genomic DNA was
treated with sodium bisulfite and subjected to PCR using primers
(Table II) designed to amplify a
region from −97 to +239 bp relative to the transcription start site
of STEAP4. The PCR products were digested with HpyCH4IV,
which recognizes sequences unique to the methylated alleles, but
cannot recognize unmethylated alleles, and the digested products
were electrophoresed on 3% agarose gels and stained with ethidium
bromide. Methylation levels were calculated as the ratio of the
gray scale value of the methylated band to that of the combined
methylated and unmethylated bands. The gray scale value was
obtained by scanning the gel with Adobe Photoshop CS3 Extended
software (Adobe Systems Incorporated, San Jose, CA, USA).
Methylated and unmethylated bisulfite-converted control DNA
(EpiTect control DNA set; Qiagen, Tokyo, Japan) served as controls
for methylated and unmethylated DNA, respectively, in MSP and
COBRA.
Drug treatment
Cells were treated with 1 or 5 µM of
5-aza-2′-deoxycytidine (5-aza-dC; Sigma-Aldrich, St. Louis, MO,
USA) for 4 days or 50 ng/ml of trichostatin A (TSA; Wako, Osaka,
Japan) for 1 day. In assessing drug synergy, cells were cultured in
the presence of 1 or 5 µM of 5-aza-dC for 4 days, and were
then treated for an additional 24 h with 50 ng/ml of TSA.
Immunohistochemistry
The HCC tissue microarray (US Biomax, Rockville, MD,
USA) was analyzed for STEAP4 protein expression. Anti-STEAP4
polyclonal antibody (Proteintech, Chicago, IL, USA) was used at a
dilution of 1:50. Immunostaining of STEAP4 was carried out with the
EnVision+ system (Dako, Tokyo, Japan).
Statistical analyses
Fisher's exact probability test, paired t-test, and
Wilcoxon signed-rank test were performed using SPSS 15.0 software
(SPSS, Inc., Chicago, IL, USA). P-values of <0.05 were
considered significant.
KEGG pathway analysis (11) was performed to identify biological
pathways significantly enriched for differentially methylated genes
using the functional annotation tool of the Database for Annotation
Visualization and Integrated Discovery (DAVID) version 6.7
(12,13). P-values were calculated using a
modified Fisher's exact test (EASE score).
Results
Genome-wide DNA methylation profiling of
primary HCC
To identify genes that are silenced by DNA
hypermethylation in HCC, we compared DNA methylation profiles
between paired tumor and non-tumor tissues from 20 patients with
primary HCC (the discovery set) using Illumina HumanMethylation27
BeadChip. The array data have been submitted to NCBI GEO under
accession number (GSE73003). The strategy of the present study is
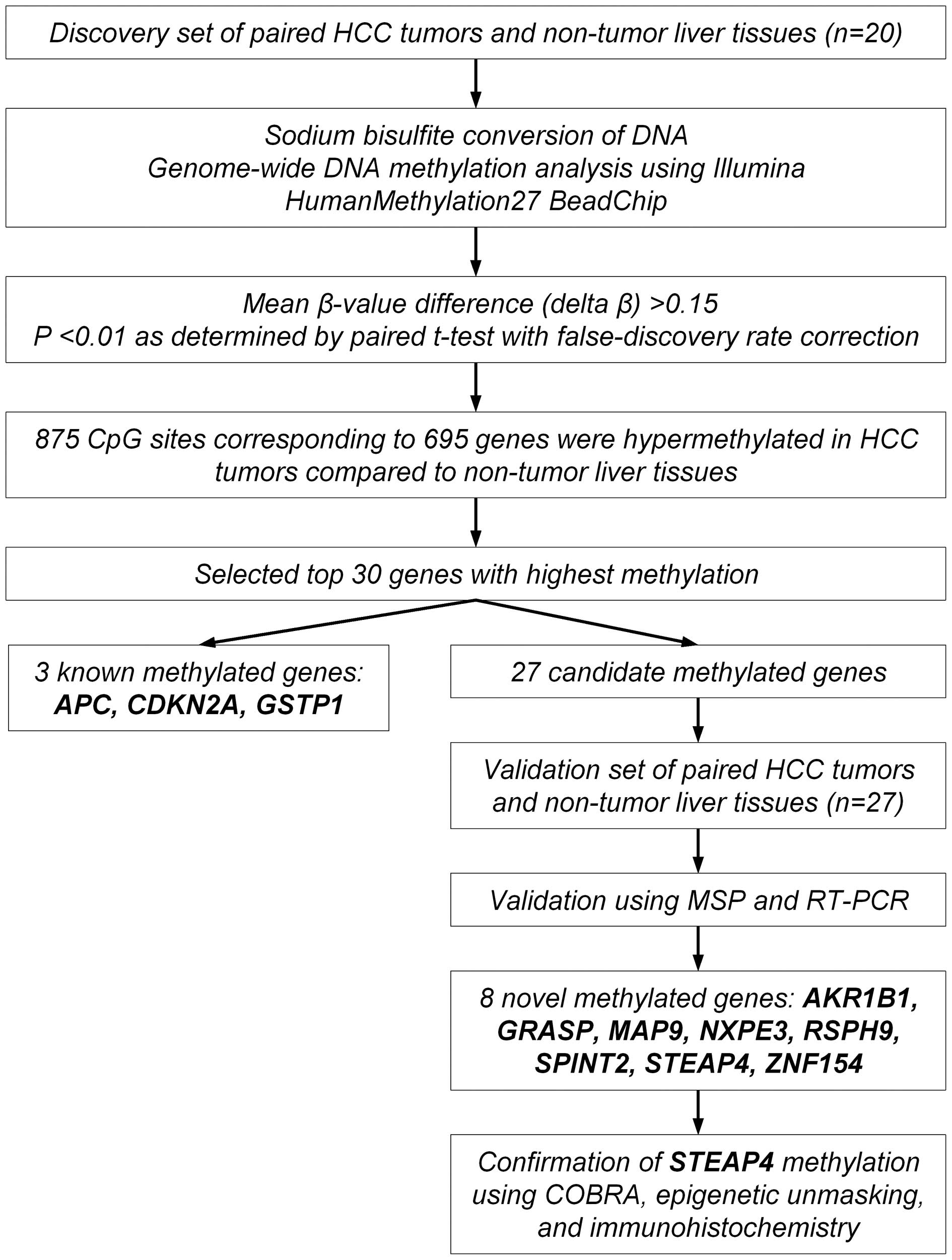
shown as a flowchart in Fig. 1.
