Introduction
Cellular DNA is constantly challenged by either
endogenous [reactive oxygen species (ROS)] or exogenous (UV)
factors. To properly transmit the genetic information to the next
generation, multiple delicate mechanisms underlying this event have
been evolved in eukaryotic cells (1,2). DNA
damage response (DDR) is such a crucial mechanism to ensure that
genetic information is faithfully transmitted between generations.
In response to DNA damages, eukaryotic cells activate a complex
signaling cascade to arrest the cell cycle for DNA repair.
Dysregulation of DDR may result in severe disorders, such as
tumorigenesis (1,3–5).
Therefore, it is critical to elucidate the mechanism of DDR in
order to treat human diseases, such as cancer. The major regulators
of DDR are phosphoinositide 3-kinase (PI3K)-related protein kinases
(PIKKs), which include ataxia telangiectasia mutated (ATM) and
ataxia telangiectasia and RAD3-related (ATR) (3,6,7). ATM
and ATR respond to different types of DNA damage; ATM mainly
responds to double-strand breaks (DSBs) while ATR mainly responds
to replication stress and UV-induced pyrimidine dimers. According
to previous studies (1,4,6–9),
Ser/Thr kinase checkpoint kinase-1 (Chk1) and -2 (Chk2) are the two
most thoroughly investigated substrates of ATR and ATM,
respectively. In the situation of DNA damage, these pathways are
able to activate p53, phosphorylate H2AX and other
downstream pathways to activate the checkpoints of DNA damage,
which eventually causes cell cycle arrest for DNA repair.
Defects in the ATR/Chk1 and ATM/Chk2 pathways are
known to increase cancer risk (1,3). Once
DSBs occur, ATM kinases can phosphorylate histone H2AX to form
γH2AX foci at the DNA break sites. The formation of γH2AX foci
subsequently promotes the gathering of repair factors on damaged
DNA sites and activates the DNA damage arrest. This indicates that
γH2AX foci play an important role in cellular DDR. Besides
activating the formation of γH2AX nuclear foci, ATM can also induce
cell cycle arrest through phosphorylation of its targets, p53 and
Chk2. Numerous studies have demonstrated that phosphorylation of
Chk2 can induce G2/M arrest by phosphorylating CDC25C, which
prevents CDC25C from dephosphorylating CDC2 (or CDK1) (1–3,6,9–11).
The phosphatase and tensin homolog (PTEN) gene
encodes a major plasma membrane lipid and protein phosphatase.
Scientists have reported that PTEN functions as a highly effective
tumor suppressor in various tissues and is frequently mutated in a
wide variety of cancers, including endometrial, breast and prostate
cancer and glioblastomas. Previous studies revealed that PTEN is a
central negative regulator of PI3K/AKT-mediated signaling cascade.
The loss of function of PTEN in various human cancers enhances AKT
activation, resulting in dysregulated cell proliferation, survival,
migration and spreading, which are all important factors in tumor
development and progression (12,13).
Recent findings have demonstrated various novel functions of PTEN
in several cellular processes. For instance, PTEN plays a critical
role in DNA damage repair and response. In 2007, Shen et al
demonstrated that cells deficient in PTEN have defective DNA DSB
repair, possibly due to the lack of or downregulation of Rad51 and
the lack of PTEN at centromeres (14). In addition, several studies have
indicated that PTEN is able to promote the formation of γ-H2AX
foci, a marker of DNA damage, which suggests that PTEN functions to
reduce DSB levels (15–17). Furthermore, previous studies have
shown that PTEN plays a unique role in DDRs through its interaction
with the Chk1 and p53 pathways (10,18–21).
In the present study, we report that PTEN is required for the
proper checkpoint activation in etoposide-treated MCF7 cells. While
etoposide treatment induces G2/M arrest in MCF-7 cells by
activating the ATM-Chk2-CDC25C pathway, knockdown of PTEN
compromises this process, thereby reducing etoposide-induced G2/M
arrest in MCF-7 cells.
Materials and methods
Materials and reagents
PTEN shRNA and related products were purchased from
Invitrogen. ECL Western Blotting kit, anti-p53, anti-β-actin,
anti-γH2AX, anti-H2AX and anti-CDC25C antibodies were purchased
from Abcam. Anti-phospho-ATM, anti-Chk2, anti-phosph-Chk2 (T68),
anti-phospho-p53 (S15) and anti-phospho-CDC25C (S216) antibodies
were purchased from Cell Signaling. Anti-phospho-ATM (S1981) and
anti-ATM antibody, FITC-donkey anti-rabbit IgG and Rhodamine-donkey
anti-mouse IgG were purchased from Sigma. Immobilon-P membranes
were purchased from Millipore.
Cell lysis and western blotting
The cells were lysed with a buffer containing 20 mM
Tris (pH, 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100,
25 mM sodium pyrophosphate, 1 mM NaF, 1 mM β-glycerophosphate, 0.1
mM sodium orthovanadate, 1 mM PMSF, 2 µg/ml leupeptin and 10
µg/ml aprotinin. Total cell lysate (50 µg) was
separated on SDS-PAGE gel and transferred onto Immobilon-P
membranes. The membranes were first blocked with 5% skim milk, and
then incubated with primary antibodies at room temperature for 2 h.
The ECL Western Blotting kit was utilized to detect the signals
(22). The concentrations of
antibodies were as follows: anti-p53 (FL-353 at 1 µg/ml),
anti-phospho-p53 (S15) (1:1,000), anti-ERK (1:500), anti-H2AX
(1:1,000), anti-γH2AX (1:1,000), anti-phospho-ATM (S1981) (1:500),
anti-ATM (1:500), anti-phospho-CHK2 (T68) (1:500), anti-CHK2
(1:1,000), anti-phospho-CDC25C (S216) (1:500), anti-CDC25C
(1:1,000) and anti-β-actin (1:1,000).
Immunofluorescence staining
Cells were treated as described in the figure
legends. The cells were fixed with pre-chilled (−20°C)
acetone-methanol for 15 min. The primary antibodies anti-γH2AX (0.5
µg/ml; Upstate) and anti-phospho-ATM (S1981) (1:100; Cell
Signaling) were then added onto the slides and incubated at 4°C
overnight. The slides were gently washed with 1X phosphate-buffered
saline (PBS) twice, and the FITC-conjugated secondary antibodies
(donkey anti-rabbit IgG and Rhodamine-donkey anti-mouse IgG; 1:200;
Jackson ImmunoResearch Laboratories) were added onto the slides and
incubated at room temperature for 1 h. Afterwards the slides were
covered with Vectashield mounting medium containing DAPI (CA94010;
Vector Laboratories, Inc., Burlingame, CA, USA). Images were
captured with a fluorescence microscope (Axiovert 200; Carl Zeiss)
(22).
Cell cycle assessment
Cells in the logarithmic growth phase were cultured
in 6-well plates for 24 h and different concentrations of etopside
were added into the plates. After 18 h of incubation, cells were
trypsinized and transferred into a 15-ml tube, followed by
centrifugation at 1,000 rpm in a 5840R cooled centrifuge
(Eppendorf, Germany) for 5 min. The cells were then washed with 1X
PBS and the centrifugation was repeated twice. The supernatant was
discarded and 1 ml propidium iodide (PI) solution (0.05 mg/ml) was
added into each tube. The samples were kept in the dark at room
temperature for 30 min, and then the phases of the cell cycle were
analyzed using a flow cytometer (22).
