Introduction
Uterine cervical cancer is the third most common
cancer and the second leading cause of cancer death in women
between 20 and 39 years of age worldwide (1). The National Comprehensive Cancer
Network (NCCN) (the version 1.2014) guidelines recommend the
primary treatment for early-stage cervical cancer (FIGO IB and IIA)
as either surgery or chemo radiotherapy. The primary
chemoradiotherapy is recommended as the preferred treatment for
patients with high-risk positive lymph node (2). However, accurate and efficient
clinical detection methods for lymph node metastasis is difficult,
so many patients receive initial unnecessary surgery with adjuvant
chemoradiotherapy (3). The
combination of surgery and chemoradiotherapy carries the worse
morbidity, particularly long-term complications (4). Furthermore, pelvic lymph node
metastasis (PLNM) has been identified as the strongest key
prognostic parameter in cervical cancer, particularly early-stage
cervical cancer (5); therefore,
effective PLNM detection is essential to select an optimal
therapy.
In clinical practice, imaging diagnostics, including
magnetic resonance imaging (MRI), positron emission tomography
(PET), and computed tomography (CT) scans, are conventional methods
of PLNM detection before treatment. Nevertheless, additional
statistical analyses demonstrate that these methods have poor
sensitivity for detecting lymph node metastasis (6,7).
Recently, Sentinel LN (SLN) biopsy in early-stage cervical cancer
yields high diagnostic value for the status of lymph node (8), but this still need general anesthesia.
Thus, a non-invasive and more accurate evaluation method is
urgently needed.
Rapid advances in molecular biotechnology have
increased attention to biomacromolecules or small RNAs, including
microRNAs and short interfering RNAs. High-throughput technologies
have been devoted to identifying lymph node-associated biomarkers
at genomic levels (9) and the
protein (10). Several recent
studies have evaluated the gene expression profiles of PLMN in
uterine cervical cancers (11–13)
but neglected long non-coding RNAs (lncRNAs), which are longer than
200 nt and do not encode proteins. LncRNAs have emerged as
potentially powerful regulators involved in various biological
processes. Accumulating evidence indicates that aberrantly
expressed lncRNAs participate in the carcinogenesis and development
of malignant tumors through binding proteins or modulating other
short regulatory RNAs (14). In
addition, lncRNAs have been identified as oncogenic RNAs that
promote tumor cell invasion in uterine cervical cancer (15) and as independent predictors of
overall survival (16,17). Hence, lncRNAs are expected to be
excellent biomarkers for PLNM in cervical cancer.
In this present study, we used a data mining method
and bioinformatics analysis to determine the PLNM-associated lncRNA
profiles, which was fortuitously represented on the commonly used
microarray platform. Using bioinformatics software and methods, we
performed an initial exploration of the potential functional
enrichment and pathway mechanisms of these lncRNAs. The candidate
lncRNAs show promise to be biomarkers of diagnosis and prognosis in
cervical cancer. This study provides a new frontier for uncovering
the potential metastasis mechanisms to lymph node in uterine
cervical cancer.
Materials and methods
GEO gene expression data
The raw PLNM gene expression data and corresponding
related clinical parameters were downloaded from the publicly
available GEO (http://www.ncbi.nlm.nih.gov/geo/) including GSE26511
and partial GSE2109 data. GSE26511 comprises 20 cervical cancer
specimens without PLNM and 19 with PLNM. For these 39 samples,
primary treatment consisted of type 3 radical hysterectomy and
pelvic lymph node dissection (18).
GSE2109 summarized 2,158 samples included in the Expression Project
for Oncology (exp0). Based on TNM stage and primary tumor site, we
selected 20 cervical cancer specimens from GSE2109, including 15
without PLNM and 5 with PLNM. All samples in these two panels were
hybridized to Affymetrix human genome U133 plus 2.0
microarrays.
GeneChip Probe Re-annotation
Based on the lncRNA classification pipeline
constructed in a previous study (19), we identified a number of lncRNAs
represented on the Affymetrix microarrays. First, the latest
version of NetAffx Annotation File (HG-U133_Plus_2 Annotations, CSV
format, Release 34, 30 MB, 10/24/13) was obtained from the
Affymetrix official website. This annotation file was mapped to the
HG-U133_Plus_2 probe sets ID. Second, for the probe sets from the
Refseq database, those IDs beginning with 'NR' were retained, and
transcript IDs labeled with 'NP' were deleted. For the probe sets
from the Ensembl database, the online software BioMart was applied
to convert Affymetrix microarray IDs to Ensembl IDs together with
the corresponding gene type. We only retained genes annotated as
'lincRNA', 'sense_intronic', 'processed_transcript', 'anti-sense',
'sense_overlapping', '3prime_overlapping_ncrna', or 'misc_RNA'.
Next, based on the above two steps, we removed probe set IDs
annotated as 'microRNA', 'snoRNAs', ' pseudo-genes' and other small
RNAs.
Differential expression analysis
The expression level of lncRNAs is lower than that
of protein-coding RNAs, and the robust multichip average (RMA) has
higher detection efficiency for lncRNAs (20). Therefore, these raw CEL files were
background-adjusted, normalized, and log-transformed using RMA
rather than Microarray Analysis Suite 5.0 (MAS 5.0) using
RMAexpress software (Windows version 1.1.0, written by Ben
Bolstad). The differentially expressed probe sets were identified
using a parametric two-sample t-test (with a random variance model)
with a significance threshold of P<0.05 and validated using
permutation testing across samples in BRB-Array Tools v4.4.0 Beta 1
(http://linus.nci.nih.gov/BRB-ArrayTools.html). We also
entered the probe set data into Cluster3.0&TreeView (originally
developed by Michael Eisen, Stanford University) to process the
Hierarchical Clustering Analysis.
To further verify the outcome of the microarray
analysis, we adopted the training-validation strategy explored by
Michiels et al (21) to
classify the microarray-based datasets. This method indicated that
the percentage of misclassification would decrease as the number of
samples in the training set increased. Thus, we defined the data
set GSE26511 as the training group to identify the differentially
expressed lncRNAs and GSE2109 as the validation group to evaluate
misclassification.
Construction of the lncRNA-mRNA
coexpression network
The Pearson correlation coefficient (PCC) and
P-value were considered in the construction of an lncRNA-mRNA co
expression network (22). In view
of the above hierachical cluster analysis resulted from GSE26511
and significant changes, we selected top 20 differential expression
lncRNAs and 189 mRNAs (fold change >1.5) to form the
coexpression network based on the Pearson correlation coefficient
(PCC, PCC≥0.60, P<0.05). The PCC was calculated using the coding
and non-coding RNAs. For the same mRNA with different probe sets or
transcripts, we used the mean value as the final gene expression
value. The lncRNA-mRNA coexpression network was created using
Cytoscape software (v2.6.3).
GO and KEGG pathway analysis of
lncRNA-coexpressed mRNAs
Gene ontology (GO) and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathway analyses were performed using Molecule
Annotation System (MAS) V3.01 (CapitalBio Corp., Beijing, China).
MAS provides a series of comprehensive functional annotation tools
to elucidate the biological meaning of differentially expressed
genes (23). GO is the product of a
collaborative effort to address the need for consistent
descriptions of gene products among several databases and covers
the following three domains: Biological Process, Cellular Component
and Molecular Function. The Fisher's exact test P-value and
EASE-score were used to denote the significance of the GO
enrichment terms correlated with the conditions, and the Fisher
P-value was used for pathways. The lower the P-value, the more
significant the GO term or pathway (24).
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis (GSEA) is used to
interpret gene expression data by determining the statistical
significance of differences in predefined gene sets between
biological states (25). In
addition, GSEA can be used to find pathways that correlate to the
expression of the gene. To probe the biological mechanisms of the
differentially expressed lncRNAs MIR100HG and AC024560.2, all
coding-gene mRNAs and these two lncRNAs were separately used to
generate the expression data set. With the help of GSEA software
V2.1.0 (Broad Institute, MIT, Cambridge, MA, USA), we constructed a
32,619 (genes) x39 (samples) expression matrix to perform GSEA. The
predefined gene set 'c2.all.v4.0.symbols.gmt' is one of 7 major
collections from the Molecular Signatures Database (MSigDB). A
normalized enrichment score (NES) was calculated as the primary
statistic of GSEA. In addition, the chromosome location of the
lncRNAs MIR100HG and AC024560.2 was visualized using the UCSC
Genome Browser (http://genome.ucsc.edu/).