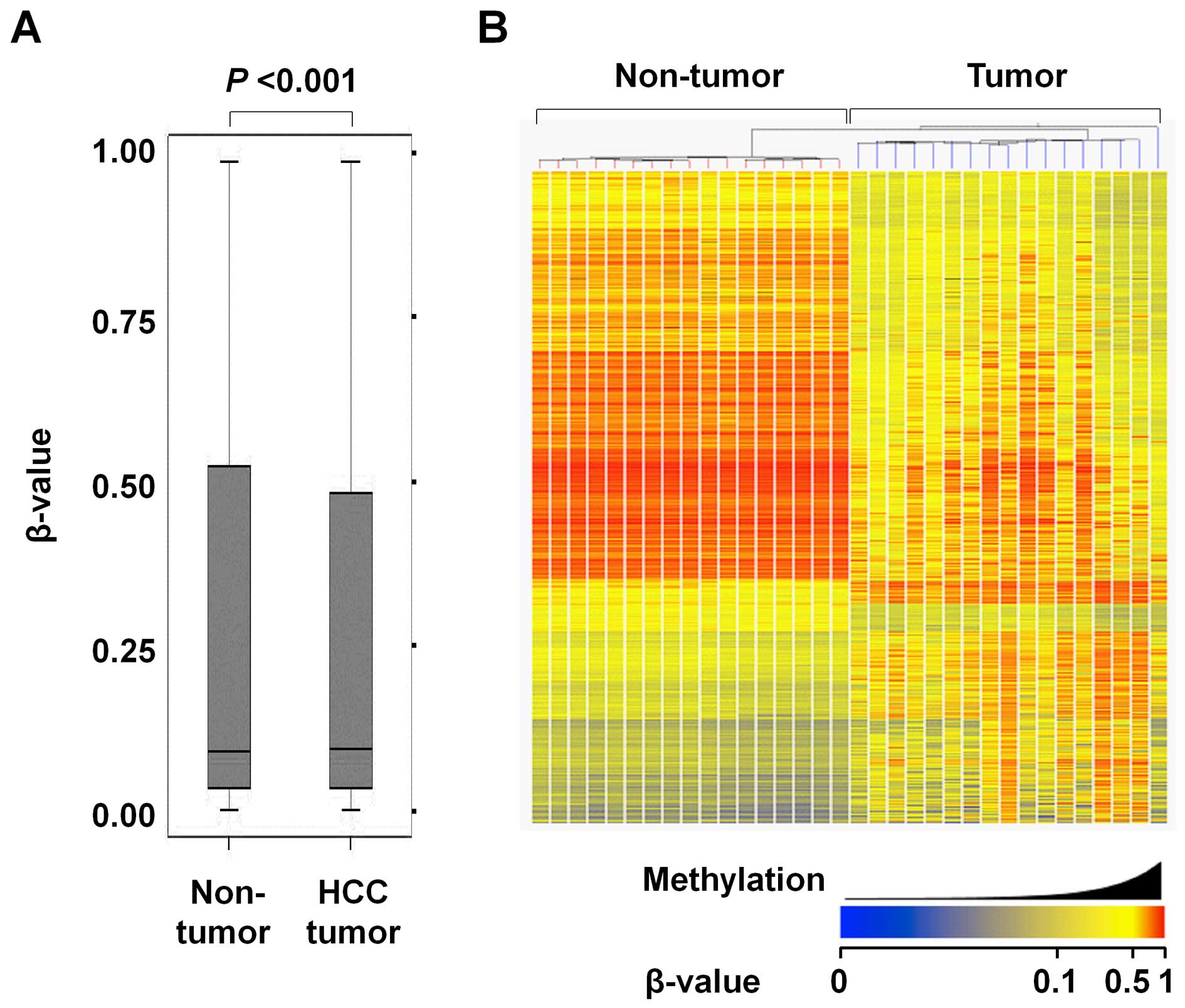
Overall, the average methylation level was slightly
but significantly higher in the HCC tumors than the matched
non-tumor liver tissues (median β-value of 0.093 and 0.091 in HCC
tumors and non-tumor liver tissues, respectively) (Fig. 2A). There were 2,670 CpG sites that
significantly differed in regards to the methylation level between
the tumor and non-tumor tissues (Δβ >0.15 and P<0.01, see
Materials and methods). Of these 2,670 CpG sites, 875 were
significantly hypermethylated and 1,795 were significantly
hypomethylated in the HCC tumors compared to the non-tumor liver
tissues.