Statistical analysis
Data were analyzed using one-way ANOVA followed by a
Tukey's test. Statistical significance was achieved if the p-value
was ≤0.05. Data values are expressed as the mean ± standard error
(SE).
Results
PTEN promotes etoposide-induced p53
phosphorylation
PTEN was initially identified as a tumor
suppressor, which has been found to be mutated in a large number of
cancers at a high frequency (23).
Mutations of PTEN are a critical step in the development of
many cancers and this gene may play an important role in DDRs
(12,14,20,21).
In order to investigate how PTEN acts on the
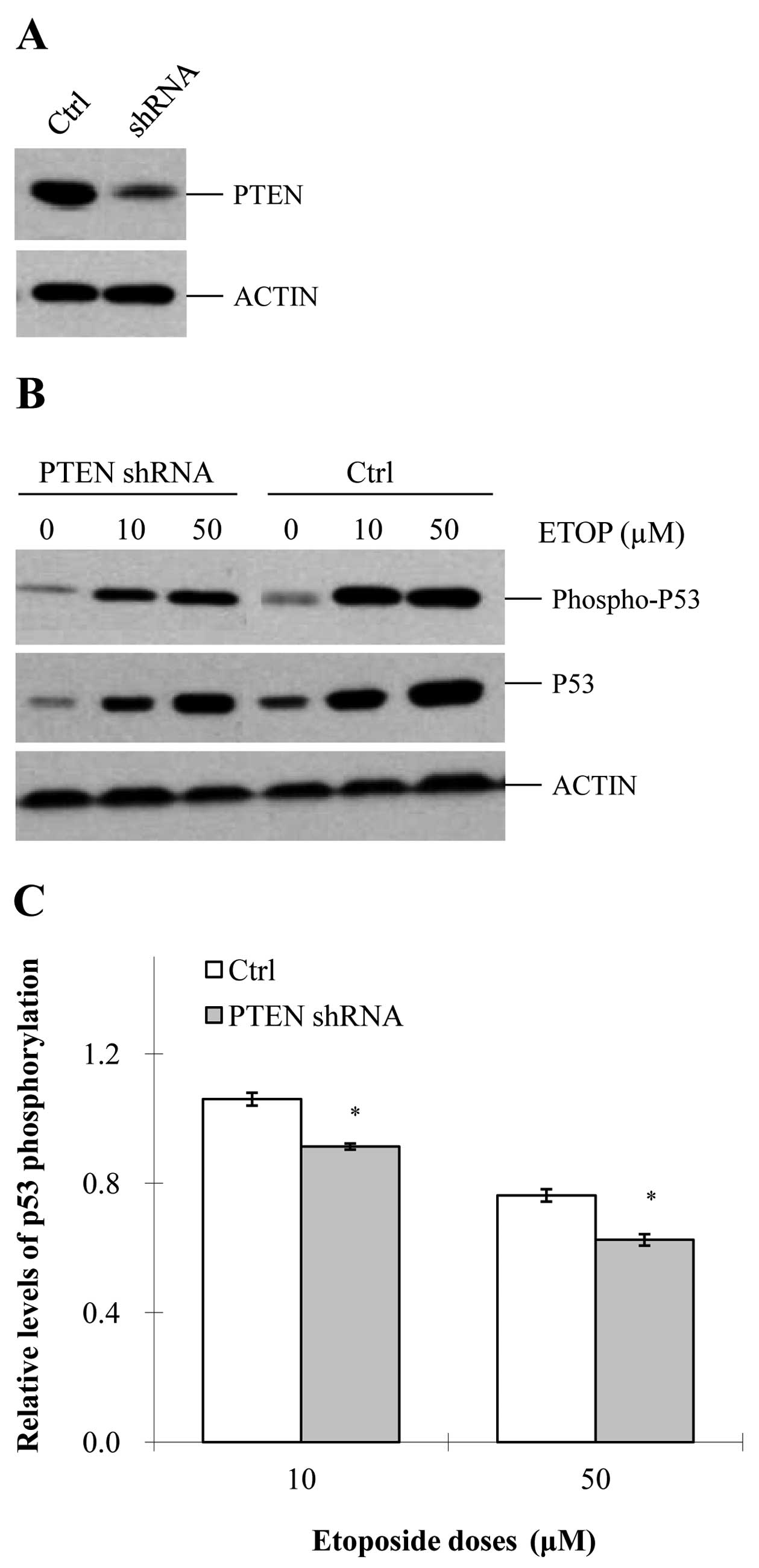
etoposide-induced DDR, we knocked down this gene in MCF-7 cells
(Fig. 1A). As observed, depletion
of PTEN did not significantly affect proliferation of the
MCF-7 cells (data not shown). It has been widely recognized that
p53 phosphorylation is a crucial event of DDR by affecting
MDM2, 14-3-3σ, DADD45 and p21Cip1 (24–27).
Therefore, we first investigated the impact of PTEN
depletion on etoposide-induced p53 phosphorylation. In both
the control and PTEN-knockdown cells, etoposide induced
p53 phosphorylation in a dose-dependent manner (Fig. 1B). However, at each specific
concentration of etoposide, the level of p53 phosphorylation
was significantly reduced in the PTEN-knockdown cells,
compared with that in the control cells (Fig. 1B and C). These data suggest that
PTEN is able to promote etoposide-induced p53
phosphorylation.
PTEN promotes etoposide-induced ATM
activation
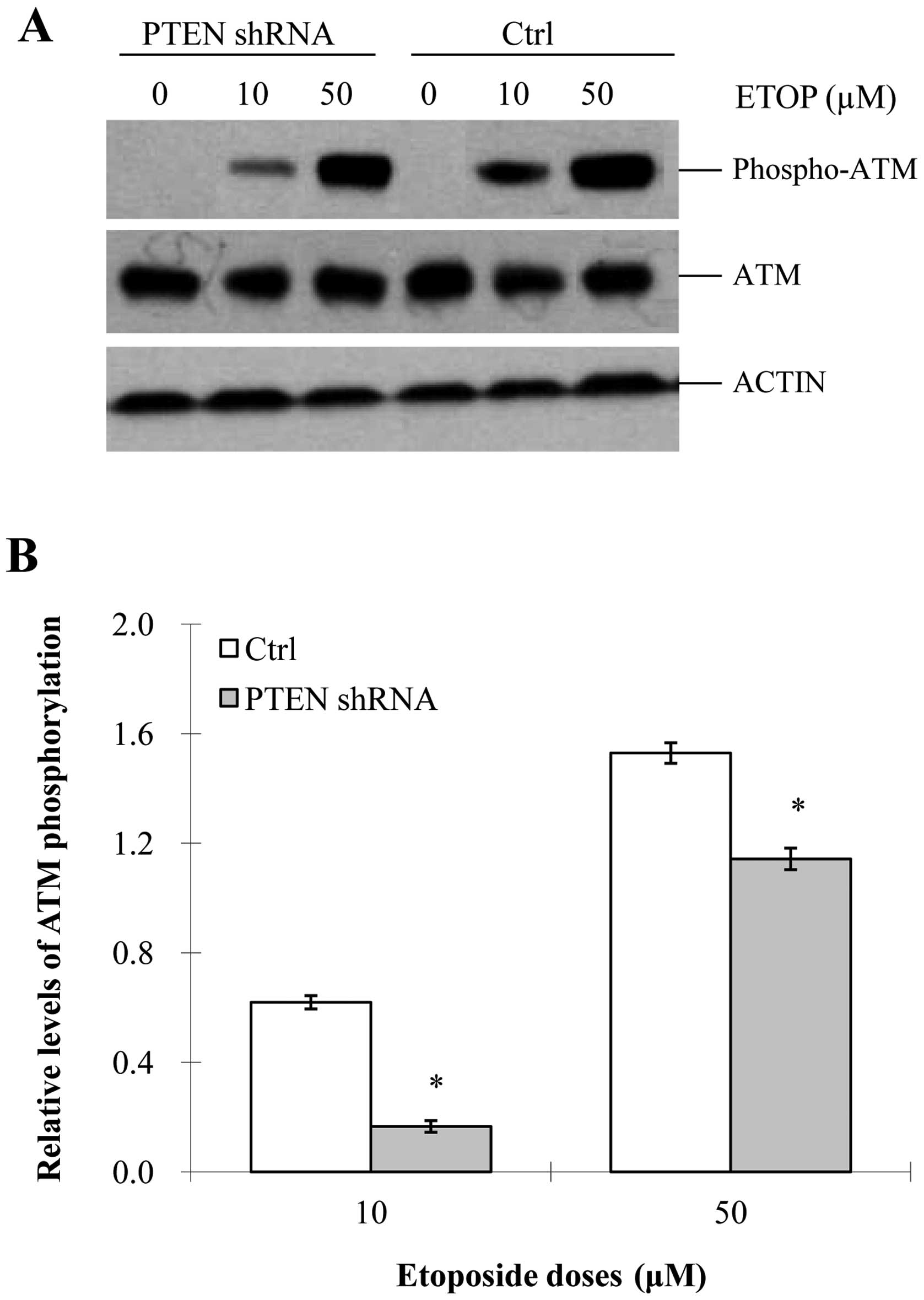
In cells, one of the key responses to DNA damage is
ATM activation, which occurs via auto-phosphorylation of ATM
(S1981). ATM activation can facilitate the activation of p53
given that S15 of p53 is the substrate of ATM (28–30).