Patients and tumor specimens
A total of 35 samples of fresh cervical cancer
tissues were randomly collected from patients who underwent surgery
at the First Affiliated Hospital of Sun Yat-sen University from May
2013 to December 2014 and provided written informed consent. All
surgical specimens were immediately frozen in liquid nitrogen and
stored at −80°C until RNA extraction. The clinical study was
approved by the Medical Ethics Committees at the First Affiliated
Hospital of Sun Yat-sen University. All samples were confirmed
pathologically, and the clinical characteristics are presented in
Table I.
 | Table IClinicopathological characteristics
of early-stage cervical cancer patients included in the qRT-PCR
analysis. |
Table I
Clinicopathological characteristics
of early-stage cervical cancer patients included in the qRT-PCR
analysis.
| No. | Age (years) | FIGO stage | Tumor size
(cm) | Number of Positive
PLN | Number of removed
PLN | LVSI |
Differentiation | Pathological
type | Stromal
invasion |
|---|
| Early-stage
cervical cancer with PLNM |
| 1 | 58 | Ib1 | 1.5 | 1 | 38 | Negative | G2 | SCC | >1/2 |
| 2 | 50 | Ib2 |
2 | 5 | 21 | Negative | G2 | SCC | <1/2 |
| 3 | 52 | Ib1 |
1 | 1 | 56 | Positive | G1 | SCC | <1/2 |
| 4 | 42 | IIa2 |
5 | 2 | 19 | Negative | G1 | SCC | >1/2 |
| 5 | 35 | IIa2 |
5 | 2 | 15 | Negative | G1 | SCC | >1/2 |
| 6 | 59 | IIa1 |
2 | 2 | 21 | Negative | G2 | SCC | >1/2 |
| 7 | 62 | IIa1 |
3 | 3 | 22 | Negative | G1 | SCC | >1/2 |
| 8 | 46 | Ib1 |
4 | 3 | 38 | Positive | G1 | SCC | >1/2 |
| 9 | 48 | IIa1 |
4 | 1 | 36 | Negative | G1 | SCC | >1/2 |
| 10 | 58 | IIa1 |
3 | 1 | 53 | Negative | G1 | SCC | >1/2 |
| 11 | 35 | IIa1 |
2 | 1 | 72 | Positive | G1 | SCC | >1/2 |
| 12 | 43 | IIa1 |
2 | 2 | 21 | Negative | G2 | SCC | <1/2 |
| 13 | 43 | Ib1 |
3 | 2 | 17 | Positive | G1 | SCC | <1/2 |
| 14 | 49 | Ib2 |
5 | 4 | 16 | Positive | G2 | SCC | >1/2 |
| 15 | 34 | IIa1 |
1 | 2 | 17 | Positive | G1 | SCC | >1/2 |
| Early-stage
cervical cancer without PLNM |
| 1 | 50 | Ib1 |
3 | 0 | 17 | Negative | G1 | SCC | <1/2 |
| 2 | 56 | Ib1 |
1 | 0 | 33 | Negative | G3 | SCC | >1/2 |
| 3 | 44 | Ib1 |
3 | 0 | 9 | Negative | G2 | SCC | >1/2 |
| 4 | 48 | Ib2 | 1.3 | 0 | 41 | Negative | G2 | SCC | <1/2 |
| 5 | 46 | IIa1 | 2.5 | 0 | 44 | Negative | G1 | SCC | >1/2 |
| 6 | 54 | IIa1 | 2.5 | 0 | 36 | Negative | G2 | SCC | >1/2 |
| 7 | 37 | Ib1 |
2 | 0 | 20 | Negative | G2 | SCC | <1/2 |
| 8 | 39 | Ib1 |
3 | 0 | 22 | Negative | G1 | SCC | <1/2 |
| 9 | 47 | Ib1 |
2 | 0 | 38 | Negative | G1 | SCC | >1/2 |
| 10 | 50 | IIa1 |
2 | 0 | 21 | Negative | G2 | SCC | >1/2 |
| 11 | 52 | Ib1 |
3 | 0 | 22 | Negative | G2 | SCC | >1/2 |
| 12 | 36 | Ib2 |
5 | 0 | 21 | Negative | G1 | SCC | <1/2 |
| 13 | 43 | Ib1 |
2 | 0 | 16 | Negative | G1 | SCC | <1/2 |
| 14 | 40 | IIa1 | 2.5 | 0 | 12 | Negative | G1 | SCC | >1/2 |
| 15 | 41 | Ib1 |
3 | 0 | 19 | Negative | G1 | SCC | <1/2 |
| 16 | 40 | Ib1 |
2 | 0 | 25 | Negative | G2 | SCC | >1/2 |
| 17 | 40 | IIa1 |
2 | 0 | 15 | Negative | G1 | SCC | >1/2 |
| 18 | 38 | Ib1 |
3 | 0 | 5 | Negative | G1 | SCC | <1/2 |
| 19 | 52 | Ib1 |
4 | 0 | 20 | Negative | G1 | SCC | >1/2 |
| 20 | 36 | Ib1 |
2 | 0 | 15 | Negative | G2 | SCC | <1/2 |
RNA extraction and quantitative real-time
polymerase chain reaction (qRT-PCR) validation
Total RNA from fresh frozen tissues was extracted
using RNAiso Plus reagent (Takara, Dalian, China), and
complementary DNA was reverse-transcribed using PrimeScript RT
Master Mix (Takara) according to the manufacturer's instructions.
Quantitative real-time polymerase chain reaction (qRT-PCR) was
performed with SYBR Premix Ex Taq (Takara). All qRT-PCRs were
performed in a 7500 Fast Real-time PCR System (Applied Biosystems,
Carlsbad, CA). The RNA primers used in qPCR are presented in
Table II.
| Table IIRNA primer sequences used in our
study. |
Table II
RNA primer sequences used in our
study.
| Primer | Sequence (5′ to
3′) |
|---|
| LINC01139 | F:
TTCTCTCACCCTTCAAACAGC |
| R:
ACCAAAGATGTCGCAGGACT |
| MIR100HG | F:
GGCGACATCAGACAGACAGA |
| R:
AGGACCAGCTGAAAGGAACA |
| AC024560.2 | F:
TGGGTCGCTCTGTATCTCTG |
| R:
CGGTGGCTGTGAGTATGAAG |
| LINC01503 | F:
TGGATTTTCATGCCTGCTG |
| R:
GGCTGCATTACCAGAAAGGT |
TCGA cohorts
In order to indicate the prognosis significance of
MIR100HG in early-stage cervical cancer, we used the MIR100HG
expression data and clinical information from the Cancer Genome
Atlas (TCGA, https://tcga-data.nci.Nih.gov/tcga/). Patients without
intact follow-up and pathological data were excluded. The clinical
data of all 131 patients from TCGA cohort are shown in Table III.
| Table IIIClinical parameters of TCGA cohort
enrolled in our study. |
Table III
Clinical parameters of TCGA cohort
enrolled in our study.
| Parameters | Data |
|---|
| Total number of
patients | 131 |
| Age (years) | |
| Median | 45 |
| Range | 20–80 |
| FIGO stage | |
| IA | 3 |
| IB | 111 |
| IIA | 17 |
| Overall
survival | |
| Deaths | 23 (17.5%) |
| Follow-up
(years) | |
| Median | 0.65 |
| Range | 0–11.6 |
Statistical analysis
Statistical analyses were performed using IBM SPSS
Statistics 19.0 for Windows (Released 2010; IBM Corp., Armonk, NY,
USA). Receiver operating characteristic (ROC) analysis was
performed to assess the sensitivity and specificity of the measured
markers. The cut-point of MIR100HG expression was defied as the
median. Kaplan-Meier and the two-sided log-rank test were used to
calculate the survival curves. Significance was defined at a
P-value of <0.05.