Fig. 2B shows a
heatmap of 2,670 significantly differentially methylated CpG sites
between the HCC tumors and non-tumor liver tissues. Good separation
of HCC tumors and non-tumor liver tissues was observed. While the
HCC tumors showed a variation in methylation profiles, the
non-tumor liver tissues did not.
KEGG pathway analysis was performed to identify
biological pathways significantly enriched for the 695
hypermethylated genes corresponding to 875 CpG sites that were
hypermethylated in the HCC tumors. Four KEGG pathways, including
neuroactive ligand-receptor interaction, focal adhesion, vascular
smooth muscle contraction, and systemic lupus erythematosus, were
significantly enriched for hypermethylated genes (Table III).
| Table IIIKEGG pathways enriched for
hypermethylated genes. |
Table III
KEGG pathways enriched for
hypermethylated genes.
| Pathway | Description | Counta | %b | P-valuec | Fold
enrichment |
|---|
| hsa04080 | Neuroactive
ligand-receptor interaction | 22 | 3.19 | 0.001 | 2.08 |
| hsa04510 | Focal adhesion | 17 | 2.46 | 0.008 | 2.05 |
| hsa04270 | Vascular smooth
muscle contraction | 11 | 1.59 | 0.016 | 2.38 |
| hsa05322 | Systemic lupus
erythematosus | 9 | 1.30 | 0.049 | 2.20 |
Selection and validation of candidate
methylated genes
We focused on further examination for the top 30
most hypermethylated genes in the HCC tumors compared to the
non-tumor liver tissues (Table
IV). The list of 30 genes included three known tumor-suppressor
genes, APC (adenomatous polyposis coli), CDKN2A
(cyclin-dependent kinase inhibitor 2A) and GSTP1
(glutathione S-transferase pi 1), which are known to be silenced by
DNA hypermethylation in HCC (14,15),
supporting the appropriateness of our methodology.
| Table IVTop 30 hypermethylated genes in the
HCC tumorsa. |
Table IV
Top 30 hypermethylated genes in the
HCC tumorsa.
| Gene | Target ID | Mean β-value in
non-tumors | Mean β-value in
tumors | Mean β-value
difference (Δ β) | Corrected
p-valueb |
|---|
| DNM3 | cg23391785 | 0.16 | 0.70 | 0.54 | 1.93E-06 |
| ZNF154 | cg21790626 | 0.08 | 0.62 | 0.54 | 1.67E-09 |