As previously reported (31,32),
etoposide activates ATM and thereby promotes a series of downstream
responses leading to G2/M arrest. This finding was further
supported by our results which showed that etoposide was able to
enhance the phosphorylation level of ATM (Fig. 2A and B). In addition, the
phosphorylated level of ATM was increased in an etoposide
dose-dependent manner. However, the activation of ATM was largely
compromised in the PTEN-knockdown cells at any specific
concentration of etoposide (Fig. 2A and
B), which indicated that PTEN is needed for the
etoposide-induced activation of ATM/ATR.
PTEN enhances the etoposide-induced
production of γH2AX
Histone H2A is one of the main histone proteins
involved in the structure of chromatin in eukaryotic cells and is
responsible for maintaining the shape and structure of the
nucleosome (33). H2AX is a variant
of H2A and its C-terminus is involved in DNA repair. A known
response of H2AX to DNA damage is the formation of γH2AX (34). According to previous studies
(35–37), ATM activation promotes the
phosphorylation of H2AX, and this phosphorylated form, named γH2AX,
may bind to SDBs and form nuclear γH2AX foci, which are critical
for DNA repair or G2/M arrest (38). Given the role of γH2AX in DDRs, in
the present study, we also investigated whether PTEN
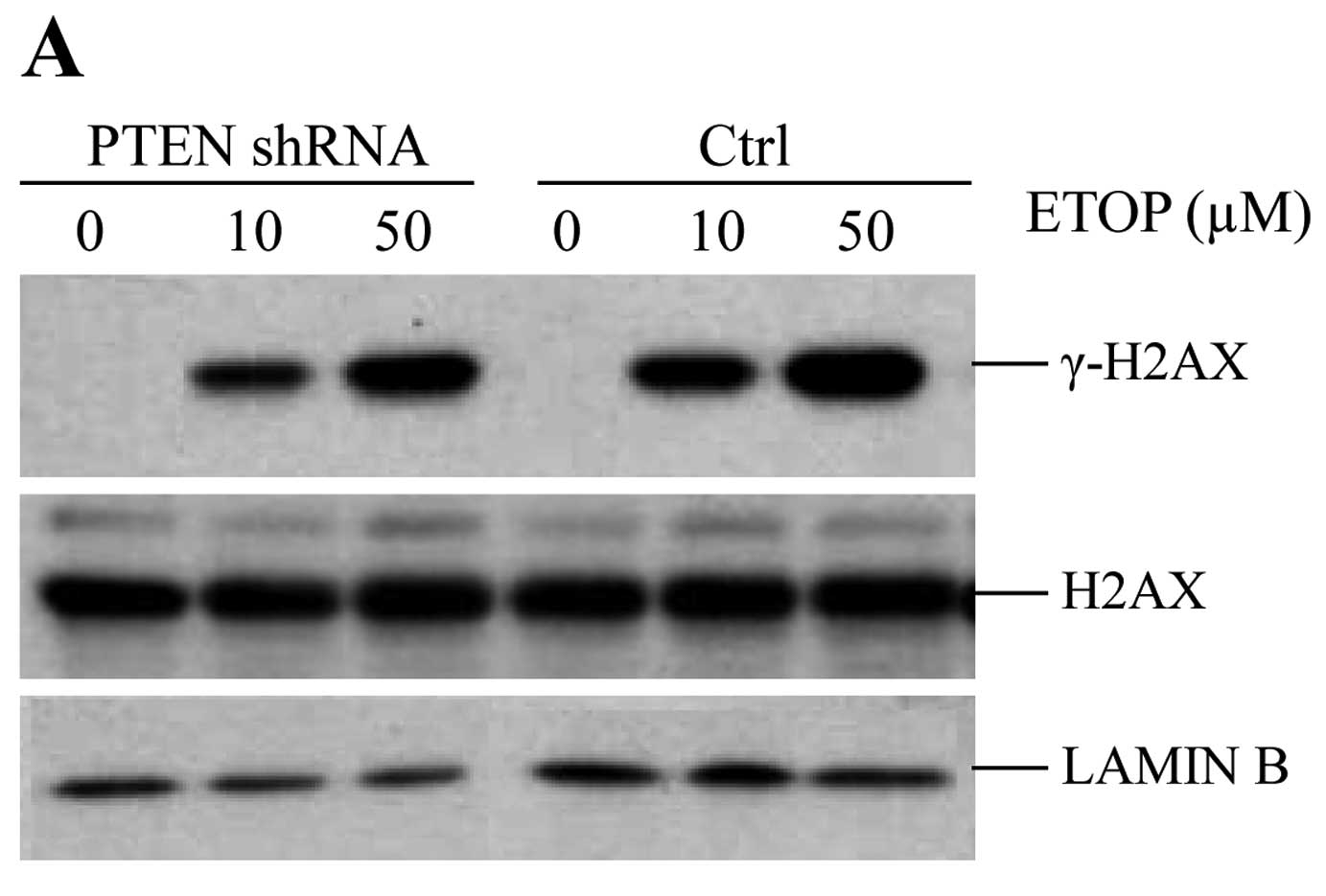
contributes to etoposide-induced production of γH2AX. As shown in
the control cells, the higher the dose of etoposide, the more γH2AX
was produced in the MCF-7 cells. However, γH2AX was significantly
reduced in the PTEN-knockdown MCF-7 cells (Fig. 3A and B). In addition, the formation
of γH2AX was dose-dependent on etoposide in both the control and
PTEN-depleted cells (Fig. 3A and
B). In order to confirm these results, we performed
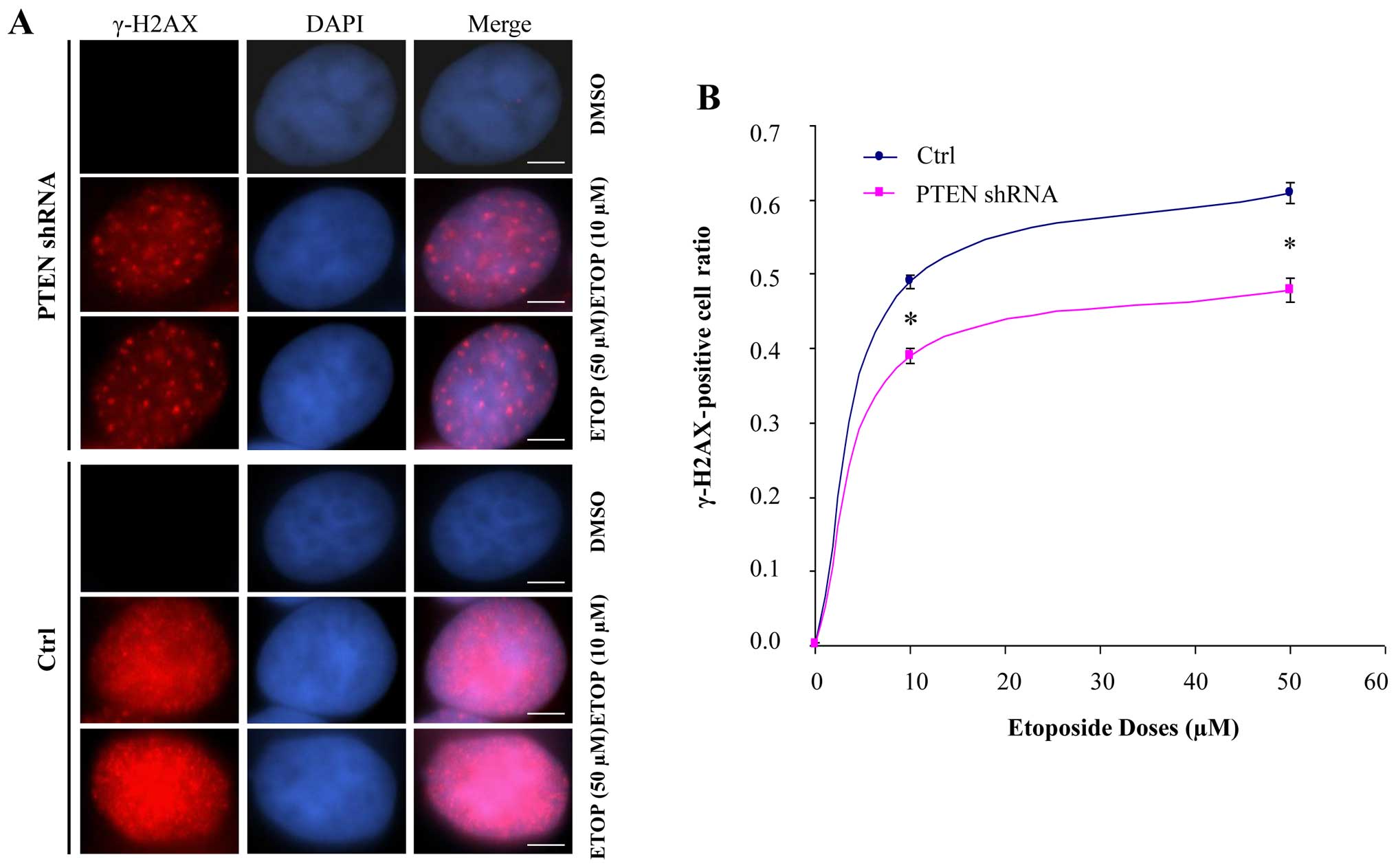
immunostaining to examine the formation of γH2AX nuclear foci in
the etoposide-treated PTEN-knockdown MCF-7 cells. The γH2AX
nuclear foci were strikingly reduced in the PTEN-knockdown
etoposide-treated cells, compared with that in the control cells
(Fig. 4A). In addition, the
production of γH2AX nuclear foci was dose-dependent on etoposide in
both groups. Furthermore, the number of γH2AX-positive cells was
