Introduction
Lung cancer is the leading cause of cancer-related
death worldwide, with an increasing mortality each year (1). Non-small cell lung cancer (NSCLC)
accounts for 80–85% of all lung cancers. NSCLC subtypes include
adenocarcinoma, squamous cell carcinoma and large cell carcinoma.
The majority of patients diagnosed with NSCLC are diagnosed at
advanced stages with local or distant metastases. Standard NSCLC
treatment includes chemotherapy and surgery which have severe side
effects and limited efficacy (2).
Targeted therapy, which specifically attacks cancer cells with
designated molecular targets, has emerged as a promising strategy
due to its high efficacy and reduced side effects (3,4).
Several drugs targeting key oncogenesis signaling molecules have
been developed and showed efficacy for specific patient groups,
such as erlotinib targeting epidermal growth factor (EGF) (5), bevacizumab targeting vascular
endothelial growth factor (VEGF) (6) and crizotinib targeting anaplastic
lymphoma kinase (ALK) (7). However,
due to the complexity of pathogenic pathways in individual
patients, it is urgent to uncover the largely unknown molecular
origins of lung cancer and provide new targets for lung cancer
therapy.
The rhotekin (RTKN) gene encodes a scaffold protein
which interacts with active GTP-bound Rho proteins and interferes
with the conversion to inactive GDP-bound Rho proteins (8). Rho proteins regulate critical cell
functions including cell growth and transformation, cytokinesis,
transcription, and smooth muscle contraction. Rho signaling pathway
dysregulation was implicated in several forms of cancer (9). Although the RTKN gene has been
reported to be associated with several cancer types such as bladder
cancer, gastric cancer and breast cancer (10–12),
the role of RTKN in lung cancer has not been investigated.
Cancer cells are characterized by uncontrolled
proliferation (13). Cell cycle
progression and DNA replication are essential events for cell
proliferation (14). Cell cycle was
finely tuned by a number of factors including cyclins and
cyclin-dependent kinases (CDKs) (15). CDK1 is a catalytic subunit of the
M-phase promoting factor (MPF), which promotes G1/S and G2/M
transitions of eukaryotic cells (16). CDK2 is part of a cyclin-dependent
protein kinase complex. CDK2 activity is essential during G1/S
transition (17). Minichromosome
maintenance protein complex (MCM) is involved in the initiation of
DNA replication. The complex formed by MCM2, 4, 6, and 7 was shown
to regulate the helicase activity of the pre-replication complex
(18,19).
Here we report that the RTKN gene expression level
was significantly higher in tumor tissue of lung cancer patients.
Further analysis in RTKN stable knock-down A549 and SPC-A-1 lung
adenocarcinoma cells indicated that RTKN knock-down exhibited
antitumor activity as evidenced by decreased cancer cell viability,
induction of cell cycle arrest, increased apoptosis, and decreased
invasion and migration. Detailed analysis showed that RTKN
knock-down decreased the cell cycle regulators CDK1 and CDK2
expression, as well as the DNA replication modulators MCM2 and MCM6
expression.
Materials and methods
Clinical patient samples
Primary tissues were collected from patients who
received surgery for lung cancer at our institution. All the
patients had given their informed consent. Dissected samples were
frozen immediately after surgery and stored at −80°C until
needed.
Cell culture
A549, H460, H128, H1299, SPC-A-1 and SK-MES human
lung adenocarcinoma cell lines were obtained from the Chinese
Academy of Sciences (Shanghai, China). The cells were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS),
100 U/ml penicillin and 100 µg/ml streptomycin (Invitrogen)
and maintained in an incubator with a humidified atmosphere of 95%
air and 5% CO2 at 37°C.
Establishment of stable RTKN knock-down
cell lines
A549 and SPC-A-1 stable knock-down cell lines were
constructed using the pLKO.1-EGFP vector-based lentiviral
transduction system. shRNA target RTKN and non-target control shRNA
(NC) were synthesized and cloned into the pLKO.1-EGFP vector using
AgeI and EcolI restriction sites. pLKO.1-EGFP-RTKN or
pLKO.1-EGFP-NC and the packing vector psPAX2 and pMD2G were
co-transfected with into 293T cells. Lentivirus particles were
produced in 293T cells. A549 and SPC-A-1 cells were transfected by
the lentiviruses and selected using the EGFP green fluorescence
marker. Three shRNA sequences targeting the RTKN gene are: RTKN-F1:
CCGGG AACTGCGGTTAGAGCTGTATCTCGAGATACAGCTCT AACCGCAGTTCTTTTTC and
AATTGAAAAAGAACTG CGGTTAGAGCT-GTATCTCGAGATACAGCTCTAACCG CAGTTC;
RTKN-F2: CCGGGAAGCAG-TGCTGTGATGA
AATCTCGAGATTTCATCACAGCACTGCTTCTTTTTC and
AATTGAAAAAGAAGCAGTGCTGTGATGAAATCT CGAGATTTCATCACAGCACTGCTTC; and
RTKN-F3: CCGGAAGAACCCTTGGAGCAAACATCTCGAGATGTT
TGCTCCAAGGGTTCTTTTTTTC and AATTGAAAAAA
AGAACCCTTGGAGCAAACATCTCGAGATGTTTGCTC CAAGGGTTCTT.
Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent,
reverse transcribed to cDNA, and quantified by real-time PCR using
SYBR Green Universal Master Mix (all reagents were from Thermo
Fisher Scientific, Waltham, MA, USA). Results were normalized by
using human GAPDH mRNA levels as internal control. Relative mRNA
levels were expressed as fold change to control. The primer pairs
used are as follows: RTKN (forward) GCCGCTGCTTACTATTGC and
(reverse) GTGCTTCCCGACTTTCTG; GAPDH (forward) CACCCACTCCTCCACCTTTG
and (reverse) CCA CCACCCTGTTGCTGTAG; CDK1 (forward) ACCATACCC
ATTGACTAAC and (reverse) ATAAGCACATCCTGAAGAC; CDK2 (forward)
CCAGGAGTTACTTCTATGCCTGA and (reverse) TTCATCCAGGGGAGGTACAAC; MCM2
(forward) CTACCAGCGTATCCGAATC and (reverse) GTTG AGGGAGCCATCATAG;
and MCM6 (forward) CCAAACA TCTGCCGAAATC and (reverse)
TCAGTGTCCCTGTAAA GTC.