| MAP9 | cg03616357 | 0.12 | 0.60 | 0.48 | 9.64E-07 |
| NETO2 | cg02755525 | 0.13 | 0.59 | 0.46 | 5.78E-05 |
| INA | cg25764191 | 0.16 | 0.62 | 0.46 | 7.22E-07 |
| SPINT2 | cg15375239 | 0.10 | 0.55 | 0.45 | 8.67E-04 |
| AKR1B1 | cg13801416 | 0.06 | 0.50 | 0.45 | 2.87E-07 |
| CDKL2 | cg24432073 | 0.08 | 0.52 | 0.45 | 1.08E-06 |
| RSPH9 | cg04600618 | 0.13 | 0.57 | 0.44 | 1.50E-07 |
| APC | cg16970232 | 0.20 | 0.64 | 0.44 | 6.98E-04 |
| LDHB | cg06437004 | 0.13 | 0.56 | 0.43 | 5.64E-06 |
| CDKN2A | cg09099744 | 0.11 | 0.55 | 0.43 | 2.20E-06 |
| ZFP41 | cg12680609 | 0.14 | 0.56 | 0.43 | 1.75E-06 |
| GSTP1 | cg02659086 | 0.07 | 0.50 | 0.43 | 4.14E-05 |
| FOXE3 | cg18815943 | 0.14 | 0.57 | 0.42 | 3.74E-04 |
| HBQ1 | cg07703401 | 0.18 | 0.60 | 0.42 | 1.11E-06 |
| GRASP | cg04034767 | 0.12 | 0.53 | 0.42 | 2.20E-06 |
| ABHD9 | cg05488632 | 0.19 | 0.61 | 0.42 | 1.44E-06 |
| BMP4 | cg14310034 | 0.10 | 0.52 | 0.41 | 1.38E-05 |
| SF3B14 | cg04809136 | 0.18 | 0.60 | 0.41 | 8.11E-04 |
| DGKE | cg01344452 | 0.12 | 0.53 | 0.41 | 8.27E-04 |
| ABCA3 | cg00949442 | 0.20 | 0.61 | 0.41 | 2.33E-05 |
| STEAP4 | cg00564163 | 0.17 | 0.58 | 0.41 | 1.35E-05 |
| POU4F1 | cg08097882 | 0.12 | 0.53 | 0.40 | 7.97E-06 |
|
DKFZp434I1020 | cg17886204 | 0.04 | 0.45 | 0.40 | 7.53E-07 |
| NXPE3 | cg06073471 | 0.04 | 0.44 | 0.40 | 1.53E-06 |
| PRDM14 | cg01295203 | 0.15 | 0.54 | 0.39 | 1.09E-06 |
| CCNJ | cg04590978 | 0.11 | 0.50 | 0.39 | 5.84E-06 |
| SPDY1 | cg04786857 | 0.18 | 0.58 | 0.39 | 2.08E-06 |
|
HIST1H4F | cg08260959 | 0.15 | 0.54 | 0.39 | 9.91E-07 |
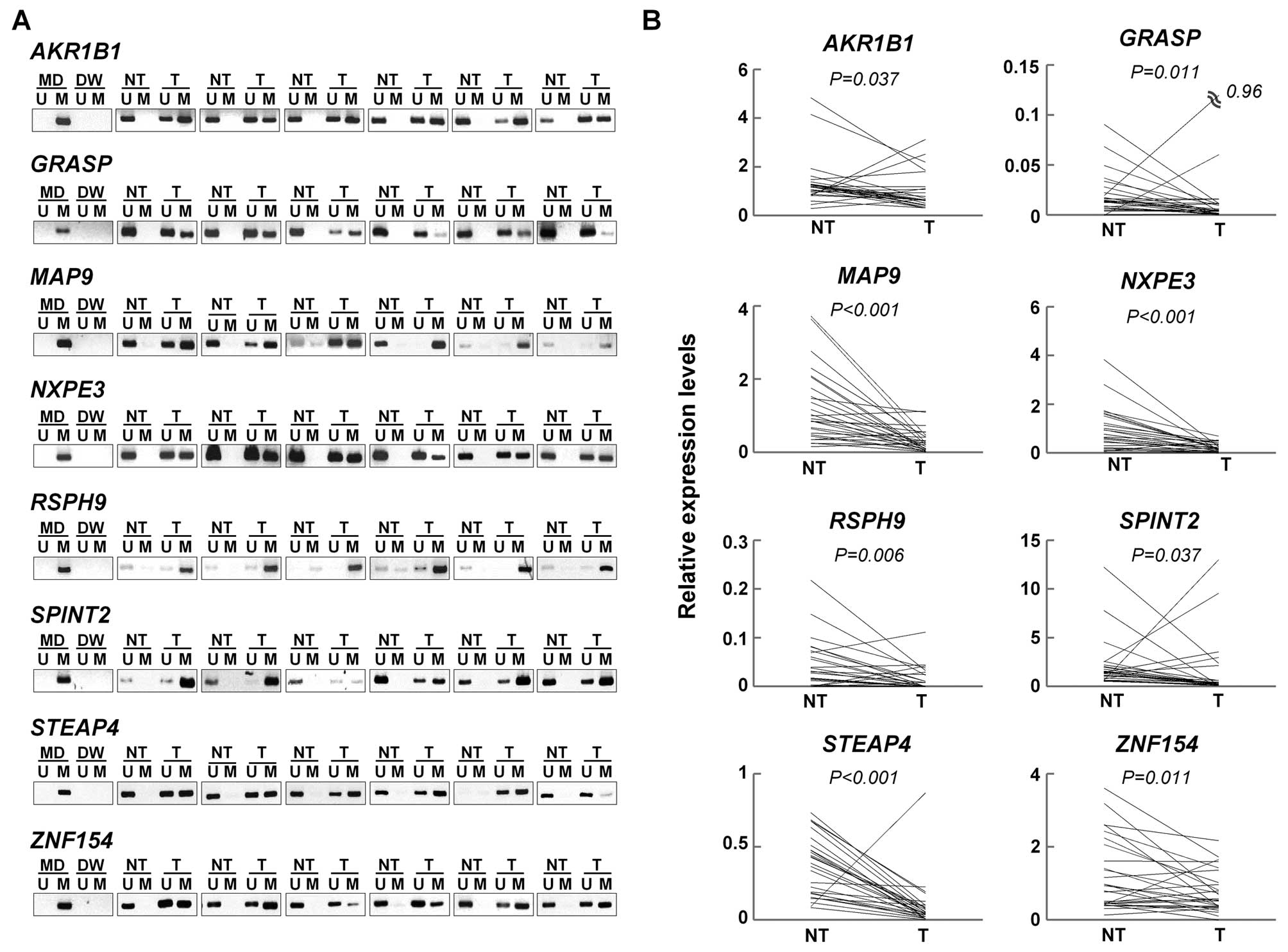
We examined whether the remaining 27 genes were
silenced by DNA hepermethylation in the paired tumor and non-tumor
tissues from an additional 27 patients with primary HCC (the
validation set) using MSP and qRT-PCR. Of the 27 genes, eight genes
(AKR1B1, GRASP, MAP9, NXPE3,
RSPH9, SPINT2, STEAP4, and ZNF154) were
significantly hypermethylated and downregulated in the HCC tumors
compared to the non-tumor liver tissues (Fig. 3 and Table V). Therefore, these eight genes were
identified and validated to be methylated genes in HCC.