significantly reduced in the PTEN-knockdown MCF-7 cells,
compared with that in the control cells. This quantitative
difference between the control and PTEN-knockdown cells was
also dose-dependent on etoposide (Fig.
4B). These data demonstrated that PTEN facilitated
etoposide-induced γH2AX formation. Given that γH2AX foci not only
functions in DNA damage repair but also activates DNA damage
checkpoints, together with the data we presented above, we
concluded that PTEN plays an important role in
etoposide-induced G2/M arrest.
PTEN facilitates etoposide-induced
G2M/arrest in MCF-7 cells
During DDR, ATM/ATR can be activated and thereby
influences their substrates such as p53 and Chk2
(29). Similar with p53,
phosphorylated Chk2 modulates downstream CDC25C and CDC2 (39–41).
As a typical substrate of ATM, Chk2 kinase can be phosphorylated
and activated by ATM (42). To
further confirm our results and examine the role of PTEN in
the DNA damage pathway, we investigated whether PTEN regulates Chk2
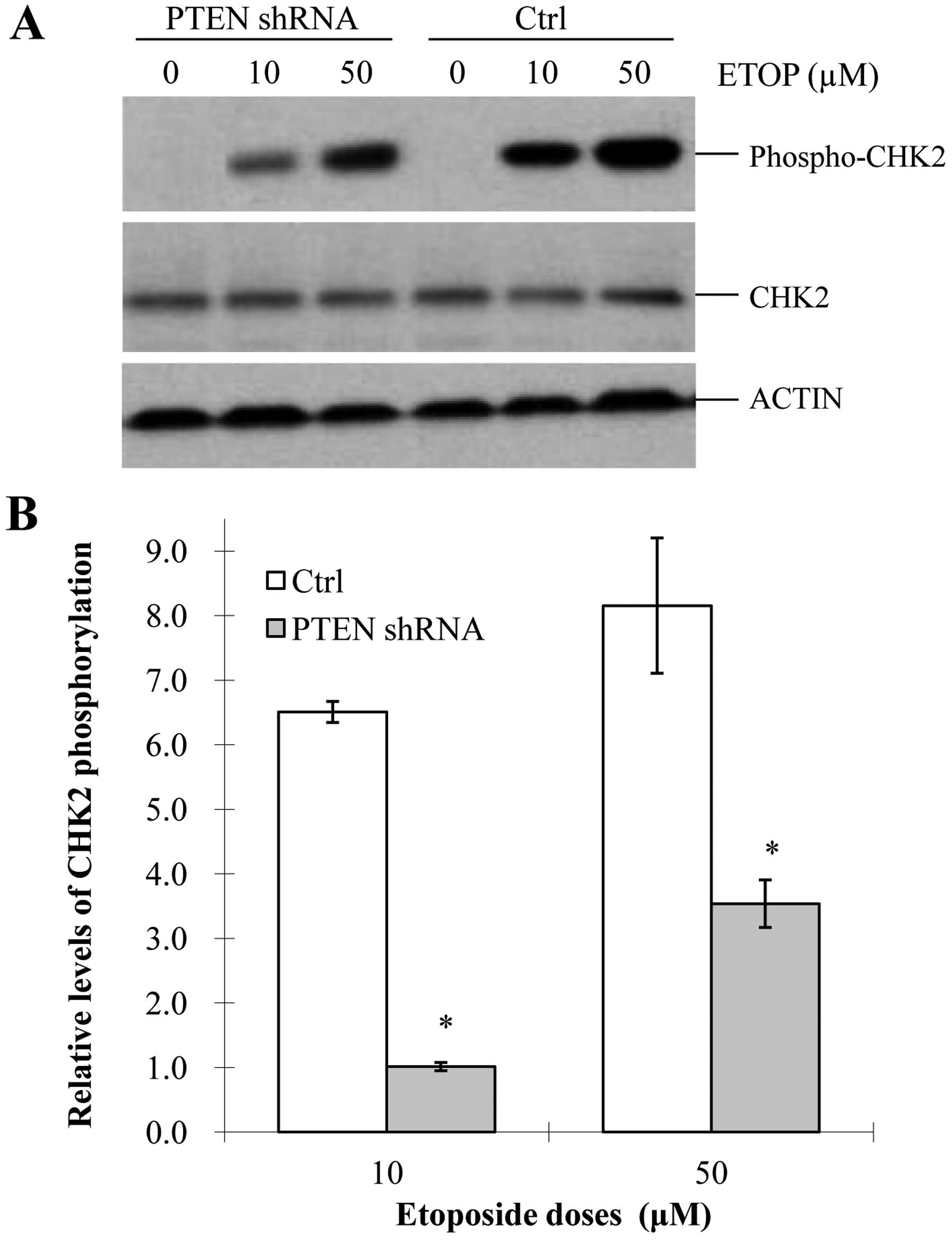
in the etoposide-treated MCF-7 cells. Our data showed that
phospho-Chk2 (T68) was strikingly reduced in the
PTEN-knockdown cells, compared with that in the control
cells (Fig. 5A and B). In addition,
the activation of Chk2 was dose-dependent on etoposide in both
groups (Fig. 5A and B). These
results demonstrated that PTEN promoted etoposide-induced
Chk2 phosphorylation, which is consistent with our above finding
that PTEN facilitated etoposide-induced ATM activation. It
is known that the activation of CDC2 is needed for cells to pass
G2/M during a cell cycle. Normally, CDC2 is dephosphorylated and
activated by CDC25C, which can be phosphorylated and activated by
Chk2. Once CDC25C is phosphorylated, cells cannot proceed through
G2/M and thus G2/M arrest occurs. Therefore, in addition to
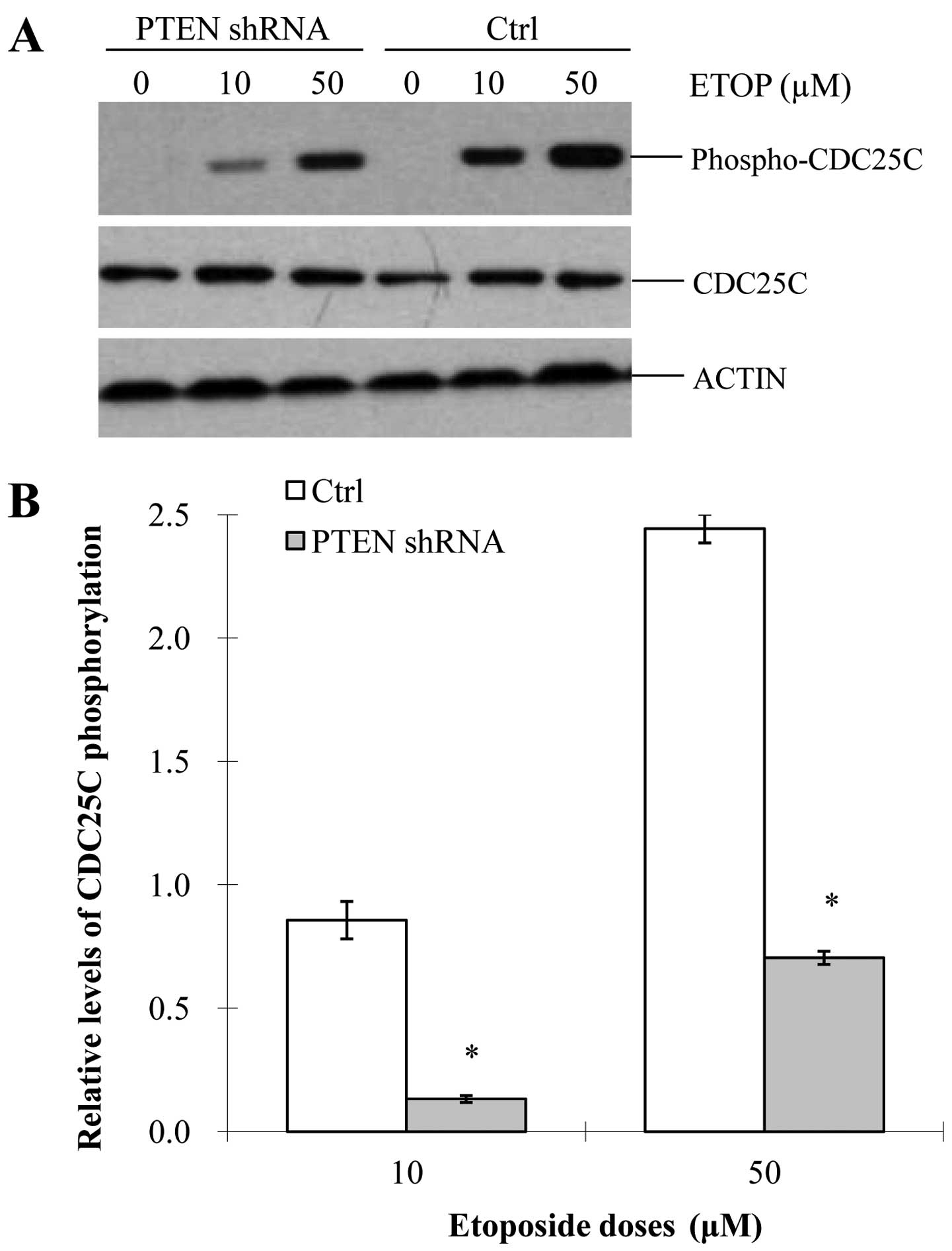
assessing etopo-side-induced Chk2 activation, we examined whether
PTEN could activate CDC25C in the MCF-7 cells. Similar to our
previous results, phosphorylated CDC25C was significantly reduced
in the PTEN-knockdown MCF-7 cells, compared with that in the
control cells (Fig. 6A and B). We
also observed that etoposide induced the phosphorylation of CDC25C