Western blotting
Cells were lysed with RIPA lysis buffer containing
protease inhibitor cocktails (both from Thermo Scientific). Protein
concentrations were determined using BCA protein assay kit (Thermo
Scientific). Equal amounts of lysates (20-40 µg of protein)
were resolved with 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE). Protein blots were transferred to a
nitrocellulose membrane and probed with the corresponding primary
antibodies. The membrane was then incubated with appropriate
horseradish peroxidase (HRP)-conjugated secondary antibodies, and
the protein expression was detected by Immobilon Western
Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA).
Antibodies used were all from Abcam (Cambridge, MA, USA): RTKN
(Ab154954), CDK1 (Ab18), CDK2 (Ab6538), MCM2 (Ab4461), MCM6
(Ab190948), and GAPDH (Ab8254).
Cell viability assay
Cell viability was determined using the Cell
Counting Kit-8 (CCK-8) assay kit (Dojindo Molecular Technologies,
Japan) following the manufacturer's instructions. Briefly, 5,000
cells were seeded into each well of 96-well plates in triplicates.
Cells were cultured for 24 h and then 10 µl of the CCK-8
solution was added to each well and incubated for 1 h. The
absorbance at 450 nm was measured using a microplate reader.
Results were calculated as percentage to cells numbers at 0 h.
Cell cycle analysis
Cells were cultured for 24 h and then suspended and
fixed with ethanol. After an overnight incubation at −20°C, cells
were washed with PBS and incubated with 1 mg/ml RNase A at 37°C for
30 min. The cells were then incubated with 50 µg/ml
propidium iodide (PI) on ice for 10 min in the dark. Cell cycle
distribution was analyzed using FACSCalibur instrument (BD
Biosciences, San Jose, CA, USA) equipped with CellQuest
software.
Apoptosis
Cell Apoptosis was determined using the Annexin
V-APC apoptosis detection kit (BD Biosciences) following the
manufacturer's instructions. Briefly, cells were detached by
trypsin, harvested and stained with Annexin V-APC for 10 min at
room temperature. Cell were then incubated with 50 µg/ml PI
on ice and analyzed using flow cytometry on the FACSCalibur
instrument. Annexin V positive cells were considered the apoptotic
fraction.
Tumor invasion assay
Cancer cell invasion was evaluated using Matrigel
and Transwells in triplicates. Cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) without serum 24 h prior to seeding.
Transwells (24-well plates) were rinsed with PBS and then each
Transwell was coated with 80 µl Matrigel at 37°C for 30 min.
Cells were detached with trypsin and resuspended with DMEM
containing 1% FBS. Cells (1×105) in 0.5 ml suspension
were seeded in each Transwell. DMEM (0.75 ml) with 10% FBS was
added into each well of the 24-well plate under the Transwell.
Cells were incubated at 37°C for 48 h and then fixed with 4%
formaldehyde. The cells were stained with 0.5% crystal violet
solution, rinsed with PBS and air dried. Residue cells in Transwell
were wiped away and the migrated cells were visualized under a
microscope. Cell numbers from triplicate wells in each group were
counted for statistical analysis.
Cell adhesion assay
Cells were detached by trypsin and resuspended.
Cells (1×105) were plated in each well of fibronectin
coated 12-well plates and culture in growth medium for 1 h at 37°C
in the incubator for evaluation of adhesion. Cell culture medium
was discarded and cells were washed with PBS twice. Cells were then
fixed with 5 ml of 4% formaldehyde for 15 min and washed with PBS.
Cells were stained with Giemsa staining buffer for 30 min and
unbound dye was rinsed with water. The plates were air dried and
cell images were captured under a microscope. Cells in 3 triplicate
wells were counted for each group.
Data analysis
All the experiments were performed in triplicates.
Results are expressed as mean ± SD. The statistical difference
between multiple treatments and control was analyzed using one-way
ANOVA. Differences between two groups were analyzed by Student's
t-test. A p-value of <0.05 was considered statistically
significant.
Results
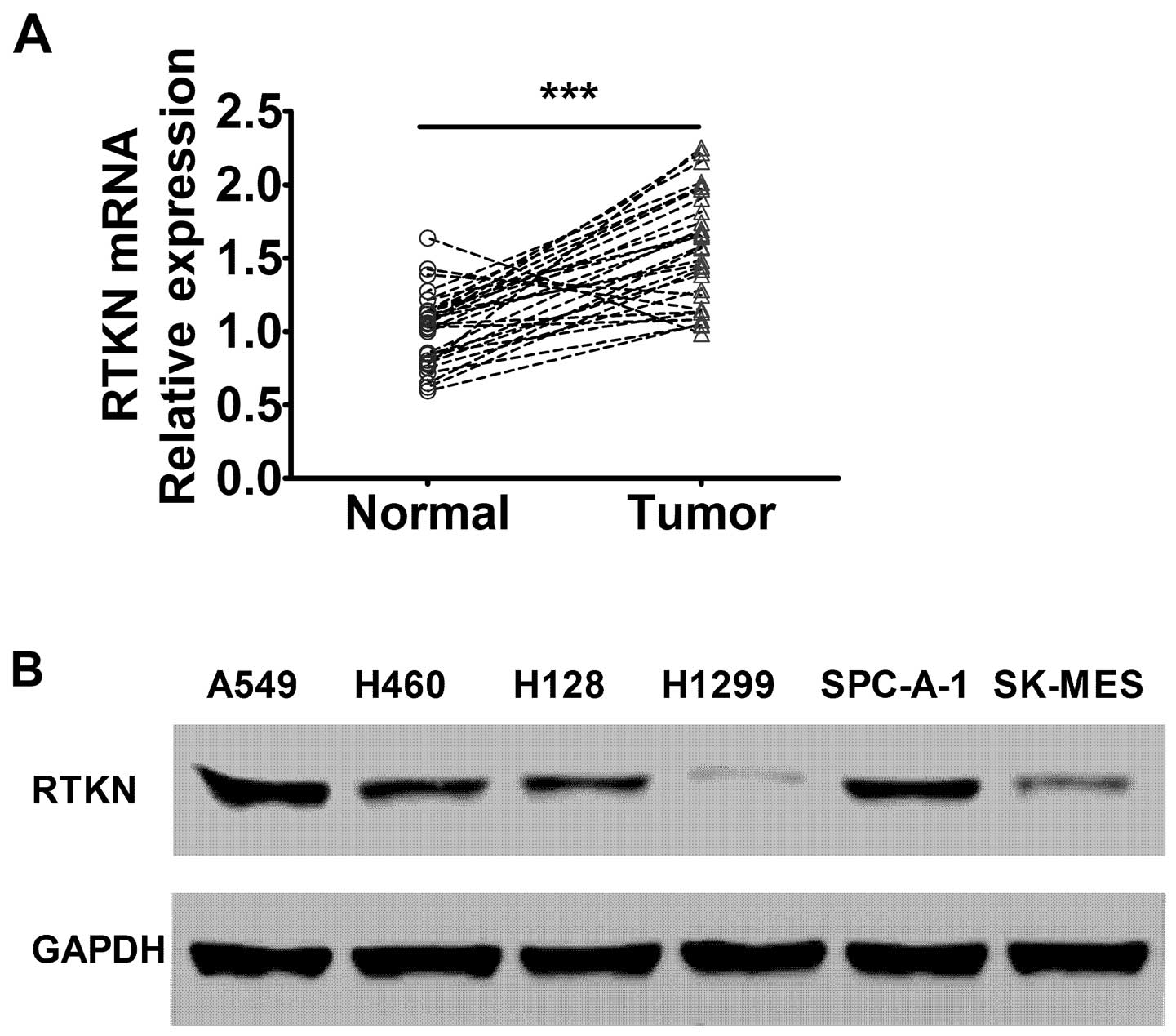
The RTKN levels are elevated in lung
cancer cells in vivo and in vitro
To study the expression pattern of RTKN gene in lung
cancer, tumor and matched benign tissue from 30 non-small cell lung
cancer cases were collected and evaluated by RT-PCR analysis. The
results showed that RTKN gene mRNA level was significantly elevated
in lung cancer tissue compared with matched benign tissue,
indicating that the RTKN gene may be associated with the
development of lung cancer (Fig.