Results
Gene expression data characteristics
The GSE26511 and GSE2109 series were used in our
study. To ensure consistent clinical data of patients included in
these two series, we listed the age and FIGO stage of patients in
Table IV. Primary analysis was
focused on GSE26511, which was a published dataset consisting of
the largest number of specimens. The entire workflow of this
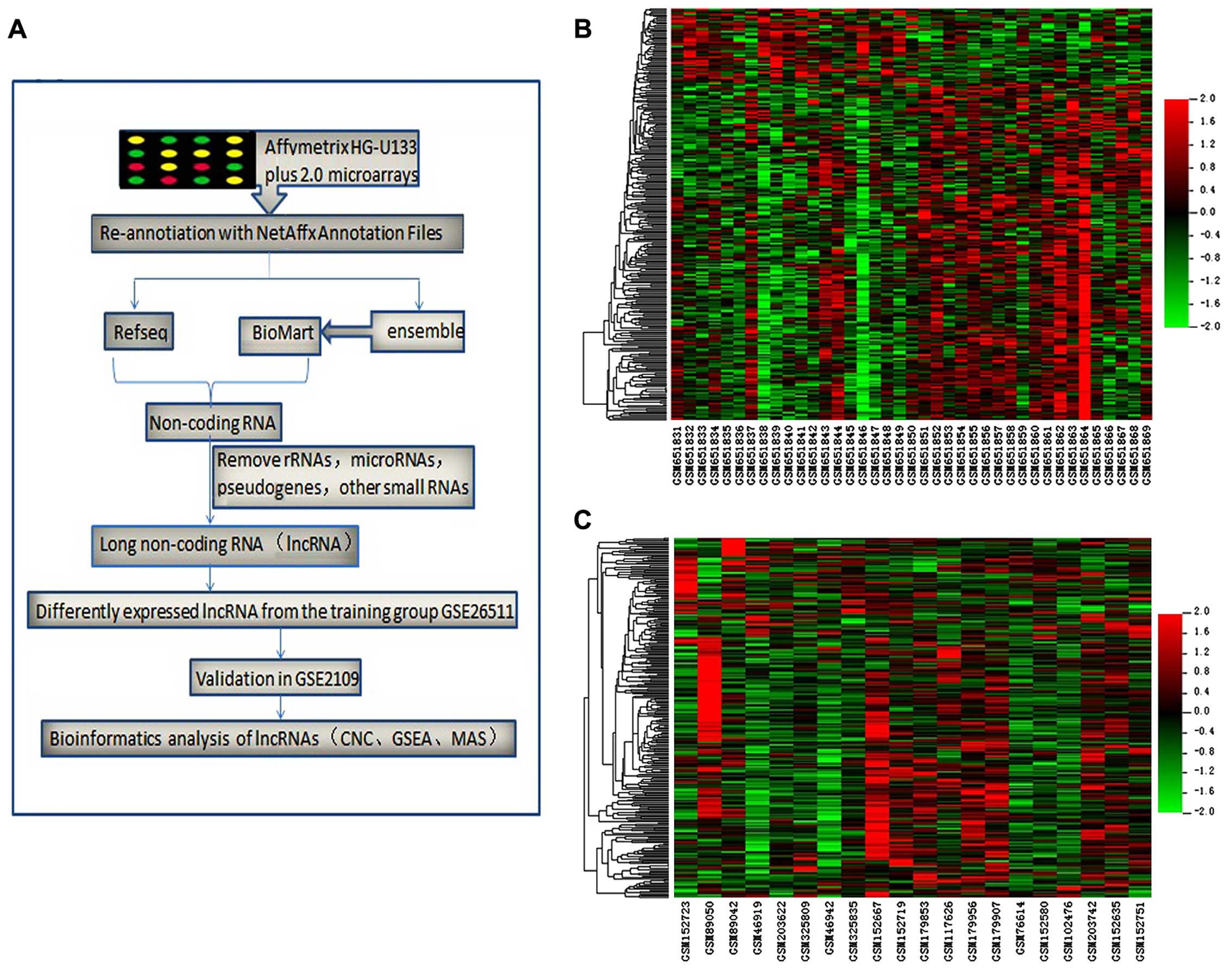
process is presented in Fig.
1A.
| Table IVCharacteristics of the gene
expression data covariates (all squamous cell carcinoma). |
Table IV
Characteristics of the gene
expression data covariates (all squamous cell carcinoma).
| GSE26511 | Pelvic lymph
nodes | Age at
diagnosis | FIGO stage | GSE2109 | Pelvic lymph
nodes | Age at
diagnosis | FIGO stage |
|---|
| GSM651831 | Negative | 56.4 | 1b1 | GSM46919 | Negative | 60–70 | 1b1 |
| GSM651832 | Negative | 45.8 | 1b1 | GSM46942 | Negative | 30–40 | 1b1 |
| GSM651833 | Negative | 49.5 | 1b1 | GSM117626 | Negative | 60–70 | 1b1 |
| GSM651834 | Negative | 34.7 | 2a | GSM152580 | Negative | 20–30 | 1b2 |
| GSM651835 | Negative | 55.5 | 1b1 | GSM152635 | Negative | 20–30 | 1b1 |
| GSM651836 | Negative | 38.5 | 1b1 | GSM152667 | Negative | 30–40 | 1b1 |
| GSM651837 | Negative | 34.9 | 1b1 | GSM152719 | Negative | 30–40 | 1b1 |
| GSM651838 | Negative | 47.4 | 1b1 | GSM179853 | Negative | 30–40 | 1b1 |
| GSM651839 | Negative | 42.3 | 1b1 | GSM179907 | Negative | 50–60 | 1b1 |
| GSM651840 | Negative | 35.8 | 1b2 | GSM179956 | Negative | 30–40 | 1b1 |
| GSM651841 | Negative | 51.6 | 2a | GSM203742 | Negative | 50–60 | 1b1 |
| GSM651842 | Negative | 72 | 1b2 | GSM325809 | Negative | 40–49 | 1b1 |
| GSM651843 | Negative | 71 | 1b2 | GSM325835 | Negative | 30–39 | 1b1 |
| GSM651844 | Negative | 35.9 | 1b2 | GSM89042 | Negative | 40–50 | 1b1 |
| GSM651845 | Negative | 68.9 | 2a | GSM102476 | Negative | 40–50 | 1b1 |
| GSM651846 | Negative | 47.4 | 1b2 | GSM76614 | Positive | 40–50 | 1b1 |
| GSM651847 | Negative | 31.5 | 1b1 | GSM89050 | Positive | 30–40 | 1b1 |
| GSM651848 | Negative | 72.7 | 2a | GSM152723 | Positive | 50–60 | 2a |
| GSM651849 | Negative | 39.9 | 1b1 | GSM152751 | Positive | 40–50 | 1b2 |
| GSM651850 | Negative | 50.7 | 1b1 | GSM203622 | Positive | 50–60 | 1b2 |
| GSM651851 | Positive | 56.2 | 1b1 | | | | |
| GSM651852 | Positive | 29.1 | 1b1 | | | | |
| GSM651853 | Positive | 32.2 | 2a | | | | |
| GSM651854 | Positive | 60.6 | 1b1 | | | | |
| GSM651855 | Positive | 49.9 | 2a | | | | |
| GSM651856 | Positive | 34.9 | 1b2 | | | | |
| GSM651857 | Positive | 32.7 | 1b2 | | | | |
| GSM651858 | Positive | 40.4 | 1b1 | | | | |
| GSM651859 | Positive | 48.5 | 1b2 | | | | |
| GSM651860 | Positive | 37.4 | 1b1 | | | | |
| GSM651861 | Positive | 37 | 1b2 | | | | |
| GSM651862 | Positive | 32 | 1b1 | | | | |
| GSM651863 | Positive | 37.4 | 1b1 | | | | |
| GSM651864 | Positive | 45.5 | 1b2 | | | | |
| GSM651865 | Positive | 72.5 | 1b1 | | | | |
| GSM651866 | Positive | 42.3 | 1b1 | | | | |
| GSM651867 | Positive | 46.3 | 1b1 | | | | |
| GSM651868 | Positive | 34.2 | 1b2 | | | | |
| GSM651869 | Positive | 50.5 | 2a | | | | |
Differentially expressed lncRNA
profile
We identified 3,432 probe sets (matching with 2,803
lncRNAs) via re-annotation of the Affymetrix human genome U133 plus
2.0 microarrays based on the Refseq and Ensembl databases. For the
GSE26511 training group, 249 probe sets (matching with 234 lncRNAs)
that were differentially expressed in cervical cancer specimens of
varying lymph node metastasis status were identified using the
method described above (data not shown). Of these 249 probe sets,
40 probe sets (31 lncRNAs) were upregulated in cervical cancer
tissues without PLNM compared to the lymph node metastasis group,
and 209 probe sets (203 lncRNAs) were downregulated. Hierarchical
clustering maps of all samples from the GSE26511 training group and
the GSE2109 validation group were constructed from these
differentially expressed lncRNAs. GSE2109 was used to reduce the
error due to the collection of specimens from cervical cancer
tissues with or without lymph node metastasis (Fig. 1B and C).