| Table VMethylation-specific PCR analysis of
candidate genes. |
Table V
Methylation-specific PCR analysis of
candidate genes.
| Gene | Non-tumor
(n=27) | Tumor (n=27) | P-valuea |
|---|
| AKR1B1 | 1 (4) | 20 (74) | <0.001 |
| GRASP | 0 (0) | 12 (44) | <0.001 |
| MAP9 | 11 (41) | 25 (93) | <0.001 |
| NXPE3 | 2 (7) | 21 (78) | <0.001 |
| RSPH9 | 10 (37) | 26 (96) | <0.001 |
| SPINT2 | 0 (0) | 15 (56) | <0.001 |
| STEAP4 | 10 (37) | 23 (85) | <0.001 |
| ZNF154 | 2 (7) | 23 (85) | <0.001 |
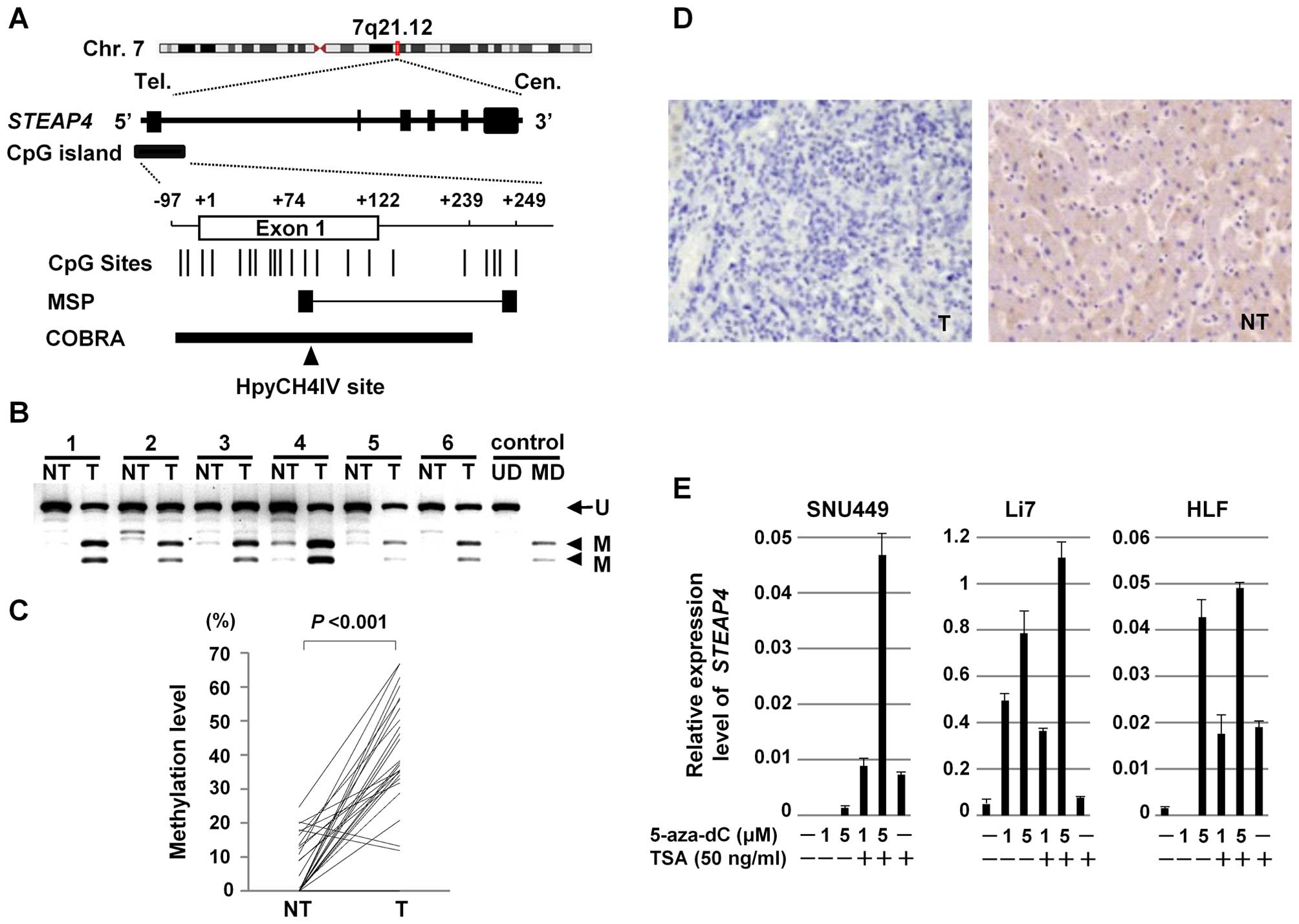
Epigenetic silencing of STEAP4
As an example, we further assessed the methylation
status of STEAP4, as little is known about the association
of STEAP4 with HCC. Using the Methyl Primer Express software
ver.1.0 (Applied Biosystems, Foster City, CA, USA), a CpG island
was found around the transcription start site of STEAP4
(Fig. 4A). To confirm the
methylation status of STEAP4, we quantified methylation
levels of STEAP4 in the paired tumor and non-tumor tissues
from 27 patients with primary HCC (the validation set) using COBRA
(Fig. 4B). The level of methylation
of STEAP4 was significantly higher in 25 (93%) of the 27 HCC
tumors, compared to their non-tumor tissue counterparts (Wilcoxon
signed-rank test, P<0.001) (Fig.