1A). We further detected the RTKN expression levels in
different lung adenocarcinoma cell lines. The results showed that
RTKN level was elevated in several lung adenocarcinoma cell lines
with A549 and SPC-A-1 showing the highest levels among the cell
lines evaluated (Fig. 1B).
The RTKN knock-down inhibits lung
adenocarcinoma cell viability and induces cell cycle arrest and
apoptosis
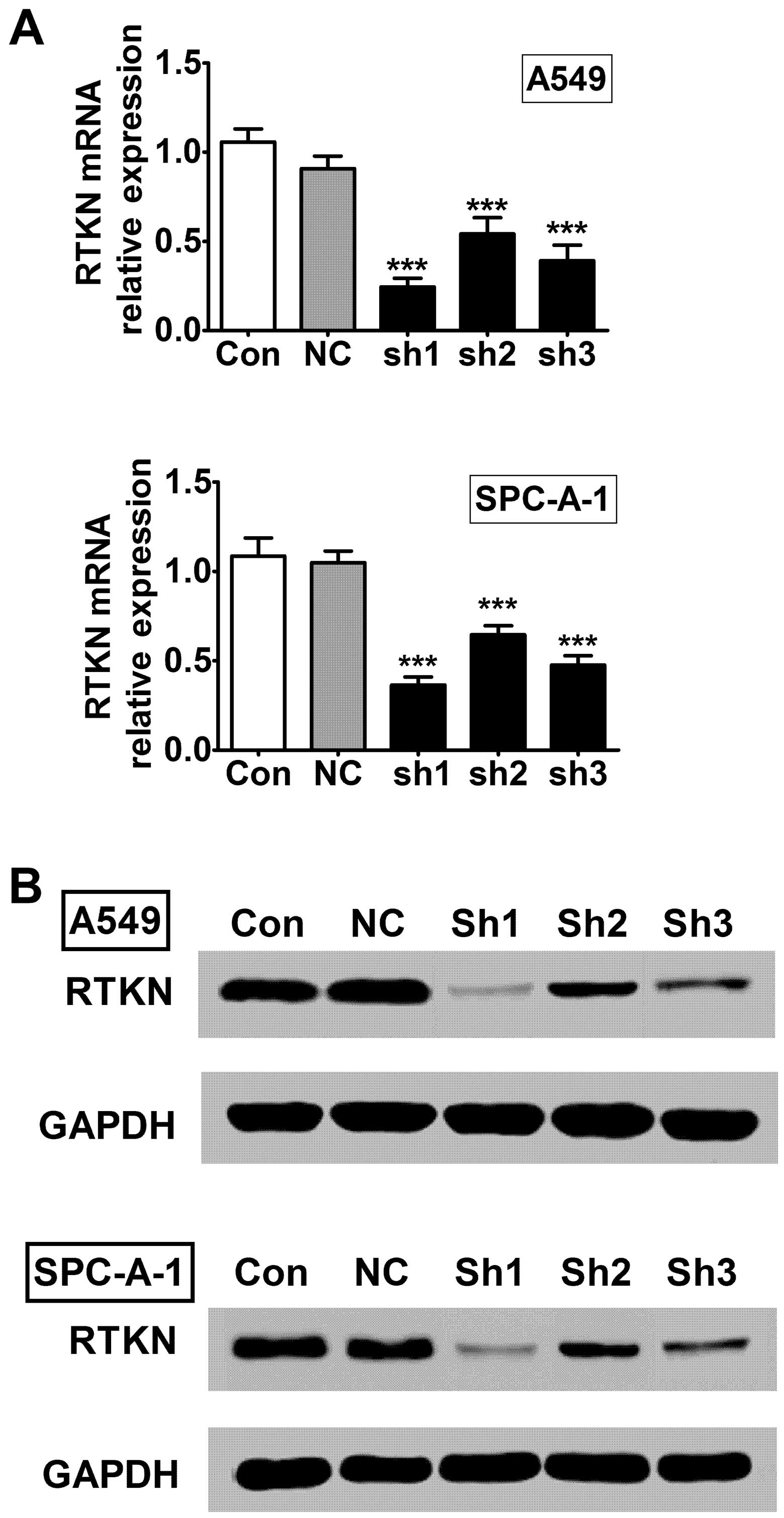
To further study the role of RTKN in lung cancer, we
established RTKN stable knock-down A549 and SPC-A-1 lung
adenocarcinoma cell lines using lentiviral system. We designed
three shRNA constructs (sh1, sh2, and sh3) and detected their
knock-down efficiencies on RTKN mRNA and protein levels in A549 and
SPC-A-1 cells. The first construct (sh1) showed the best inhibiting
efficiency in both cell lines compared with other two constructs
(Fig. 2), therefore it was selected
for further experiments.