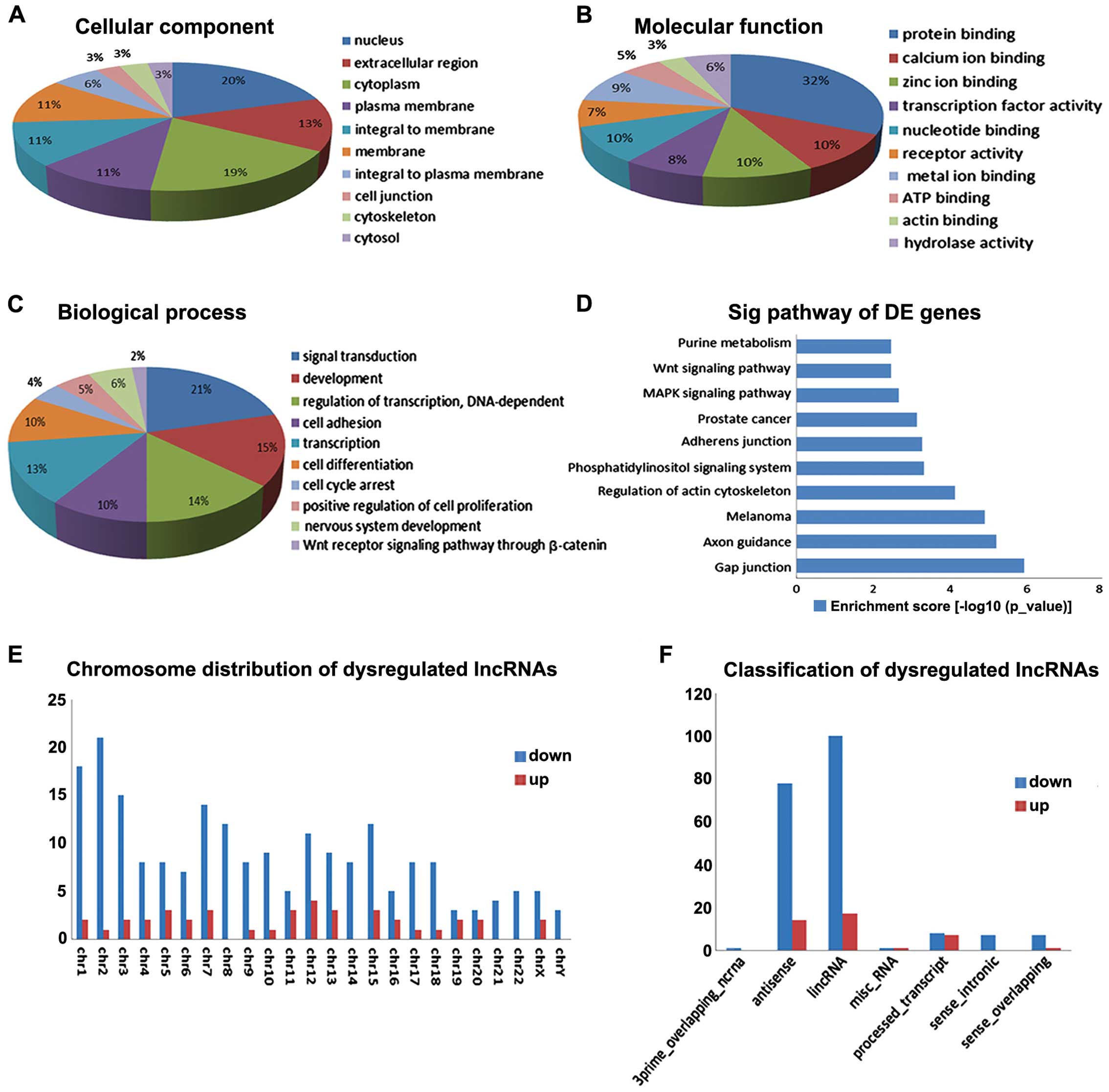
LncRNA classification and
distribution
The genetic location of biomolecules plays an
important role in diverse biological and molecular functions
(14). Using the UCSC genome
browser and Ensembl database, the differentially expressed lncRNAs
were characterized as lincRNA, 3prime_overlapping_ncRNA, antisense,
processed_transcript, and sense_intronic.sense_overlapping based on
the correlation between lncRNAs and their associated coding genes.
A total of 100 and 17 lncRNAs were included in the up- and
down-regulated lncRNA classifications, respectively. Each
chromosome had different numbers of up- and down-regulated lncRNAs
(Fig. 2E and F).
The overview of mRNA profile
Similar to the lncRNA analysis method, 2,980 probe
sets (matching 228 lncRNAs) were identified as differentially
expressed in cervical cancer specimens with PLNM. Among these
mRNAs, 1,641 probe sets (matching 1,170 mRNAs) were upregulated in
cervical cancer tissues without PLNM compared to the lymph node
metastasis group, and 1,339 probe sets (matching with 1,111 mRNAs)
were downregulated. In the subsequent analysis, we selected 335
mRNAs with a differential expression fold change >1.5 to
construct a clear and significant network.
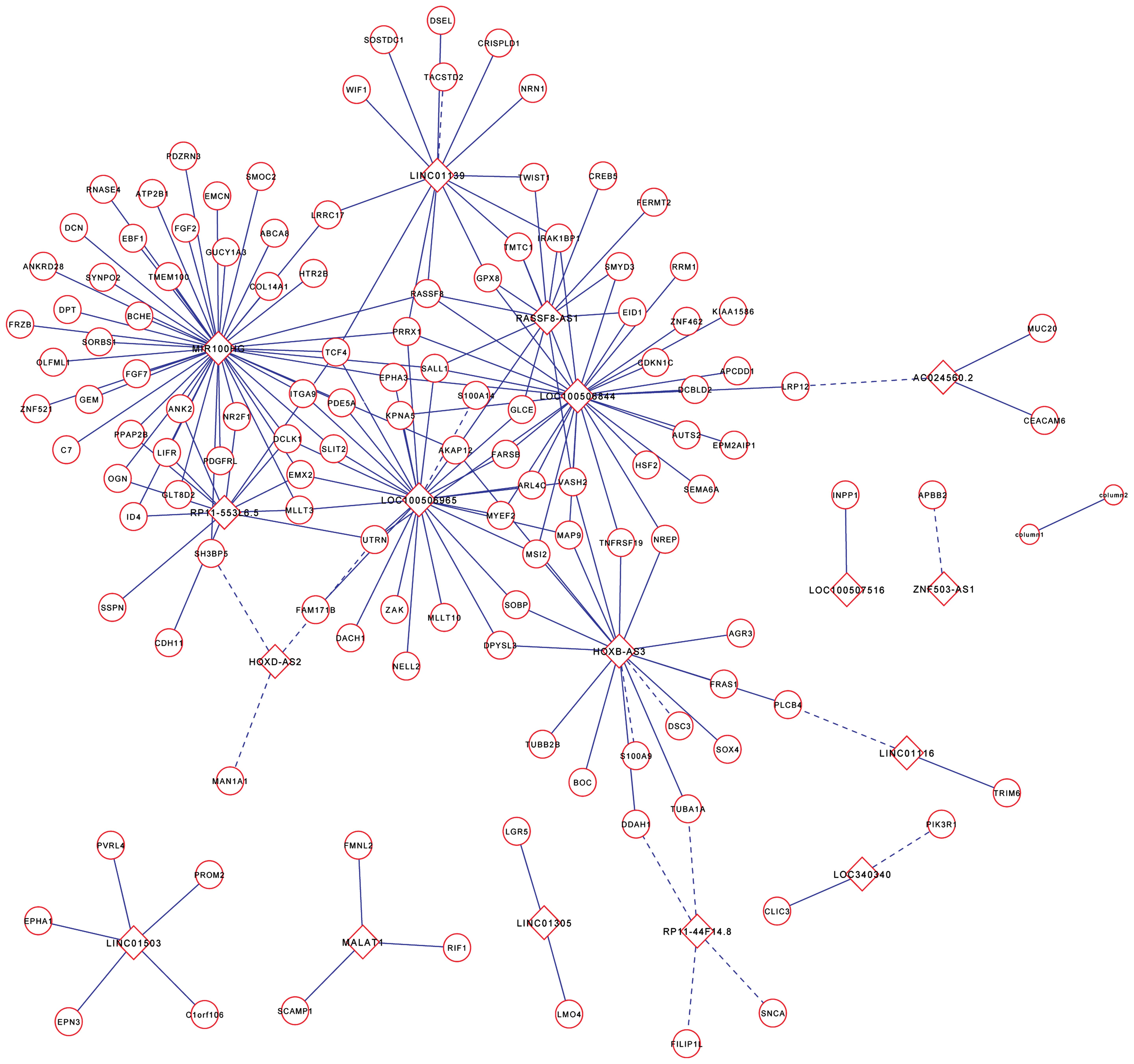
LncRNA-mRNA coexpression network
Although lncRNA functions have been implicated in
many diseases, the biological functions of a large proportion of
lncRNAs remain unknown. The tumor metastasis is a multifactor,
multistep and multigene interactive process. Affymetrix human
genome U133 plus 2.0 microarrays provided the expression levels of
lncRNAs and mRNAs, and we constructed an lncRNA-mRNA coexpression
network to interpret the potential biological roles of
PLNM-associated lncRNAs in early-stage cervical cancer (26). The coexpression network revealed
that one mRNA could target several lncRNAs and vice versa,
suggesting a potential role of lncRNA and mRNA interactions in the
process of lymph node metastasis in cervical cancer (Fig. 3).
GO and pathway analysis
To further evaluate the differentially expressed
lncRNAs, we performed GO and pathway analyses of mRNAs co-expressed
with lncRNAs. For biological process (Fig. 2C), i) signal transduction; ii)
development; iii) regulation of transcription, DNA dependence; iv)
cell adhesion; v) transcription; vi) cell differentiation; vii)
cell cycle arrest; viii) positive regulation of cell proliferation;
ix) nervous system development; x) and the Wnt receptor signaling
pathway through β-catenin were involved. For cellular component and
molecular function, nucleus and protein binding were included
separately (Fig. 2A and B). KEGG
pathway analysis indicated that the mRNAs co-expressed with the
lncRNAs were mainly involved in the following: i) Gap junction; ii)
Axon guidance; iii) melanoma; iv) Regulation of actin cytoskeleton;
v) Phosphatidylinositol signaling system; vi) Adherens junction;
vii) Prostate cancer; viii) MAPK signaling pathway; ix) Wnt
signaling pathway; x) and Purine metabolism (Fig. 2D). The gap junction pathway plays a
physiologically relevant regulator role in cervical tumor cells
(27). We also identified the Wnt
receptor signaling pathway through β-catenin, in agreement with the
results of Noordhuis et al (18). These data imply that PLNM-lncRNAs
may contribute to the lymph node metastasis process through the
above mechanisms and pathways.