4C).
To confirm the silencing of STEAP4 in the HCC
tumors, we compared the expression of the STEAP4 protein using
immunohistochemistry on tissue microarrays. Representative images
are shown in Fig. 4D. Whereas the
STEAP4 protein was expressed in all of the 30 non-tumor liver
tissues, it was expressed in 26 of the 40 HCC tumors (Fisher's
exact probability test, P<0.001).
We then assessed the effect of demethylation on the
expression of STEAP4. Three HCC cell lines (SNU449, Li7, and
HLF) that lack STEAP4 expression were treated with 5-aza-dC,
a methyltransferase inhibitor, and expression levels of
STEAP4 mRNA were assayed with qRT-PCR. Expression of
STEAP4 was restored with 5-aza-dC treatment in a
dose-dependent manner in all three HCC cells (Fig. 4E), suggesting that aberrant DNA
methylation suppressed the expression of STEAP4.
Additionally, it was observed that treatment with a histone
deacetylase inhibitor, TSA, enhanced the expression of STEAP4 by
5-aza-dC in all three cell lines (Fig.
4E). This finding suggests that histone deacetylation may also
contribute to the transcriptional repression of STEAP4.
Discussion
In the present study, AKR1B1, GRASP,
MAP9, NXPE3, RSPH9, SPINT2,
STEAP4 and ZNF154 were identified as genes that are
silenced by DNA hypermethylation in HCC. Except for GRASP
and SPINT2, to our knowledge, this is the first study to
describe the hypermethylation of AKR1B1, MAP9,
NXPE3, RSPH9, STEAP4 and ZNF154 in HCC
and the relevance of these genes with HCC. Our methodology appears
to be appropriate, since APC, CDKN2A and
GSTP1, which are known methylated genes in HCC, were also
identified by our approach.
GRASP [GRP1 (general receptor for
phosphoinositides 1)-associated scaffold protein; also known as
Tamalin] encodes a protein that functions as a molecular scaffold
and contains several putative protein-protein interaction motifs.
It regulates the membrane trafficking pathway (16,17).
Although a recent study showed the hypermethylation of GRASP
in hepatitis B-virus related HCC (18), its functional relevance for the
development of HCC remains unknown.
SPINT2 (serine peptidase inhibitor, Kunitz
type, 2) encodes Kunitz-type serine protease inhibitor called
hepatocyte growth factor activator inhibitor type 2 (HAI-2). Recent
studies have suggested that SPINT2 is a candidate
tumor-suppressor gene that is frequently hypermethylated and
underexpressed in human cancers, including hepatocellular
carcinomas (19,20), gastric carcinomas (21), ovarian cancer (22), cervical cancer (23), renal cell carcinoma (24), and esophageal squamous cell
carcinoma (25). Studies showed
that ectopic expression of SPINT2 significantly inhibited
cell migration and invasiveness of HCC cells in vitro and
suppressed tumorigenicity in vivo (20).
AKR1B1 (aldo-keto reductase family 1, member
B1) encodes aldose reductase, which participates in glucose
metabolism and osmoregulation and is believed to play a protective
role against toxic aldehydes derived from lipid peroxidation and
steroidogenesis. AKR1B1 is mainly expressed in the adrenal
grand and its expression is decreased in adrenocortical cancer
(26).
STEAP4 (STEAP family member 4), also known as
STAMP2, is a member of the six transmembrane epithelial antigen of
prostate (STEAP) family and functions as a metalloreductase. STEAP4
is involved in adipocyte development and metabolism, and it is
essential for maintenance of systemic metabolic homeostasis
(27). Studies suggest that STEAP4
may contribute to normal physiology of the prostate as well as
prostate cancer progression. STEAP4 was reported to be
overexpressed in primary prostate cancer (28), whereas it was also reported that the
STEAP4 promoter region is methylated in androgen-independent
prostate cancer cells, but not in androgen-dependent prostate
cancer cells (29).
ZNF154 (zinc finger protein 154) encodes a
protein that belongs to the zinc finger Kruppel family of
transcriptional regulators. Although the function of ZNF154 is
unknown, hypermethylation of this gene was recently reported in
bladder cancer (30) and ovarian
cancer (31).
MAP9 (microtubule-associated protein 9; also known
as ASAP) is a microtubule-associated protein required for spindle
function, mitotic progression, and cytokinesis (32). Expression of MAP9 is downregulated
in colorectal cancer compared to normal tissues (33). RSPH9 (radial spoke head 9
homolog) encodes a protein thought to be a component of the radial
spoke head in motile cilia and flagella. Mutations in this gene
have been found in patients with primary ciliary dyskinesia
(34). However, the relevance of
RSPH9 with cancer has not been reported. The function of
NXPE3 (neurexophilin and PC-esterase domain family, member
3) is unknown.