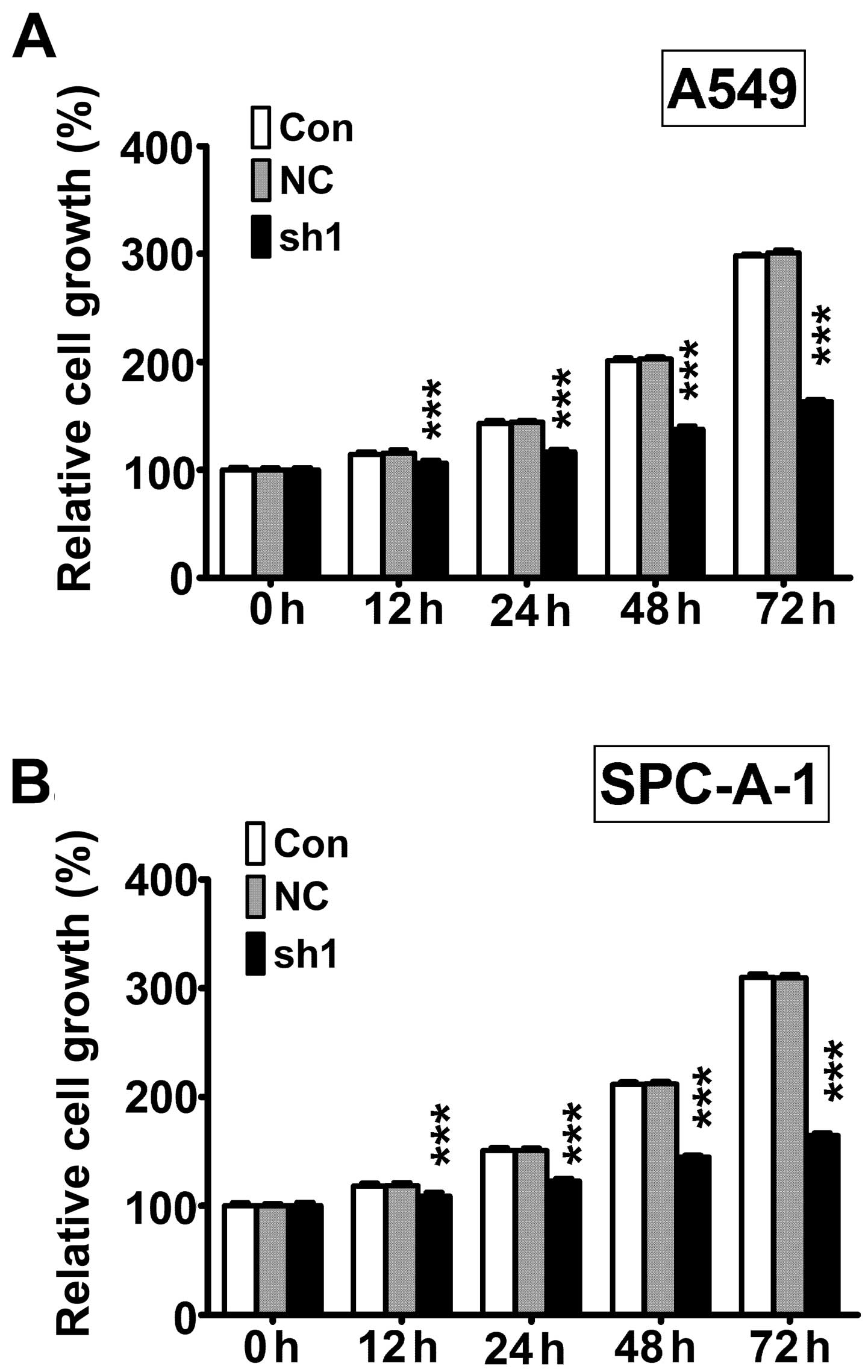
Next, we measured cell viability using CCK-8 assay.
The results showed that cell viability was significantly lower
after 12, 24, 48, and 72 h in RTKN knock-down (sh1) cells compared
with non-transfected control cells (Con) and non-target scramble
control shRNA transfected cells (NC) in both A549 (Fig. 3A) and SPC-A-1 (Fig. 3B) cell lines.
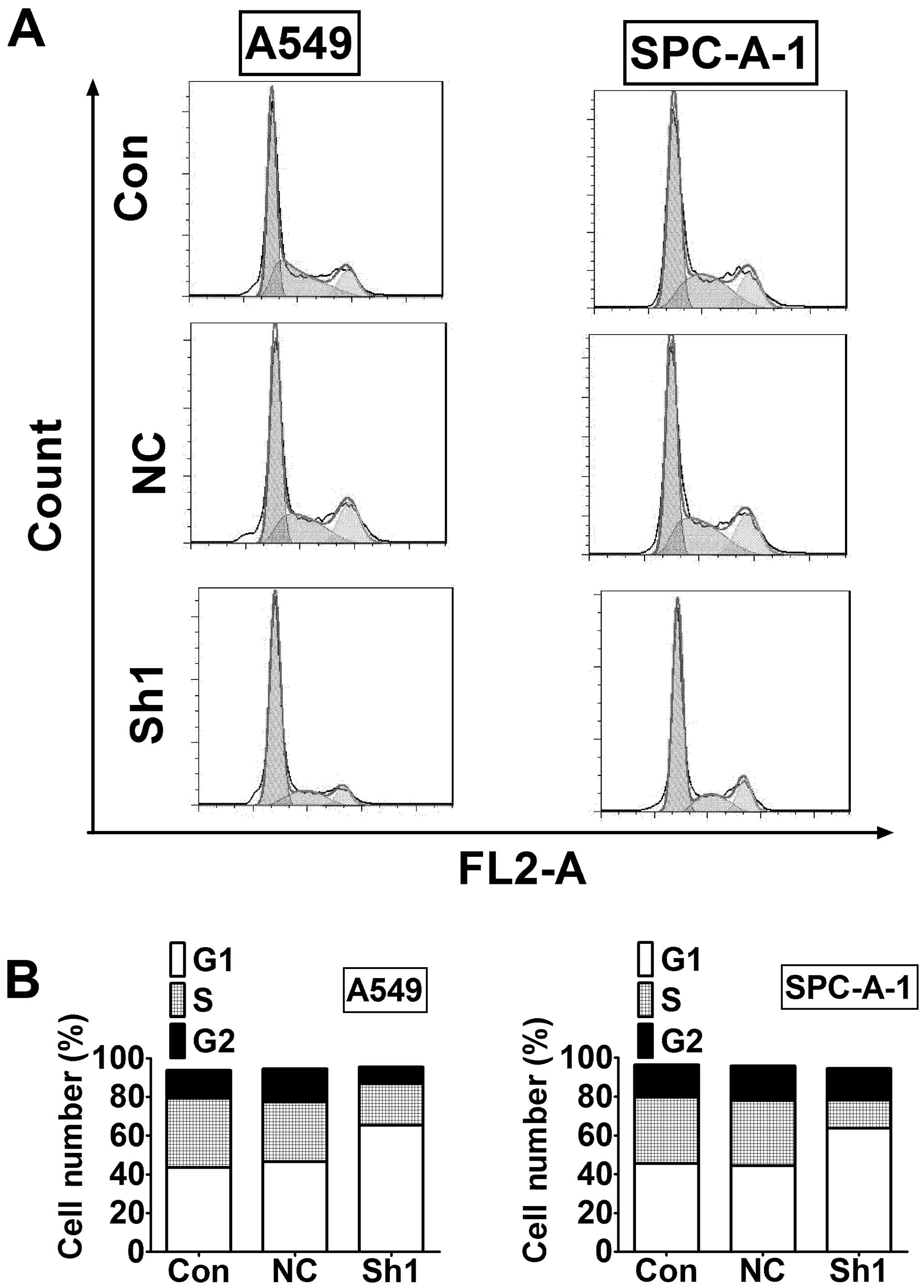
To explore the mechanism of reduced cell viability
after RTKN inhibition, we examined cell cycle distribution using
flow cytometry. The results showed that G1 phase was greatly
increased in RTKN knock-down cells compared to control cells in
both A549 and SPC-A-1 cells (Fig.