in a dose-dependent manner. To further confirm the role of
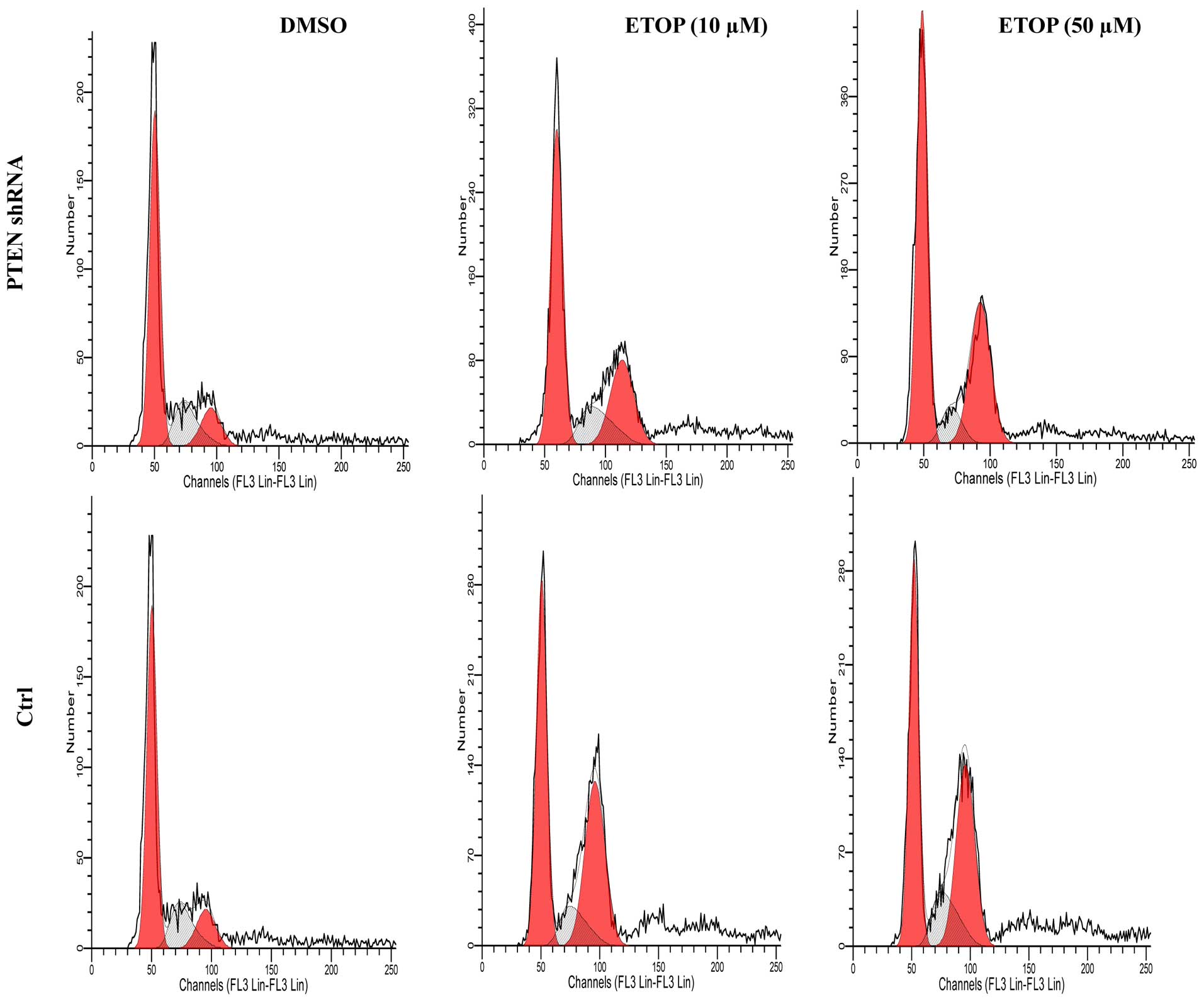
PTEN in G2M arrest, we performed flow cytometry to analyze
the cell cycle profile in the PTEN-knockdown cells. As shown
in Fig. 7 and Table I, etoposide induced G2/M arrest in a
dose-dependent manner in both the control and PTEN-knockdown
cells. However, when PTEN was knocked down in the
etoposide-treated cells, the rate of G2/M arrest was significantly
reduced. These data, together with other data shown above, suggest
that PTEN may enhance etoposide-induced G2/M arrest through
activation of the ATM-p53/Chk2-CDC2C pathway.
 | Table IKnockdown of PTEN reduces the
etoposide-induced G2/M arrest in MCF-7 cells. |
Table I
Knockdown of PTEN reduces the
etoposide-induced G2/M arrest in MCF-7 cells.
| Cell cycle
phase | DMSO
| ETOP (10 µM)
| ETOP (50 µM)
|
|---|
| Control | PTEN shRNA | Control | PTEN shRNA | Control | PTEN shRNA |
|---|
| G1 | 63.7±5.3 | 61.5±5.2 | 36.7±3.2 |
47.4±3.9a | 24.8±4.3 |
42.8±1.3a |
| S | 14.3±5.7 | 21.5±4.4 | 9.0±3.0 |
17.2±2.4a | 6.2±1.9 |
11.2±1.6a |
| G2/M | 18.7±9.3 | 17.0±1.1 | 54.4±1.4 |
35.4±3.4a | 69.0±6.2 |
48.1±2.1a |
Discussion
PTEN is one of the most commonly mutated
tumor suppressors in human cancers (12,14,19).
During tumor progression, mutations of PTEN can result in
increased cell proliferation and decreased cell death. PTEN is
involved in the regulation of cell proliferation, at a normal rate.
PTEN has been one of the targets for drug candidates such as the
oncomiR, miR-21 (12,43–45).
It is widely recognized that PTEN dephosphorylates
phosphatidylinositol (3,4,5)-trisphosphate [PtdIns (3,4,5)P3
or PIP3] (46), which
leads to the inhibition of the AKT signaling pathway. It was
reported that PTEN interacts with p53 and PTK2 and modulates
the progression of various cancers (23,47).
Over the last decades, it has been commonly accepted
that DNA damage response (DDR) is one of the most important factors
to maintain genomic integrity. In fact, DDR is not only essential
to DNA repair, but is also critical to distinct cellular events,
such as cell proliferation, apoptosis and gene transcription. The
AKT signaling pathway has been demonstrated to function in these