Of these eight genes, we further examine the
methylation status of STEAP4 using a variety of methods,
including COBRA and the treatment with a methyltransferase
inhibitor and a histone deacetylase inhibitor. We confirmed the
hypermethylation of the promoter region of STEAP4 in HCC.
Moreover, qRT-PCR and immunohistochemistry showed that the
expression of STEAP4 was downregulated at the mRNA and
protein levels in HCC. These combined results suggest that the
silencing of STEAP4 by aberrant promoter hypermethylation
may be associated with the development and progression of HCC. We
are now going to study the relationship between the reduced
expression of STEAP4 in HCC tumors and clinicopathological
features with a larger number of samples. It is also required to
study the functional role of STEAP4 in
hepatocarcinogenesis.
Our pathway analysis suggested that hypermethylated
genes may be involved in the pathways of neuroactive
ligand-receptor interaction, focal adhesion, vascular smooth muscle
contraction, and systemic lupus erythematosus in HCC. However, the
relevance of hypermethylated genes with these pathways is largely
unknown.
Functional studies are needed to clarify the roles
of hypermethylated genes that were identified in the present study
in the development and progression of HCC, as they could be useful
markers for the diagnosis or be targets for the therapy of
HCCs.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Lou C, Du Z, Yang B, Gao Y, Wang Y and
Fang S: Aberrant DNA methylation profile of hepatocellular
carcinoma and surgically resected margin. Cancer Sci. 100:996–1004.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsuda Y, Ichida T, Matsuzawa J, Sugimura
K and Asakura H: p16(INK4) is inactivated by extensive CpG
methylation in human hepatocellular carcinoma. Gastroenterology.
116:394–400. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang YJ, Ahsan H, Chen Y, Lunn RM, Wang
LY, Chen SY, Lee PH, Chen CJ and Santella RM: High frequency of
promoter hypermethylation of RASSF1A and p16 and its relationship
to aflatoxin B1-DNA adduct levels in human hepatocellular
carcinoma. Mol Carcinog. 35:85–92. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tchou JC, Lin X, Freije D, Isaacs WB,
Brooks JD, Rashid A, De Marzo AM, Kanai Y, Hirohashi S and Nelson
WG: GSTP1 CpG island DNA hypermethylation in hepatocellular
carcinomas. Int J Oncol. 16:663–676. 2000.PubMed/NCBI
|
|
6
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Lee JS, Conner EA, Schroeder I, Factor VM and Thorgeirsson SS:
Mechanistic and prognostic significance of aberrant methylation in
the molecular pathogenesis of human hepatocellular carcinoma. J
Clin Invest. 117:2713–2722. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishida N, Kudo M, Nagasaka T, Ikai I and
Goel A: Characteristic patterns of altered DNA methylation predict
emergence of human hepatocellular carcinoma. Hepatology.
56:994–1003. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300.
1995.
|
|
9
|
Zen K, Yasui K, Nakajima T, Zen Y, Zen K,
Gen Y, Mitsuyoshi H, Minami M, Mitsufuji S, Tanaka S, et al: ERK5
is a target for gene amplification at 17p11 and promotes cell
growth in hepatocellular carcinoma by regulating mitotic entry.
Genes Chromosomes Cancer. 48:109–120. 2009. View Article : Google Scholar
|
|
10
|
Dohi O, Yasui K, Gen Y, Takada H, Endo M,
Tsuji K, Konishi C, Yamada N, Mitsuyoshi H, Yagi N, et al:
Epigenetic silencing of miR-335 and its host gene MEST in
hepatocellular carcinoma. Int J Oncol. 42:411–418. 2013.
|
|
11
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
12
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
13
|
Huang W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar :
|
|
14
|
Nishida N, Nagasaka T, Nishimura T, Ikai
I, Boland CR and Goel A: Aberrant methylation of multiple tumor
suppressor genes in aging liver, chronic hepatitis, and
hepatocellular carcinoma. Hepatology. 47:908–918. 2008. View Article : Google Scholar
|
|
15
|
Edamoto Y, Hara A, Biernat W, Terracciano
L, Cathomas G, Riehle HM, Matsuda M, Fujii H, Scoazec JY and Ohgaki
H: Alterations of RB1, p53 and Wnt pathways in hepatocellular
carcinomas associated with hepatitis C, hepatitis B and alcoholic
liver cirrhosis. Int J Cancer. 106:334–341. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sugi T, Oyama T, Muto T, Nakanishi S,
Morikawa K and Jingami H: Crystal structures of autoinhibitory PDZ
domain of Tamalin: Implications for metabotropic glutamate receptor
trafficking regulation. EMBO J. 26:2192–2205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Venkataraman A, Nevrivy DJ, Filtz TM and
Leid M: Grp1-associated scaffold protein (GRASP) is a regulator of
the ADP ribosylation factor 6 (Arf6)-dependent membrane trafficking
pathway. Cell Biol Int. 36:1115–1128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tao R, Li J, Xin J, Wu J, Guo J, Zhang L,
Jiang L, Zhang W, Yang Z and Li L: Methylation profile of single
hepatocytes derived from hepatitis B virus-related hepatocellular
carcinoma. PLoS One. 6:e198622011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fukai K, Yokosuka O, Chiba T, Hirasawa Y,
Tada M, Imazeki F, Kataoka H and Saisho H: Hepatocyte growth factor
activator inhibitor 2/placental bikunin (HAI-2/PB) gene is
frequently hypermethylated in human hepatocellular carcinoma.