Bioinformatics analysis of
lncRNA-MIR100HG and AC024560.2
To identify lncRNAs that play an important role in
lymph node metastasis in early-stage cervical cancer, we analyzed
the detailed information for two of the top lncRNAs from the
upregulated group and downregulated groups (Table V). The lncRNA linc01503 had 4
transcripts, increasing the uncertainty of the primer and
transcription products. MIR100HG was co-expressed with more coding
genes than linc01139 in the upregulated group. In light of these
findings, we selected MIR100HG and AC024560.2 as promising lncRNAs
for further investigation.
| Table VThe top four differentially expressed
lncRNAs in cervical cancer specimens with lymph node
metastasis. |
Table V
The top four differentially expressed
lncRNAs in cervical cancer specimens with lymph node
metastasis.
| ProbeSet | Gene symbol | Regulation in
N+ | Fold-change | Parametric
P-value | Co-expression
coding genes | Transcripts | Biotype | bp | Chr |
|---|
| 235599_at | LINC01139 | Down | 2.05 | 0.0001141 | 14 | 1 | lincRNA | 1540 | Chr1 |
| 225381_at | MIR100HG | Down | 1.96 |
0.002493 | 47 | 1 |
sense_overlapping | 3082 | Chr11 |
| 238804_at | AC024560.2 | Up | 1.61 | 0.0002909 | 3 | 1 | lincRNA | 1471 | Chr3 |
| 229296_at | LINC01503 | Up | 1.50 | 0.0099363 | 5 | 4 | lincRNA | 901 | Chr9 |
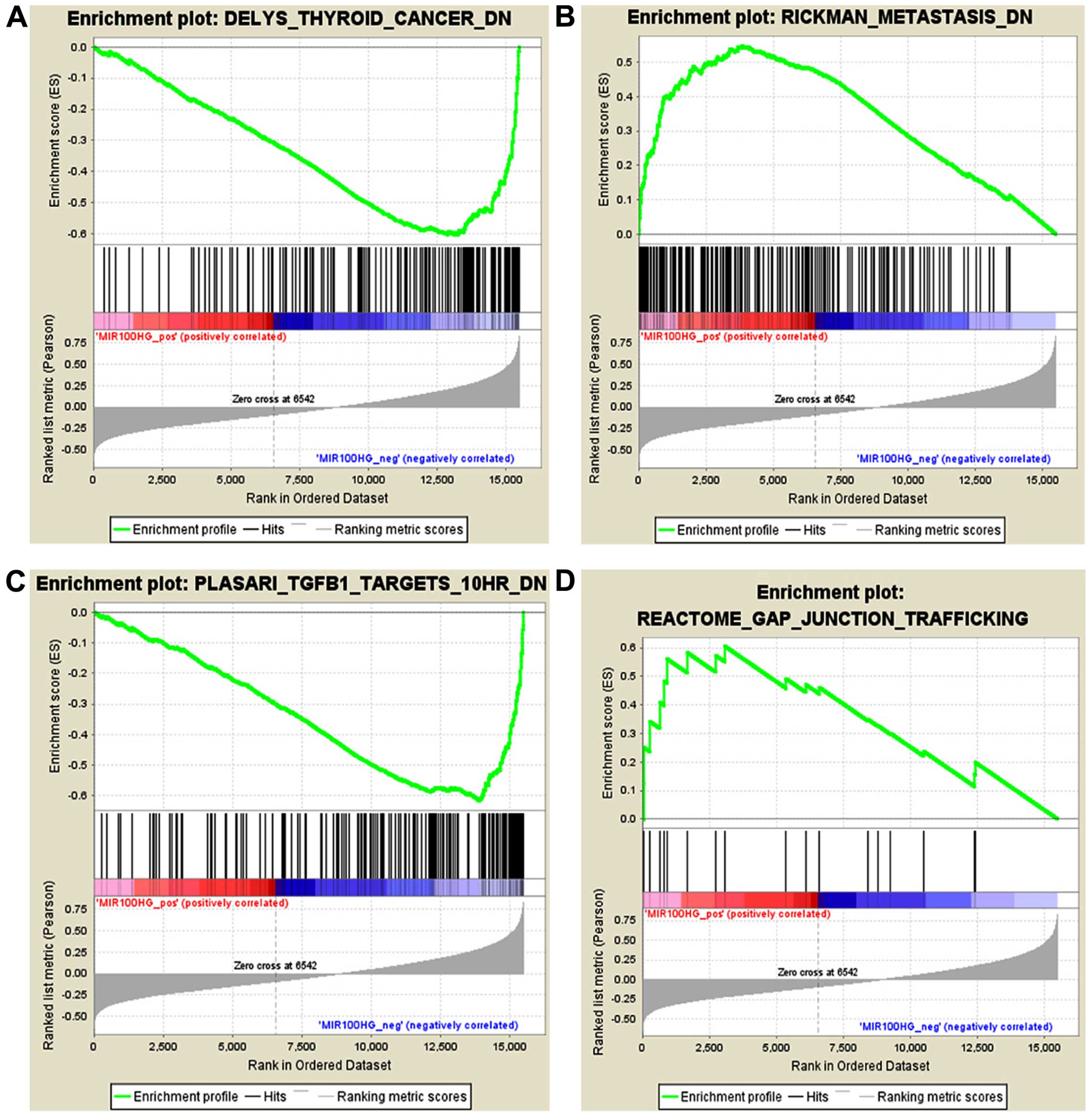
The mir-100-let-7a-2 cluster host gene, MIR100HG, is
located on chromosome 11q24.1 and is a regulator of hematopoiesis
and oncogenes in the development of myeloid leukemia (28). We performed GSEA using the
expression level of MIR100HG and the entire mRNA expression
dataset. Based on the GSEA results, we identified 1,127 and 2,362
gene sets that were positively or negatively associated,
respectively, with MIR100HG among the 3,489 'curated gene sets'.
The curated gene sets were collected from various sources such as
online pathway databases, publications in PubMed, and knowledge of
domain experts. Gene sets with the highest NES in the positive and
negative correlation groups included RICKMAN_METASTASIS_DN and,
DELYS_THYROID_ CANCER_DN, respectively (Fig. 4A and B). We also estimate that
MIR100HG participates in the gap junction pathway and TGF-β
pathway, with NES values of 1.6079853 and −2.2482386, respectively
(Fig. 4C and D). Disruption of the
TGF-β/Smad signaling pathway may be conducive to the malignant
progression of cervical dysplasia in human cervical cancer
(29). These analysis results are
consistent with our earlier data and further indicate that MIR100HG
may participate in the regulation of lymph node metastasis in
early-stage cervical cancer through several pathways.