cellular processes while PTEN was also reported to be partially
involved in progression of DDR (48–50).
The contributions of molecular factors or pathways to DDR are
variable and cell type-specific. Although some mechanisms
underlying DDR are well understood, more factors associated with
DDR, such as PTEN, remain to be further explored and elucidated.
PTEN has certain impacts on some DNA repair pathways, for example
the AKT pathway (50–53). To date, the effects of PTEN on the
ATM pathway, particularly on etoposide-induced ATM-Chk2-CDC2
activation, have not yet been systematically investigated. Indeed,
PTEN can be an important player in DDR given that it can
functionally interact with a number of key important factors such
as p53.
Etoposide is a medicine commonly used as a
chemotherapy agent to treat cancers, such as Kaposi's sarcoma,
Ewing's sarcoma, lung and testicular cancer, lymphoma,
non-lymphocytic leukemia and glioblastoma multiforme. It is also
used in a conditioning regimen prior to bone marrow or blood stem
cell transplant (54). The cancer
cell killing activity of etoposide is indeed dependent on the
activation of the ATM pathway followed by G2/M arrest (31,55,56).
Considering that the ATM/p53 pathway is critical in DDR,
scientists have made efforts to investigate this pathway. Recently,
BMI1, ERK1 and ERK2, have been reported to affect etoposide-induced
ATM activation, indicating that they are also required for
etoposide-induced G2/M arrest (22,57).
In the present study, we provide evidence that PTEN
facilitates etoposide-induced ATM activation, and also contributes
to etoposide-induced changes of other factors in the ATM pathway,
including activation of Chk2 and inactivation of CDC25C, eventually
leading to G2/M arrest. Our data demonstrated that PTEN is required
for proper activation of the ATM pathway in response to etoposide
treatment. It is not clear yet how PTEN functions in activation of
the ATM pathway and ATM pathway-mediated G2/M arrest. It is
possible that PTEN facilitates the etoposide-induced activation of
the ATM pathway through interacting with ATM directly or other
proteins such as AKT, p53 and BRCA1 indirectly. Further
studies are required to clarify these questions.