4).
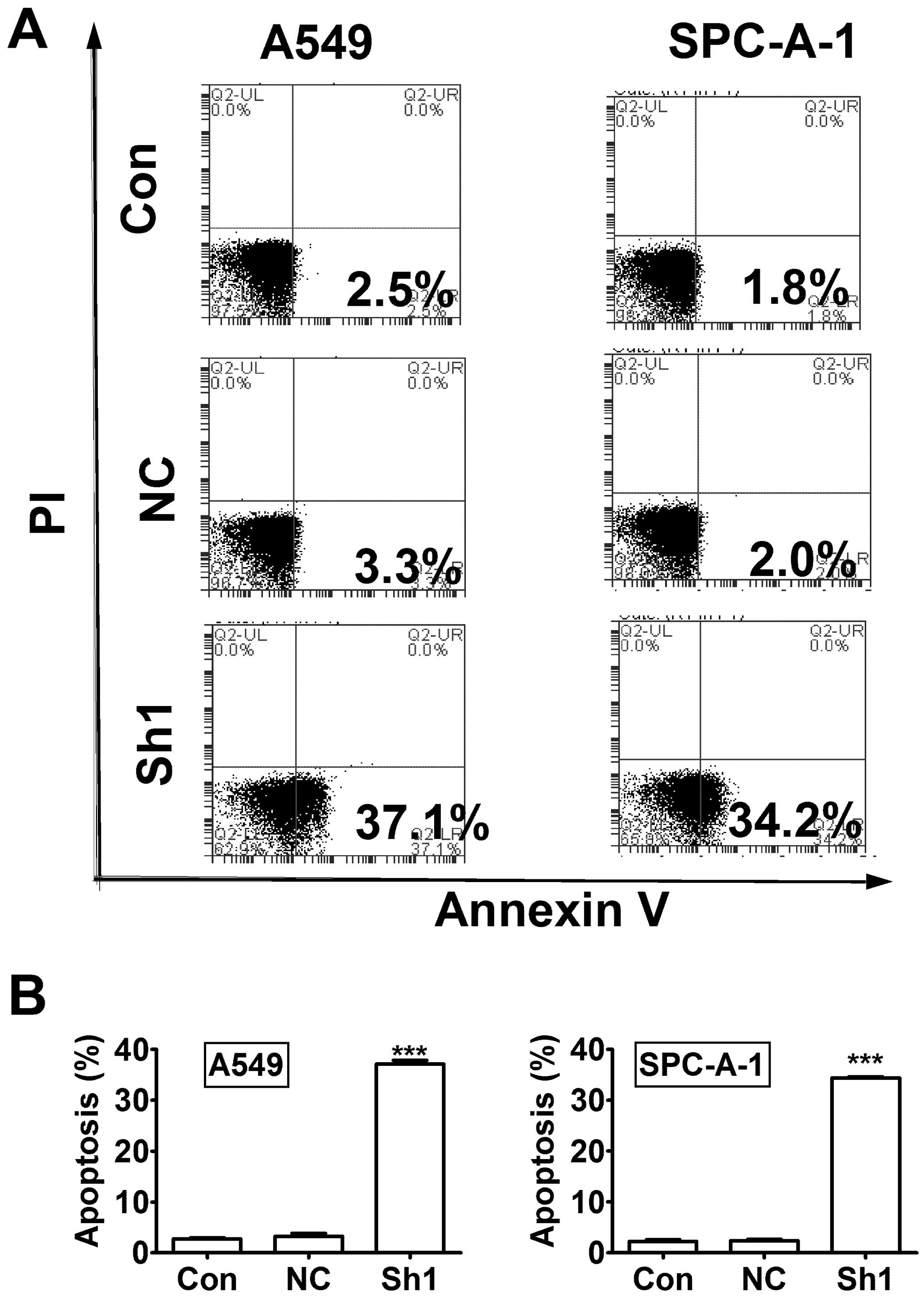
Next, we evaluated cell apoptosis using Annexin
V-APC staining and flow cytometry. The results showed significant
increase of apoptosis percentage in RTKN knock-down A549 and
SPC-A-1 cells compared to control knock-down cells (Fig. 5, P<0.001).