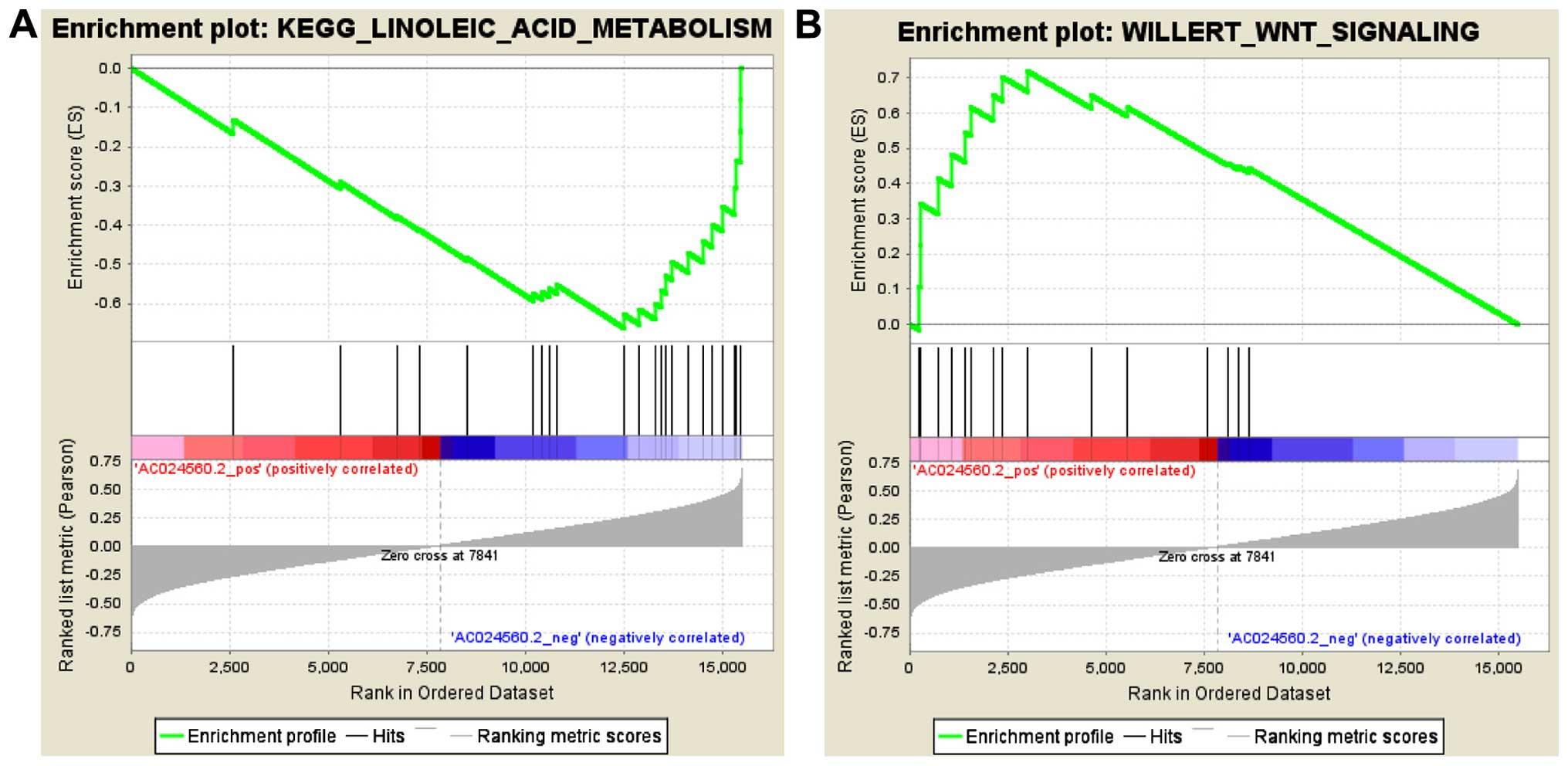
AC024560.2 is an lncRNA located on chromosome 3 and
is described as Homo sapiens long non-coding RNA
OTTHUMT00000340266.1. We used the expression level of AC024560.2 in
GSEA using the entire mRNA expression dataset. GSEA resulted in the
identification of 2,823 and 666 gene sets that were positively or
negatively associated, respectively, with AC024560.2 among the
3,489 'curated gene sets'. Gene sets with the highest NES in the
positive and negative correlation groups included
WILLERT_WNT_SIGNALING (NES=1.9029536) and
KEGG_LINOLEIC_ACID_METABOLISM (NES= −1.7962813), respectively
(Fig. 5A and B). A review of the
literature revealed that the activation of the Wnt/β-catenin
pathway promotes proliferation and tumor formation in cervical
cancer cells (30), consistent with
the GSEA report.
Clinical relevance
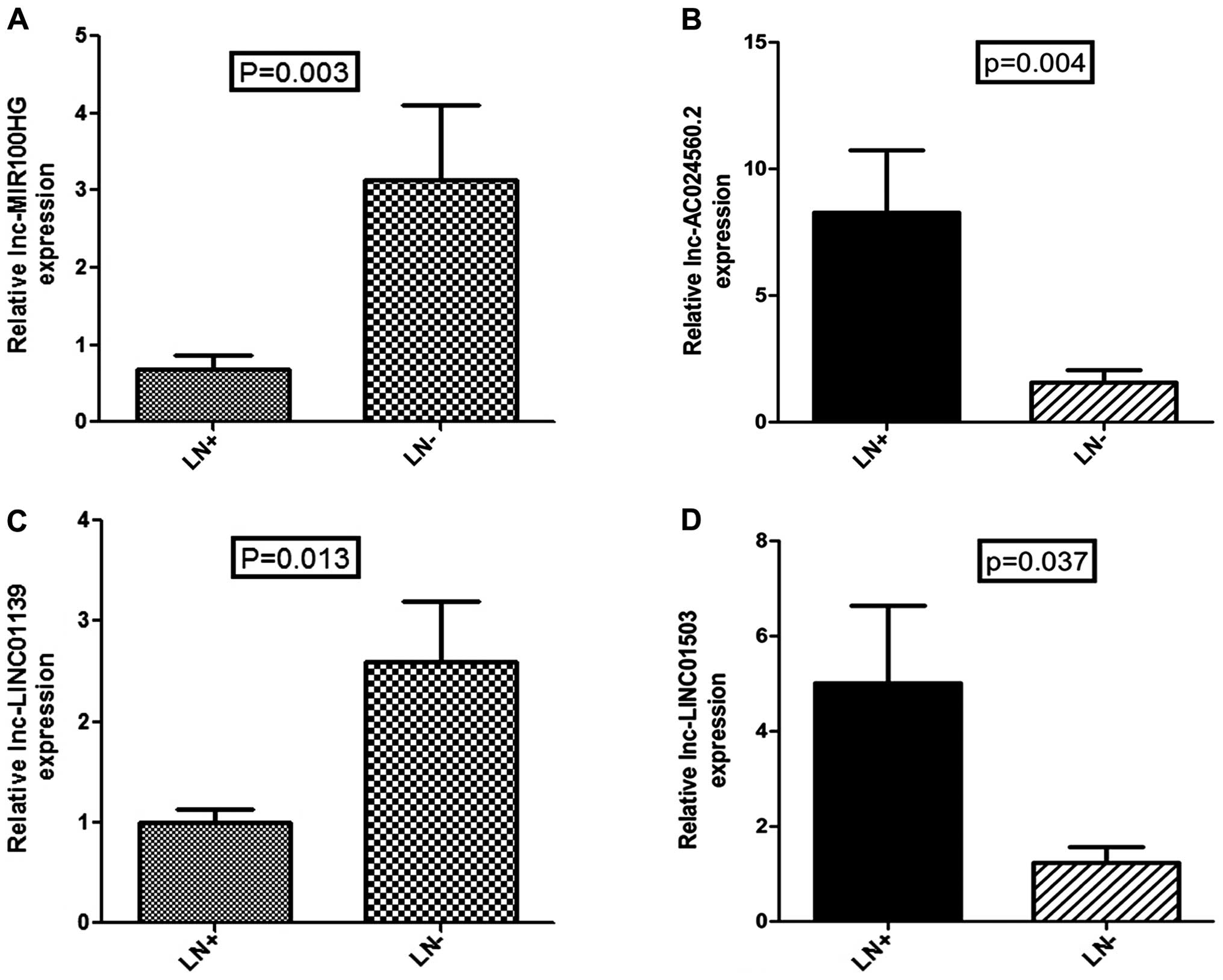
To validate the results of data mining and determine
the clinical relevance of lncRNA dysregulation, we detected the
expression levels of the above four lncRNAs in 35 clinical tissues
with and without PLNM by qRT-PCR (Fig.