Taken together, our finding that PTEN regulates DNA
damage through activation of the ATM-Chk2 pathway provides a new
perspective with which to understand the relationship between PTEN
and initiation/progression of cancer cells; it also provides a new
potential therapeutic target for cancer treatment. In addition, the
fact that PTEN affects etoposide-induced G2/M arrest in cancer
cells provides us with important information when patients are
treated with etoposide, which is critical for precision
medicine.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81201568), the
National Natural Science Foundation of Guangdong Province (grant
no. 2014A030313749), and the Shenzhen Program of Innovation and
Entrepreneurship for Overseas Elites (grant no.
KQCX20120814150420241).
References
|
1
|
Spryszyńska S, Smok-Pieniążek A, Ferlińska
M, Roszak J, Nocuń M and Stępnik M: The influence of ATM, ATR,
DNA-PK inhibitors on the cytotoxic and genotoxic effects of
dibenzo[def,p]chrysene on human hepatocellular cancer cell line
HepG2. Mutat Res Genet Toxicol Environ Mutagen. 791:12–24. 2015.
View Article : Google Scholar
|
|
2
|
Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y,
Zhang J, Yuan J, Wang M, Chen D, Sun Y, et al: ATM-mediated
stabilization of ZEB1 promotes DNA damage response and
radioresistance through CHK1. Nat Cell Biol. 16:864–875. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Benada J and Macurek L: Targeting the
checkpoint to kill cancer cells. Biomolecules. 5:1912–1937. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reddy V, Wu M, Ciavattone N, McKenty N,
Menon M, Barrack ER, Reddy GP and Kim SH: ATM inhibition
potentiates death of androgen receptor-inactivated prostate cancer
cells with telomere dysfunction. J Biol Chem. 290:25522–25533.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seol HJ, Yoo HY, Jin J, Joo KM, Kong DS,
Yoon SJ, Yang H, Kang W, Lim DH, Park K, et al: Prognostic
implications of the DNA damage response pathway in glioblastoma.
Oncol Rep. 26:423–430. 2011.PubMed/NCBI
|
|
6
|
Neumann J, Yang Y, Köhler R, Giaisi M,
Witzens-Harig M, Liu D, Krammer PH, Lin W and Li-Weber M: Mangrove
dolabrane-type of diterpenes tagalsins suppresses tumor growth via
ROS-mediated apoptosis and ATM/ATR-Chk1/Chk2-regulated cell cycle
arrest. Int J Cancer. 137:2739–2748. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwok M, Davies N, Agathanggelou A, Smith
E, Petermann E, Yates E, Brown J, Lau A and Stankovic T: Synthetic
lethality in chronic lymphocytic leukaemia with DNA damage response
defects by targeting the ATR pathway. Lancet. 385(Suppl 1):
S582015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andrs M, Korabecny J, Nepovimova E, Jun D,
Hodny Z, Moravcova S, Hanzlikova H and Kuca K: The development of
ataxia telangiectasia mutated kinase inhibitors. Mini Rev Med Chem.
14:805–811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Penedos A, Johnson AL, Strong E, Goldman
AS, Carballo JA and Cha RS: Essential and checkpoint functions of
budding yeast ATM and ATR during meiotic prophase are facilitated
by differential phosphorylation of a meiotic adaptor protein, Hop1.
PLoS One. 10:e01342972015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang XP, Liu F and Wang W: Two-phase
dynamics of p53 in the DNA damage response. Proc Natl Acad Sci USA.
108:8990–8995. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Palmitelli M, de Campos-Nebel M and
González-Cid M: Progression of chromosomal damage induced by
etoposide in G2 phase in a DNA-PKcs-deficient context. Chromosome
Res. 23:719–732. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ming M and He YY: PTEN in DNA damage
repair. Cancer Lett. 319:125–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ming M, Feng L, Shea CR, Soltani K, Zhao
B, Han W, Smart RC, Trempus CS and He YY: PTEN positively regulates
UVB-induced DNA damage repair. Cancer Res. 71:5287–5295. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen WH, Balajee AS, Wang J, Wu H, Eng C,
Pandolfi PP and Yin Y: Essential role for nuclear PTEN in
maintaining chromosomal integrity. Cell. 128:157–170. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rosser CJ, Tanaka M, Pisters LL, Tanaka N,
Levy LB, Hoover DC, Grossman HB, McDonnell TJ, Kuban DA and Meyn
RE: Adenoviral-mediated PTEN transgene expression sensitizes
Bcl-2-expressing prostate cancer cells to radiation. Cancer Gene
Ther. 11:273–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pappas G, Zumstein LA, Munshi A, Hobbs M
and Meyn RE: Adenoviral-mediated PTEN expression radiosensitizes
non-small cell lung cancer cells by suppressing DNA repair
capacity. Cancer Gene Ther. 14:543–549. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gupta A, Yang Q, Pandita RK, Hunt CR,
Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, et al: Cell
cycle checkpoint defects contribute to genomic instability in PTEN
deficient cells independent of DNA DSB repair. Cell Cycle.
8:2198–2210. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paez J and Sellers WR: PI3K/PTEN/AKT
pathway. A critical mediator of oncogenic signaling. Cancer Treat
Res. 115:145–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee C, Kim JS and Waldman T: PTEN gene
targeting reveals a radiation-induced size checkpoint in human
cancer cells. Cancer Res. 64:6906–6914. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Puc J, Keniry M, Li HS, Pandita TK,
Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE,
et al: Lack of PTEN sequesters CHK1 and initiates genetic
instability. Cancer Cell. 7:193–204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ali IU: Gatekeeper for endometrium: The
PTEN tumor suppressor gene. J Natl Cancer Inst. 92:861–863. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei F, Xie Y, Tao L and Tang D: Both ERK1
and ERK2 kinases promote G2/M arrest in etoposide-treated MCF7
cells by facilitating ATM activation. Cell Signal. 22:1783–1789.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakanishi A, Kitagishi Y, Ogura Y and
Matsuda S: The tumor suppressor PTEN interacts with p53 in
hereditary cancer (Review). Int J Oncol. 44:1813–1819.
2014.PubMed/NCBI
|
|
24
|
Conde-Perez A, Gros G, Longvert C,
Pedersen M, Petit V, Aktary Z, Viros A, Gesbert F, Delmas V, Rambow
F, et al: A caveolin-dependent and PI3K/AKT-independent role of
PTEN in β-catenin transcriptional activity. Nat Commun. 6:80932015.
View Article : Google Scholar
|
|
25
|
Puzio-Kuter AM, Laddha SV, Castillo-Martin
M, Sun Y, Cordon-Cardo C, Chan CS and Levine AJ: Involvement of
tumor suppressors PTEN and p53 in the formation of multiple
subtypes of liposarcoma. Cell Death Differ. 22:1785–1791. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ushio Y, Tada K, Shiraishi S, Kamiryo T,
Shinojima N, Kochi M and Saya H: Correlation of molecular genetic
analysis of p53, MDM2, p16, PTEN, and EGFR and survival of patients
with anaplastic astrocytoma and glioblastoma. Front Biosci.
8:e281–e288. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou M, Gu L, Findley HW, Jiang R and
Woods WG: PTEN reverses MDM2-mediated chemotherapy resistance by
interacting with p53 in acute lymphoblastic leukemia cells. Cancer
Res. 63:6357–6362. 2003.PubMed/NCBI
|
|
28
|
Bradbury JM and Jackson SP: ATM and ATR.
Curr Biol. 13:R4682003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Xu ZP, Huang Y, Hamrick HE,
Duerksen-Hughes PJ and Yu YN: ATM and ATR: Sensing DNA damage.