The RTKN knock-down blocks signaling
pathways in cell growth
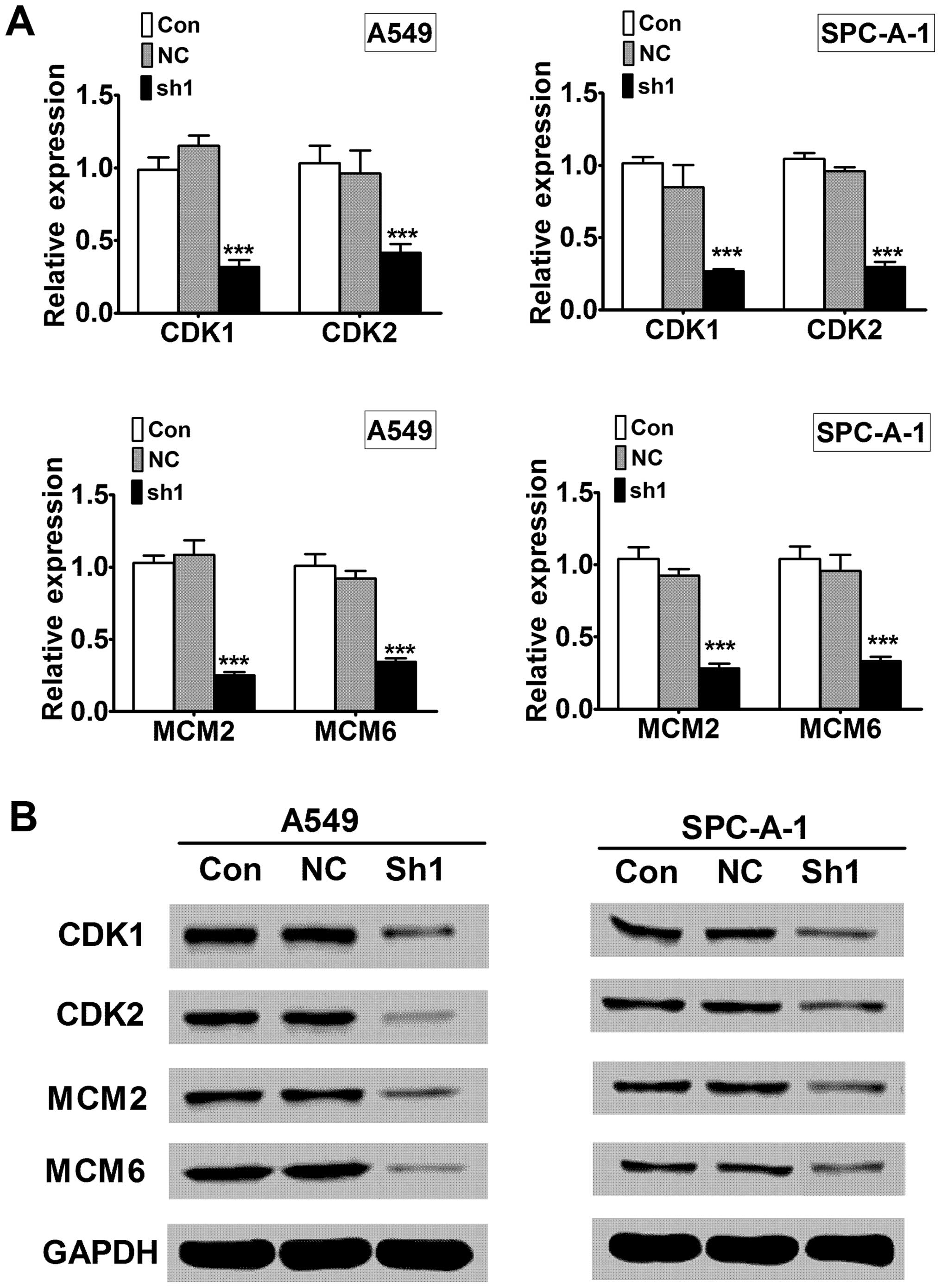
To investigate the underlying mechanism of RTKN
shRNA induced growth inhibition, we examined signaling molecules in
cell cycle progression (CDK1 and CDK2) and DNA replication (MCM2
and MCM6). Quantitative RT-PCR showed that RTKN knock-down
decreased CDK1, CDK2, MCM2, and MCM6 mRNA levels compared to the
control knock-down (Fig. 6A,
P<0.001). Western blot analysis showed that the protein levels
of CDK1, CDK2, MCM2 and MCM6 were also decreased in RTKN knock-down
cells compared to control knock-down cells (Fig. 6B).
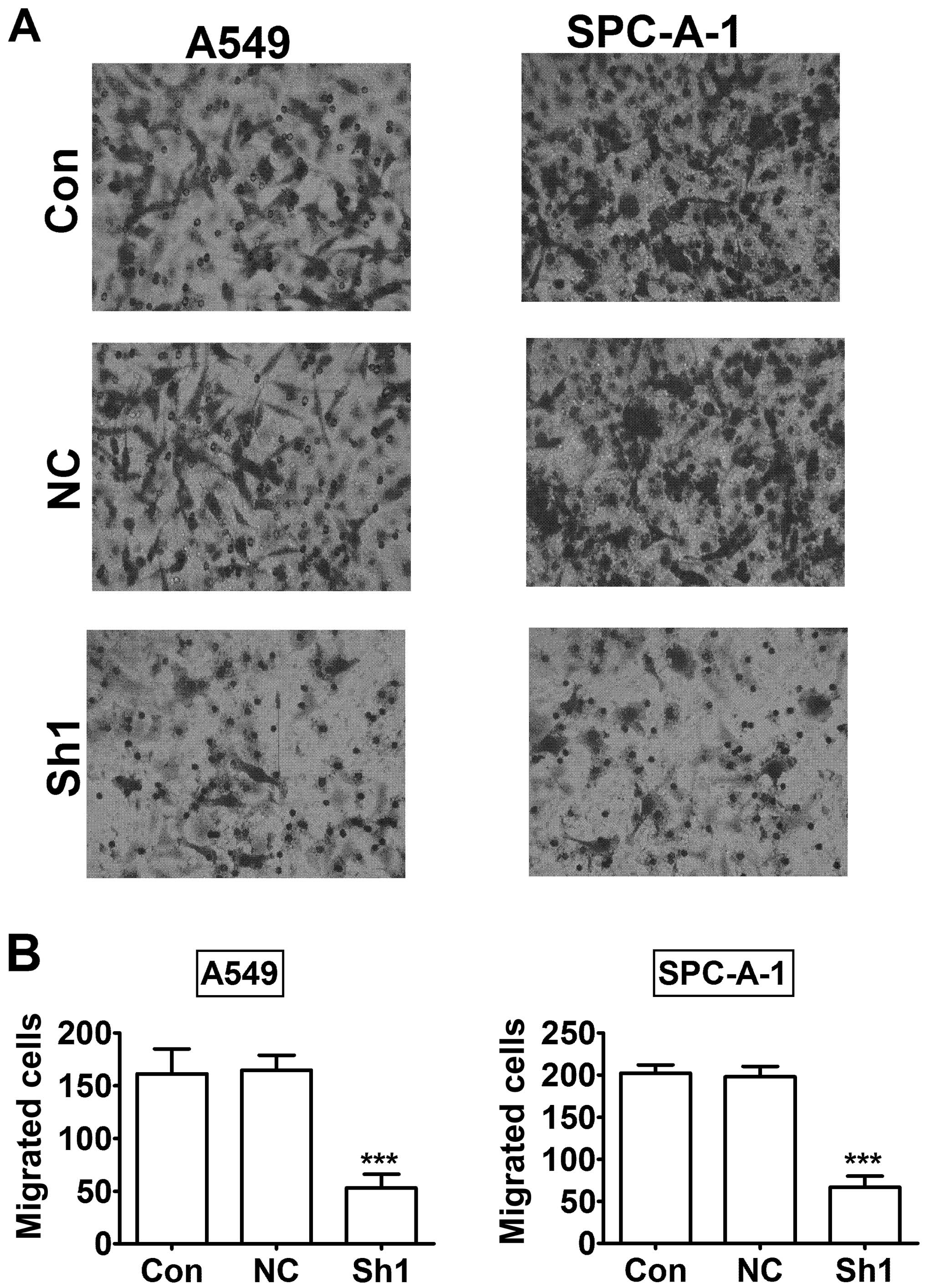
The RTKN knock-down inhibits lung cancer
cell invasion and adhesion
To further investigate the role of RTKN in lung
cancer cells, we examined A549 and SPC-A-1 lung cancer cell
invasion and adhesion after RTKN knock-down. The Transwell tumor
invasion assay showed that migrated cells were significantly
decreased in RTKN knock-down A549 and SPC-A-1 cells compared to
control knock-down cells (Fig. 7,
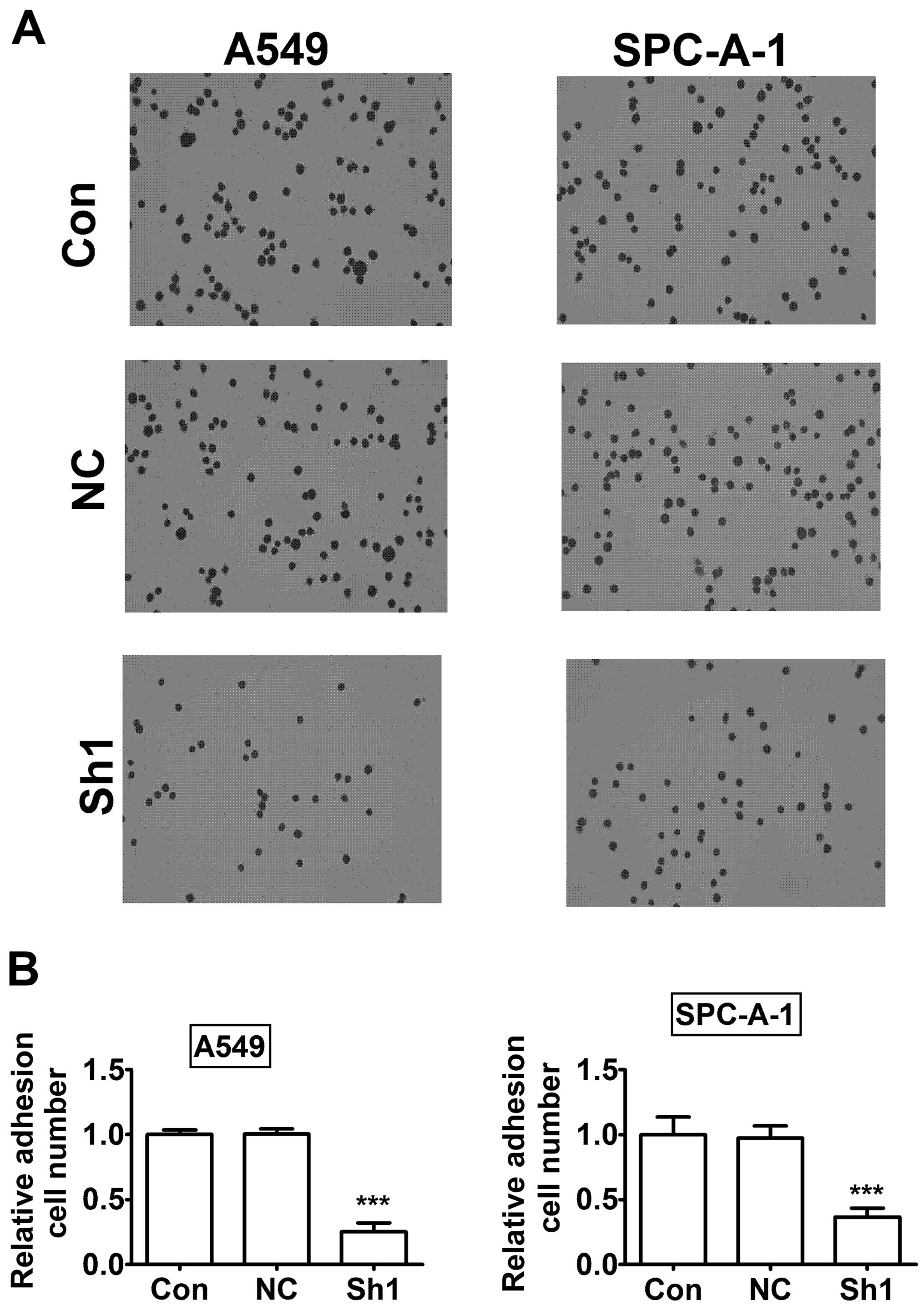
P<0.001). Next, we investigated the role of RTKN in cell
adhesion.
The results showed a significant reduction in cell
adhesion in RTKN knock-down A549 and SPC-A-1 cells compared to
control knock-down cells (Fig. 8,
P<0.001). Taken together, the results indicated that the RTKN
knock-down decreased lung cancer cell invasion and adhesion.
Discussion
Non-small cell lung cancer is the major type of lung
cancer with adenocarcinoma as the main subtype. Targeted cancer
therapy showed high efficacy and low toxicity compared with
traditional chemotherapy. Identifying specific tumor genesis
pathways could promote personalized cancer therapy and greatly
improve cancer treatment outcome. We studied tumor tissue from lung
cancer patients and found that the RTKN gene was highly expressed
in tumor tissue compared with benign tissue. We further
investigated the role of RTKN in lung cancer and underlying
mechanisms using RTKN stable knockdown lung adenocarcinoma cell
lines. Our results showed that knock-down of RTKN exhibited
antitumor activity in lung cancer cells.
Rho GTPase is a master regulator of cytoskeleton in
multiple cell functions such as cell migration, adhesion and
cytokinesis. Upon binding GTP, Rho exerts its functions through
downstream Rho effectors, such as ROCK, mDia, Citron, PKN,
Rhophilin and Rhotekin (RTKN) (9,20–22).