6). The qRT-PCR result was consistent with our analysis, in
that expression of all 4 lncRNAs had statistical difference with
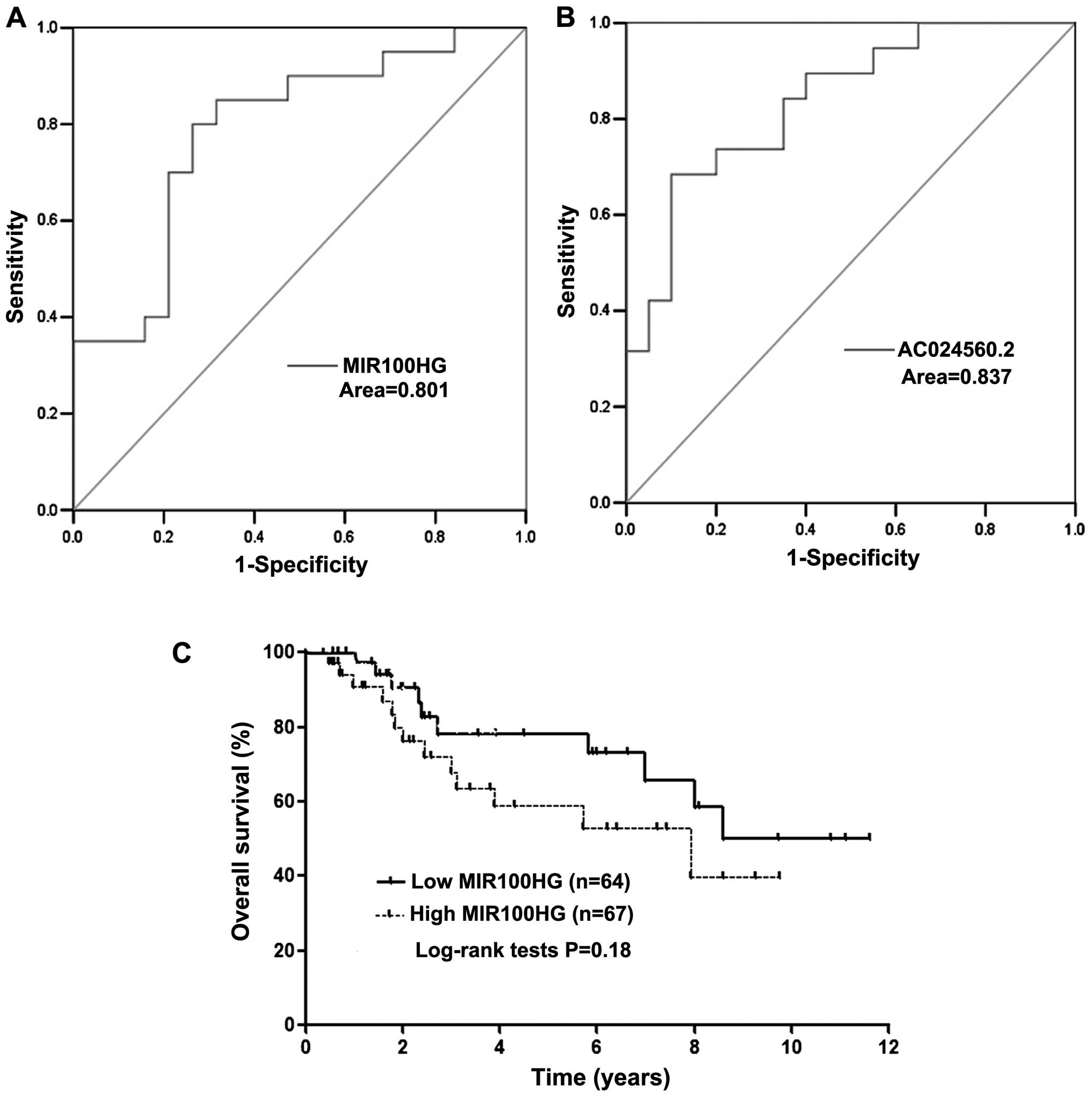
the same trend (P<0.05). Next, we used existing data to access
the discriminatory power for lncRNA MIR100HG and AC024560.2 by
constructing ROC curves. Areas under ROC curves of two lncRNA
signatures were 0.801 and 0.837, respectively (Fig. 7A and B). This suggested that
MIR100HG and AC024560.2 achieve a fine diagnostic accuracy in
diagnosing lymph node metastasis of cervical cancer. According to
the TCGA cohort, the Kaplan-Meier analysis demonstrated that the
patients with lncRNA MIR100HG high expression had poor prognosis
(Fig. 7C). The prognosis value of
AC024560.2 is not shown due to the lack of exact genetic match
information in TCGA cohort.
Discussion
PLNM has critically significant implications for
individual therapy in early-stage cervical cancer. The evaluation
of PLNM status using an accurate, noninvasive method is a major
focus of research. As part of the development of a molecular
diagnosis for PLNM, various types of proteins and microRNAs that
participate in the regulation of PLNM through various pathways have
been identified. However, the clinical application of these
molecules has been limited by sensitivity, sample size, regions and
other factors. LncRNAs are emerging as crucial regulators of
genomic activity and the expression of protein-coding genes and
other non-coding transcripts, including microRNAs (14). A role of lncRNAs in oncogenesis,
tumor progression, tumor cell apoptosis and cell period arrest has
been confirmed in several human cancers (31). Therefore, lncRNA function may be
relevant to predicting PLNM. The present study is the first to
comprehensively investigate the lncRNA expression profile
associated with PLNM in early-stage cervical cancer patients.
Although gene expression chip and protein
microarrays have been widely used by researchers, the design of
lncRNA expression microarrays is not mature. Therefore, mining
lncRNA information from the most commonly used commercial
microarrays in human cancer profiling, including the Affymetrix
HG-U133 Plus 2.0 array, is an important supplement for this field.
This approach is accurate, feasible, and low cost and has been
adopted in several studies (19,32).
Herein, we used this method for reference and successfully mined
the ideal lncRNA expression profile associated with PLNM.
After screening, 2,803 lncRNA transcripts were
filtered from the Affymetrix microarray, and 234 lncRNAs that were
significantly associated with pelvic lymph node metastasis were
identified. We validated these results by qRT-PCR. Using the
lncRNA-mRNA CNC network and molecular analysis system, we assessed
the major biological functions and molecular mechanisms in which
PLNM-associated lncRNAs might be involved. lncRNA MIR100HG and
AC024560.2 were further analyzed based on their location,
co-expression with coding genes, gene set enrichment and clinical
discriminatory power.
In our MAS report, the enrichment score of gap
junctions was highest. Gap junctions are a specialized
intercellular connection between two cells composed of proteins
from the connexin family in vertebrates. Gap junctions allow
various molecules, ions and electrical impulses to directly pass
through the channel (33). The
relationship between the aberrant expression of the gap junction
protein connexin and lymph node metastasis has been verified for
several cancers, including ovarian adenocarcinoma, human ductal
breast cancer, colorectal cancer, and oral squamous cell carcinoma.
Some membrane connexins are independent prognostic factors in
various cancers. In uterine cervical cancers, some connexin
proteins that mediate gap junction intercellular communication have
been associated with carcinogenesis and tumor progression (34). The expression levels of gap junction
beta-3 protein (GJA1) and gap junction alpha-10 protein (GJA10)
were also significantly different in our mRNA analysis (P<0.05).
These findings provide a theoretical basis to further explore this
biological pathway.
The lncRNA MIR100HG was originally identified as
highly expressed in acute megakaryoblastic leukemia (AMKL).
MIR100HG acts as a mediator of hematopoiesis and oncogenes in the
progression of AMKL, an aggressive form of hematological cancer
(28). Moreover, there is an
intronic coding region (BLID) in MIR100HG gene, which functions as
a proapoptotic molecule through a caspase-dependent mitochondrial
pathway of cell death (35). This
activity provides a starting point to explore the expression
pattern of MIR100HG in cervical cancer. GSEA revealed that the gene
set RICKMAN_METASTASIS_UP had a higher normal enrichment score and
positive correlation with the profile. This result confirms the key
value of MIR100HG in cancer lymph node metastasis. Furthermore,
MIR100HG is closely correlated with the gap junction and TGF-β
pathways. Most of the mRNAs that were co-expressed with MIR100HG
participate in the gap junction pathway. The TGF-β pathway is an
important PLNM-associated pathway in cervical cancer and was
analyzed by Noordhuis et al (18). Thus, our findings are consistent
with previous research results. We explained the biological pathway
of PLNM in cervical cancer from a novel molecular mechanism
perspective and established a more comprehensive understanding
based on molecular profile information.
lncRNA-AC024560.2 is a novel lncRNA that has not
been previously associated with cancer. GSEA indicated an
association of AC024560.2 with the linoleic acid metabolism
pathway. Downregulation of peroxisome proliferator-activated
receptors (PPARs) acting as nuclear receptors for linoleic acid
metabolites participating in the apoptosis signaling pathway in
colorectal cancer was reported as early as 2003 (36). The role of the Wnt signaling
pathway, which is related to AC024560.2, was reported previously in
cancer (18). Therefore, as
regulators of biological function for certain proteins and
microRNAs, these lncRNAs may play an important and valuable role in
PLNM of early-stage cervical cancer. To increase the clinical
significance of our findings, we analyzed the diagnostic capacity
of MIR100HG and AC024560.2 using ROC curves. Our results provide
strong evidence for the prediction of lymph node status in cervical
cancer using these two lncRNAs.
However, there are limitations to our study. First,
the sample size of the GSE2109 validation group was small, and thus
hierarchical clustering was less obvious. Second, the lncRNAs
selected here might not represent the entire lncRNA profile
involved in PLNM, because the Affymetrix human genome U133 plus 2.0
microarrays did not include all the lncRNAs present. Third, only
131 patients of early-stage cervical squamous cell carcinoma from
TCGA cohort were accorded with the inclusive criteria, so that the
difference between survival curves was not significant. In
addition, only one pathological type of cervical cancer (squamous
cell cancer) was studied because of the limitation of GEO data.
Finally and unfortunately, we had no more experimental evidence to
further prove our analysis report. The focus of our study was the
value of bioinformatics analysis in addressing important clinical
topics and discovering potential role of lncRNAs in lymph node
metastasis.
In summary, we successfully identified 234
differentially expressed PLNM-associated lncRNAs in early-stage
cervical cancer using the data mining method. The qRT-PCR was
carried out to further detect the lncRNA expression patterns in
clinical cervical cancer tissues. Using the LncRNA-mRNA
Coexpression network, we detailed the possible function of these
lncRNAs from different perspectives, including molecular mechanism
and biological pathways. Two promising lncRNAs MIR100HG and
AC024560.2 were evaluated using GSEA reports and other databases to
uncover their location, biological function and discrimination
power. Our results fully revealed the significance of
bioinformatics in analyzing clinical issues and will serve as a
guide for future research. Our study also increases the
understanding of lncRNAs in pathogenesis of lymph node metastasis
in early-stage cervical cancer and may be a reference basis for the
treatment of cervical cancer metastasis.