World J Gastroenterol. 10:155–160. 2004.PubMed/NCBI
|
|
30
|
Shiloh Y: ATM and ATR: Networking cellular
responses to DNA damage. Curr Opin Genet Dev. 11:71–77. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nam C, Doi K and Nakayama H: Etoposide
induces G2/M arrest and apoptosis in neural progenitor cells via
DNA damage and an ATM/p53-related pathway. Histol Histopathol.
25:485–493. 2010.PubMed/NCBI
|
|
32
|
Kim KY, Cho YJ, Jeon GA, Ryu PD and Myeong
JN: Membrane-bound alkaline phosphatase gene induces antitumor
effect by G2/M arrest in etoposide phosphate-treated cancer cells.
Mol Cell Biochem. 252:213–221. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Redon C, Pilch D, Rogakou E, Sedelnikova
O, Newrock K and Bonner W: Histone H2A variants H2AX and H2AZ. Curr
Opin Genet Dev. 12:162–169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sánchez-Flores M, Pásaro E, Bonassi S,
Laffon B and Valdiglesias V: γH2AX assay as DNA damage biomarker
for human population studies: Defining experimental conditions.
Toxicol Sci. 144:406–413. 2015. View Article : Google Scholar
|
|
35
|
Rao VA, Conti C, Guirouilh-Barbat J,
Nakamura A, Miao ZH, Davies SL, Saccá B, Hickson ID, Bensimon A and
Pommier Y: Endogenous gamma-H2AX-ATM-Chk2 checkpoint activation in
Bloom's syndrome helicase deficient cells is related to DNA
replication arrested forks. Mol Cancer Res. 5:713–724. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Savic V, Yin B, Maas NL, Bredemeyer AL,
Carpenter AC, Helmink BA, Yang-Iott KS, Sleckman BP and Bassing CH:
Formation of dynamic gamma-H2AX domains along broken DNA strands is
distinctly regulated by ATM and MDC1 and dependent upon H2AX
densities in chromatin. Mol Cell. 34:298–310. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Riballo E, Kühne M, Rief N, Doherty A,
Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al: A
pathway of double-strand break rejoining dependent upon ATM,
Artemis, and proteins locating to gamma-H2AX foci. Mol Cell.
16:715–724. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mah LJ, El-Osta A and Karagiannis TC:
gammaH2AX: A sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Agarwal C, Tyagi A and Agarwal R: Gallic
acid causes inactivating phosphorylation of cdc25A/cdc25C-cdc2 via
ATM-Chk2 activation, leading to cell cycle arrest, and induces
apoptosis in human prostate carcinoma DU145 cells. Mol Cancer Ther.
5:3294–3302. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ahn J and Prives C: Checkpoint kinase 2
(Chk2) monomers or dimers phosphorylate Cdc25C after DNA damage
regardless of threonine 68 phosphorylation. J Biol Chem.
277:48418–48426. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang H, Hu M, Zhao R, Li P and Li M:
Dihydromyricetin suppresses the proliferation of hepatocellular
carcinoma cells by inducing G2/M arrest through the
Chk1/Chk2/Cdc25C pathway. Oncol Rep. 30:2467–2475. 2013.PubMed/NCBI
|
|
42
|
Bahassi M, Myer DL, McKenney RJ, Hennigan
RF and Stambrook PJ: Priming phosphorylation of Chk2 by polo-like
kinase 3 (Plk3) mediates its full activation by ATM and a
downstream checkpoint in response to DNA damage. Mutat Res.
596:166–176. 2006. View Article : Google Scholar
|
|
43
|
Amir S, Ma AH, Shi XB, Xue L, Kung HJ and
Devere White RW: Oncomir miR-125b suppresses p14ARF to
modulate p53-dependent and p53-independent apoptosis in prostate
cancer. PLoS One. 8:e610642013. View Article : Google Scholar
|
|
44
|
Mavridis K, Gueugnon F, Petit-Courty A,
Courty Y, Barascu A, Guyetant S and Scorilas A: The oncomiR miR-197
is a novel prognostic indicator for non-small cell lung cancer
patients. Br J Cancer. 112:1527–1535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fletcher CE, Dart DA, Sita-Lumsden A,
Cheng H, Rennie PS and Bevan CL: Androgen-regulated processing of
the oncomir miR-27a, which targets Prohibitin in prostate cancer.
Hum Mol Genet. 21:3112–3127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vazquez F and Devreotes P: Regulation of
PTEN function as a PIP3 gatekeeper through membrane interaction.
Cell Cycle. 5:1523–1527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul
D, McMurray JS, Fang X, Yung WK, Siminovitch KA, et al: Src family
protein-tyrosine kinases alter the function of PTEN to regulate
phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem.
278:40057–40066. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shehata M, Schnabl S, Demirtas D, Hilgarth
M, Hubmann R, Ponath E, Badrnya S, Lehner C, Hoelbl A, Duechler M,
et al: Reconstitution of PTEN activity by CK2 inhibitors and
interference with the PI3-K/Akt cascade counteract the
antiapoptotic effect of human stromal cells in chronic lymphocytic
leukemia. Blood. 116:2513–2521. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Stocker H, Andjelkovic M, Oldham S,
Laffargue M, Wymann MP, Hemmings BA and Hafen E: Living with lethal
PIP3 levels: Viability of flies lacking PTEN restored by a PH
domain mutation in Akt/PKB. Science. 295:2088–2091. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yu J, Zhang SS, Saito K, Williams S,
Arimura Y, Ma Y, Ke Y, Baron V, Mercola D, Feng GS, et al: PTEN
regulation by Akt-EGR1-ARF-PTEN axis. EMBO J. 28:21–33. 2009.
View Article : Google Scholar :
|
|
51
|
Alyasiri NS, Mehdi SJ, Alam MS, Ali A,
Mandal AK, Gupta S, Singh I and Rizvi MM: PTEN-mediated AKT
activation contributes to the reduced apoptosis among Indian oral
squamous cell carcinoma patients. J Cancer Res Clin Oncol.
138:103–109. 2012. View Article : Google Scholar
|
|
52
|
Abbott RT, Tripp S, Perkins SL,
Elenitoba-Johnson KS and Lim MS: Analysis of the
PI-3-Kinase-PTEN-AKT pathway in human lymphoma and leukemia using a
cell line microarray. Mod Pathol. 16:607–612. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wen YG, Wang Q, Zhou CZ, Qiu GQ, Peng ZH
and Tang HM: Mutation analysis of tumor suppressor gene PTEN in
patients with gastric carcinomas and its impact on PI3K/AKT
pathway. Oncol Rep. 24:89–95. 2010.PubMed/NCBI
|
|
54
|
Hande KR: Etoposide: Four decades of
development of a topoisomerase II inhibitor. Eur J Cancer.
34:1514–1521. 1998. View Article : Google Scholar
|
|
55
|
Théard D, Coisy M, Ducommun B, Concannon P
and Darbon JM: Etoposide and adriamycin but not genistein can
activate the checkpoint kinase Chk2 independently of ATM/ATR.
Biochem Biophys Res Commun. 289:1199–1204. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fu X, Wan S, Lyu YL, Liu LF and Qi H:
Etoposide induces ATM-dependent mitochondrial biogenesis through
AMPK activation. PLoS One. 3:e20092008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wei F, Ojo D, Lin X, Wong N, He L, Yan J,
Xu S, Major P and Tang D: BMI1 attenuates etoposide-induced G2/M
checkpoints via reducing ATM activation. Oncogene. 34:3063–3075.
2015. View Article : Google Scholar
|