The RTKN gene was only identified recently and its physiological
functions remain largely unknown (8,23).
RTKN has been reported to be associated with several cancer types
such as bladder cancer (12),
gastric cancer (11), and breast
cancer (10).
However, the role of RTKN in lung cancer and its
molecular mechanisms have not been reported. We identified RTKN as
a potential oncogenic factor in lung cancer, as evidenced by the
findings that the RTKN knock-down inhibited lung cancer cell
viability, invasion and adhesion.
Suppression of apoptosis and disregulation of cell
viability are key characteristics of cancer cells which enables
uncontrolled expansion and invasion (14). Our results demonstrated increased
cell apoptosis in RTKN knock-down lung cancer cells compared to
control knock-down cells, therefore indicating antitumor activity
of RTKN inhibition. Cell cycle progression was finely tuned by
cyclin complexes, in which cyclin-dependent kinases play important
roles (15). CDK1 and CDK2 are key
regulators of G1-S transition (24). Our results showed that cells were
arrested in G1 phase in RTKN knockdown cells. Consistently, further
analysis revealed that CDK1 and CDK2 levels are decreased in RTKN
knock-down cells, indicating that RTKN affected cell cycle
regulatory proteins and cell growth. DNA replication is the major
event in cell viability (25). MCM
complex is required for DNA replication and recruited to the origin
recognition complex (ORC) during late mitosis/early G1 phase
(18,19). Our results revealed that MCM2 and
MCM6 levels are decreased in RTKN knockdown lung cancer cells,
indicating that RTKN affected the DNA replication machinery and
thus cell proliferation.
Further experiments are needed to determine the
protein levels through western blotting and the cellular
distribution through immunohistochemistry staining for RTKN in
human cancer samples. Also, it is important to establish animal
tumor models to further validate the antitumor effects of RTKN
inhibition or knockdown in vivo.
In summary, our results from both clinical specimens
and cultured lung cancer cells revealed that the RTKN level was
elevated in lung cancer and that RTKN inhibition exerts antitumor
effects in lung cancer cells. These finding suggested that RTKN may
be a potential therapeutic target in lung cancer treatment.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peters S, Adjei AA, Gridelli C, Reck M and
Kerr K: Metastatic non-small-cell lung cancer (NSCLC): ESMO
Clinical Practice Guidelines for diagnosis, treatment and
follow-up. Ann Oncol. 23(Suppl 7): vii56–vii64. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kumar M, Ernani V and Owonikoko TK:
Biomarkers and targeted systemic therapies in advanced non-small
cell lung cancer. Mol Aspects Med. Jul 14–2015.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Black A and Morris D: Personalized
medicine in metastatic non-small-cell lung cancer: Promising
targets and current clinical trials. Curr Oncol. 19(Suppl 1):
S73–S85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pérez-Soler R, Chachoua A, Hammond LA,
Rowinsky EK, Huberman M, Karp D, Rigas J, Clark GM, Santabárbara P
and Bonomi P: Determinants of tumor response and survival with
erlotinib in patients with non-small-cell lung cancer. J Clin
Oncol. 22:3238–3247. 2004. View Article : Google Scholar
|
|
6
|
Sandler A, Gray R, Perry MC, Brahmer J,
Schiller JH, Dowlati A, Lilenbaum R and Johnson DH:
Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell
lung cancer. N Engl J Med. 355:2542–2550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pender A and Popat S: The efficacy of
crizotinib in patients with ALK-positive non-small cell lung
cancer. Ther Adv Respir Dis. 9:97–104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reid T, Furuyashiki T, Ishizaki T,
Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P and Narumiya
S: Rhotekin, a new putative target for Rho bearing homology to a
serine/threonine kinase, PKN, and rhophilin in the rho-binding
domain. J Biol Chem. 271:13556–13560. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bishop AL and Hall A: Rho GTPases and
their effector proteins. Biochem J. 348:241–255. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen M, Bresnick AR and O'Connor KL:
Coupling S100A4 to Rhotekin alters Rho signaling output in breast
cancer cells. Oncogene. 32:3754–3764. 2013. View Article : Google Scholar
|
|
11
|
Liu CA, Wang MJ, Chi CW, Wu CW and Chen
JY: Overexpression of rho effector rhotekin confers increased
survival in gastric adenocarcinoma. J Biomed Sci. 11:661–670. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan J, Ma LJ, Xia SJ, Yu L, Fu Q, Wu CQ,
Huang XH, Jiang JM and Tang XD: Association between clinical
characteristics and expression abundance of RTKN gene in human
bladder carcinoma tissues from Chinese patients. J Cancer Res Clin
Oncol. 131:157–162. 2005. View Article : Google Scholar
|
|
13
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Norbury C and Nurse P: Animal cell cycles
and their control. Annu Rev Biochem. 61:441–470. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ubersax JA, Woodbury EL, Quang PN, Paraz
M, Blethrow JD, Shah K, Shokat KM and Morgan DO: Targets of the
cyclin-dependent kinase Cdk1. Nature. 425:859–864. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Labib K, Tercero JA and Diffley JF:
Uninterrupted MCM2-7 function required for DNA replication fork
progression. Science. 288:1643–1647. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lygerou Z and Nurse P: Cell cycle. License
withheld - geminin blocks DNA replication. Science. 290:2271–2273.
2000.
|
|
20
|
Sahai E and Marshall CJ: ROCK and Dia have
opposing effects on adherens junctions downstream of Rho. Nat Cell
Biol. 4:408–415. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Watanabe N, Kato T, Fujita A, Ishizaki T
and Narumiya S: Cooperation between mDia1 and ROCK in Rho-induced
actin reorganization. Nat Cell Biol. 1:136–143. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ishizaki T, Naito M, Fujisawa K, Maekawa
M, Watanabe N, Saito Y and Narumiya S: p160ROCK, a
Rho-associated coiled-coil forming protein kinase, works downstream
of Rho and induces focal adhesions. FEBS Lett. 404:118–124. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu Q, Yu L, Liu Q, Zhang J, Zhang H and
Zhao S: Molecular cloning, expression characterization, and mapping
of a novel putative inhibitor of rho GTPase activity, RTKN, to
D2S145-D2S286. Genomics. 66:328–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morgan DO: Cyclin-dependent kinases:
Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stillman B: Cell cycle control of DNA
replication. Science. 274:1659–1664. 1996. View Article : Google Scholar : PubMed/NCBI
|