Acknowledgments
This study was supported by grants from the Natural
Science Foundation of Guangdong Province, China (no.
2015A030313073) and Sun Yat-Sen University Clinical Research 5010
Program (no. 2007010). We thank the Gene Expression Omnibus (GEO)
and TCGA database for providing their platforms and contributing
their valuable data sets. The BRB-Array tools used in the study
were developed by Richard Simon and the BRB-Array Tools Development
Team.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carlson RW, Larsen JK, McClure J,
Fitzgerald CL, Venook AP, Benson AB III and Anderson BO:
International adaptations of NCCN Clinical Practice Guidelines in
Oncology. J Natl Compr Canc Netw. 12:643–648. 2014.PubMed/NCBI
|
|
3
|
Selman TJ, Mann C, Zamora J, Appleyard TL
and Khan K: Diagnostic accuracy of tests for lymph node status in
primary cervical cancer: A systematic review and meta-analysis.
CMAJ. 178:855–862. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Landoni F, Maneo A, Colombo A, Placa F,
Milani R, Perego P, Favini G, Ferri L and Mangioni C: Randomised
study of radical surgery versus radiotherapy for stage Ib-IIa
cervical cancer. Lancet. 350:535–540. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hosaka M, Watari H, Mitamura T, Konno Y,
Odagiri T, Kato T, Takeda M and Sakuragi N: Survival and
prognosticators of node-positive cervical cancer patients treated
with radical hysterectomy and systematic lymphadenectomy. Int J
Clin Oncol. 16:33–38. 2011. View Article : Google Scholar
|
|
6
|
Dong Y, Wang X, Wang Y, Liu Y, Zhang J,
Qian W and Wu S: Validity of 18F-fluorodeoxyglucose positron
emission tomography/computed tomography for pretreatment evaluation
of patients with cervical carcinoma: A retrospective
pathology-matched study. Int J Gynecol Cancer. 24:1642–1647. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nogami Y, Banno K, Irie H, Iida M, Kisu I,
Masugi Y, Tanaka K, Tominaga E, Okuda S, Murakami K, et al: The
efficacy of preoperative positron emission tomography-computed
tomography (PET-CT) for detection of lymph node metastasis in
cervical and endometrial cancer: Clinical and pathological factors
influencing it. Jpn J Clin Oncol. 45:26–34. 2015. View Article : Google Scholar
|
|
8
|
Diaz JP, Gemignani ML, Pandit-Taskar N,
Park KJ, Murray MP, Chi DS, Sonoda Y, Barakat RR and Abu-Rustum NR:
Sentinel lymph node biopsy in the management of early-stage
cervical carcinoma. Gynecol Oncol. 120:347–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grigsby PW, Watson M, Powell MA, Zhang Z
and Rader JS: Gene expression patterns in advanced human cervical
cancer. Int J Gynecol Cancer. 16:562–567. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang W, Jia HL, Huang JM, Liang YC, Tan H,
Geng HZ, Guo LY and Yao SZ: Identification of biomarkers for lymph
node metastasis in early-stage cervical cancer by tissue-based
proteomics. Br J Cancer. 110:1748–1758. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim T, Choi J, Kim WY, Choi CH, Lee J, Bae
D, Son D, Kim J, Park BK, Ahn G, et al: Gene expression profiling
for the prediction of lymph node metastasis in patients with
cervical cancer. Cancer Sci. 99:31–38. 2008.
|
|
12
|
Huang L, Zheng M, Zhou QM, Zhang MY, Jia
WH, Yun JP and Wang HY: Identification of a gene-expression
signature for predicting lymph node metastasis in patients with
early stage cervical carcinoma. Cancer. 117:3363–3373. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Biewenga P, Buist MR, Moerland PD, Ver
Loren van Themaat E, van Kampen AHC, ten Kate FJW and Baas F: Gene
expression in early stage cervical cancer. Gynecol Oncol.
108:520–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhan A and Mandal SS: Long noncoding RNAs:
Emerging stars in gene regulation, epigenetics and human disease.
Chem Med Chem. 9:1932–1956. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun NX, Ye C, Zhao Q, Zhang Q, Xu C, Wang
SB, Jin ZJ, Sun SH, Wang F and Li W: Long noncoding RNA-EBIC
promotes tumor cell invasion by binding to EZH2 and repressing
E-cadherin in cervical cancer. PLoS One. 9:e1003402014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao S, Liu W, Li F, Zhao W and Qin C:
Decreased expression of lncRNA GAS5 predicts a poor prognosis in
cervical cancer. Int J Clin Exp Pathol. 7:6776–6783.
2014.PubMed/NCBI
|
|
17
|
Huang L, Liao LM, Liu AW, Wu JB, Cheng XL,
Lin JX and Zheng M: Overexpression of long noncoding RNA HOTAIR
predicts a poor prognosis in patients with cervical cancer. Arch
Gynecol Obstet. 290:717–723. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Noordhuis MG, Fehrmann RSN, Wisman GBA,
Nijhuis ER, van Zanden JJ, Moerland PD, Ver Loren, van Themaat E,
Volders HH, Kok M, ten Hoor KA, et al: Involvement of the TGF-beta
and beta-catenin pathways in pelvic lymph node metastasis in
early-stage cervical cancer. Clin Cancer Res. 17:1317–1330. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Sun S, Pu JKS, Tsang ACO, Lee D,
Man VOY, Lui WM, Wong STS and Leung GKK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang F, Zhang L, Huo XS, Yuan JH, Xu D,
Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, et al: Long noncoding
RNA high expression in hepatocellular carcinoma facilitates tumor
growth through enhancer of zeste homolog 2 in humans. Hepatology.
54:1679–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Michiels S, Koscielny S and Hill C:
Prediction of cancer outcome with microarrays: A multiple random
validation strategy. Lancet. 365:488–492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Adler J and Parmryd I: Quantifying
colocalization by correlation: The Pearson correlation coefficient
is superior to the Mander's overlap coefficient. Cytometry A.
77:733–742. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu JB, Dai CM, Su XY, Cao L, Qin R and
Kong QB: Gene microarray assessment of multiple genes and signal
pathways involved in androgen-dependent prostate cancer becoming
androgen independent. Asian Pac J Cancer Prev. 15:9791–9795. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al The Gene Ontology Consortium: Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liao Q, Liu C, Yuan X, Kang S, Miao R,
Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al: Large-scale prediction
of long non-coding RNA functions in a coding-non-coding gene
co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Macdonald AI, Sun P, Hernandez-Lopez H,
Aasen T, Hodgins MB, Edward M, Roberts S, Massimi P, Thomas M,
Banks L, et al: A functional interaction between the MAGUK protein
hDlg and the gap junction protein connexin 43 in cervical tumour
cells. Biochem J. 446:9–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Emmrich S, Streltsov A, Schmidt F,
Thangapandi VR, Reinhardt D and Klusmann JH: LincRNAs MONC and
MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol
Cancer. 13:1712014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Iancu IV, Botezatu A, Goia-Ruşanu CD,
Stănescu A, Huică I, Nistor E, Anton G and Pleşa A: TGF-beta
signalling pathway factors in HPV-induced cervical lesions. Roum
Arch Microbiol Immunol. 69:113–118. 2010.
|
|
30
|
Chen Q, Cao HZ and Zheng PS: LGR5 promotes
the proliferation and tumor formation of cervical cancer cells
through the Wnt/β-catenin signaling pathway. Oncotarget.
5:9092–9105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang G, Lu X and Yuan L: LncRNA: A link
between RNA and cancer. Biochim Biophys Acta. 1839:1097–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu Y, Chen HY, Yu CY, Xu J, Wang JL, Qian
J, Zhang X and Fang JY: A long non-coding RNA signature to improve
prognosis prediction of colorectal cancer. Oncotarget. 5:2230–2242.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lampe PD and Lau AF: The effects of
connexin phosphorylation on gap junctional communication. Int J
Biochem Cell Biol. 36:1171–1186. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Steinhoff I, Leykauf K, Bleyl U, Dürst M
and Alonso A: Phosphorylation of the gap junction protein
Connexin43 in CIN III lesions and cervical carcinomas. Cancer Lett.
235:291–297. 2006. View Article : Google Scholar
|
|
35
|
Broustas CG, Gokhale PC, Rahman A,
Dritschilo A, Ahmad I and Kasid U: BRCC2, a novel BH3-like
domain-containing protein, induces apoptosis in a caspase-dependent
manner. J Biol Chem. 279:26780–26788. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shureiqi I, Jiang W, Zuo X, Wu Y, Stimmel
JB, Leesnitzer LM, Morris JS, Fan HZ, Fischer SM and Lippman SM:
The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid
down-regulates PPAR-delta to induce apoptosis in colorectal cancer
cells. Proc Natl Acad Sci USA. 100:9968–9973. 2003. View Article : Google Scholar : PubMed/NCBI
|