Cancer Res. 63:8674–8679. 2003.PubMed/NCBI
|
|
20
|
Tung EK, Wong CM, Yau TO, Lee JM, Ching YP
and Ng IO: HAI-2 is epigenetically downregulated in human
hepatocellular carcinoma, and its Kunitz domain type 1 is critical
for anti-invasive functions. Int J Cancer. 124:1811–1819. 2009.
View Article : Google Scholar
|
|
21
|
Dong W, Chen X, Xie J, Sun P and Wu Y:
Epigenetic inactivation and tumor suppressor activity of
HAI-2/SPINT2 in gastric cancer. Int J Cancer. 127:1526–1534. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamura K, Abarzua F, Kodama J, Hongo A,
Nasu Y, Kumon H and Hiramatsu Y: Expression of hepatocyte growth
factor activator inhibitors (HAI-1 and HAI-2) in ovarian cancer.
Int J Oncol. 34:345–353. 2009.PubMed/NCBI
|
|
23
|
Nakamura K, Abarzua F, Hongo A, Kodama J,
Nasu Y, Kumon H and Hiramatsu Y: Hepatocyte growth factor activator
inhibitor-2 (HAI-2) is a favorable prognosis marker and inhibits
cell growth through the apoptotic pathway in cervical cancer. Ann
Oncol. 20:63–70. 2009. View Article : Google Scholar
|
|
24
|
Morris MR, Gentle D, Abdulrahman M, Maina
EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, et al:
Tumor suppressor activity and epigenetic inactivation of hepatocyte
growth factor activator inhibitor type 2/SPINT2 in papillary and
clear cell renal cell carcinoma. Cancer Res. 65:4598–4606. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yue D, Fan Q, Chen X, Li F, Wang L, Huang
L, Dong W, Chen X, Zhang Z, Liu J, et al: Epigenetic inactivation
of SPINT2 is associated with tumor suppressive function in
esophageal squamous cell carcinoma. Exp Cell Res. 322:149–158.
2014. View Article : Google Scholar
|
|
26
|
Lefrançois-Martinez AM, Bertherat J, Val
P, Tournaire C, Gallo-Payet N, Hyndman D, Veyssière G, Bertagna X,
Jean C and Martinez A: Decreased expression of cyclic adenosine
mono-phosphate-regulated aldose reductase (AKR1B1) is associated
with malignancy in human sporadic adrenocortical tumors. J Clin
Endocrinol Metab. 89:3010–3019. 2004. View Article : Google Scholar
|
|
27
|
Wellen KE, Fucho R, Gregor MF, Furuhashi
M, Morgan C, Lindstad T, Vaillancourt E, Gorgun CZ, Saatcioglu F
and Hotamisligil GS: Coordinated regulation of nutrient and
inflammatory responses by STAMP2 is essential for metabolic
homeostasis. Cell. 129:537–548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Korkmaz CG, Korkmaz KS, Kurys P, Elbi C,
Wang L, Klokk TI, Hammarstrom C, Troen G, Svindland A, Hager GL, et
al: Molecular cloning and characterization of STAMP2, an
androgen-regulated six transmembrane protein that is overexpressed
in prostate cancer. Oncogene. 24:4934–4945. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tamura T and Chiba J: STEAP4 regulates
focal adhesion kinase activation and CpG motifs within STEAP4
promoter region are frequently methylated in DU145, human
androgen-independent prostate cancer cells. Int J Mol Med.
24:599–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reinert T, Borre M, Christiansen A,
Hermann GG, Ørntoft TF and Dyrskjøt L: Diagnosis of bladder cancer
recurrence based on urinary levels of EOMES, HOXA9, POU4F2, TWIST1,
VIM, and ZNF154 hypermethylation. PLoS One. 7:e462972012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sánchez-Vega F, Gotea V, Petrykowska HM,
Margolin G, Krivak TC, DeLoia JA, Bell DW and Elnitski L: Recurrent
patterns of DNA methylation in the ZNF154, CASP8, and VHL promoters
across a wide spectrum of human solid epithelial tumors and cancer
cell lines. Epigenetics. 8:1355–1372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saffin JM, Venoux M, Prigent C, Espeut J,
Poulat F, Giorgi D, Abrieu A and Rouquier S: ASAP, a human
microtubule-associated protein required for bipolar spindle
assembly and cytokinesis. Proc Natl Acad Sci USA. 102:11302–11307.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rouquier S, Pillaire MJ, Cazaux C and
Giorgi D: Expression of the microtubule-associated protein
MAP9/ASAP and its partners AURKA and PLK1 in colorectal and breast
cancers. Dis Markers. 2014:7981702014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Castleman VH, Romio L, Chodhari R, Hirst
RA, de Castro SC, Parker KA, Ybot-Gonzalez P, Emes RD, Wilson SW,
Wallis C, et al: Mutations in radial spoke head protein genes RSPH9
and RSPH4A cause primary ciliary dyskinesia with
central-micro-tubular-pair abnormalities. Am J Hum Genet.
84:197–209. 2009. View Article : Google Scholar : PubMed/NCBI
|