Introduction
Proto-oncogenes of the Myc family play important
roles in cellular growth, differentiation and cytometaplasia by
encoding different oncogenic factors. c-Myc, a well-studied and
critical Myc family member, is present in a variety of cancer cells
including lung and breast cancer (1,2). c-Myc
also has a vital function in different cancers, including stomach,
colon and bladder cancer (3,4).
Increasing numbers of target genes activated by c-Myc have been
identified (5,6). The c-Myc proto-oncogene encodes a
transcription factor, Myc, which forms a heterodimer for its
biological effects.
Myc-interacting zinc finger protein 1 (Miz-1;
Zbtb17) is a zinc finger transcription factor that forms a complex
with various oncoproteins such as Myc, Gfi-1 and Bcl-6 for its
biological activity (7–9). There are three main mechanisms by
which conjugation of Miz-1 and Myc plays a role in oncogenesis.
Firstly, the Myc/Miz-1 protein complex participates
in the Hedgehog (Hh) signaling pathway. Miz-1 combines with Smo,
the initiator of the Hh signaling pathway through subsequent
downstream signaling factor Gli2, which then translocates into the
nucleus its functional site (10).
Ras driven tumorigenesis can be restrained by the
Mule/Huwe1/Arf-BP1 pathway factors, via suppressing
c-Myc/Miz-1-mediated regulation of p21 and p15 (11). Binding of Myc to Miz-1 suppresses
TGF-β-dependent signaling via reducing CKI expression and cellular
senescence (12,13). Myc also inhibits expression of
p21CIP1 in response to DNA damage and to inducers of
differentiation in cultured cells via Miz-1 (14,15).
Secondly, Miz-1 associates with Myc on a number of
target promoters, including two promoters of cell cycle inhibitors
p21 and p15 (14). Similarly,
CDKN1C (p57KIP2) is repressed by Myc in a
Miz-1-dependent manner (12,16).
Miz-1 can also act as a platform for transcription factors
containing Bcl-6 onco-protein and Gli1 to repress cell cycle
inhibitors. In addition, cell adhesion molecules, most notably
integrins, also target the Miz-1/Myc complex (17). Moreover, Miz-1 was found to
upregulate antiapoptotic Bcl-2 to stimulate IL-7 in a
Miz-1-deficient T lymphocyte model (18).
Thirdly, Myc/Miz-1 promotes tumor formation and
development in many types of cancers. In skin carcinoma, studies
have shown that Miz-1 inhibits the expression of tumor-suppressor
protein p21, which may be related to ubiquitin ligating enzyme
HectH9/Hunel (11,19). During lymphomagenesis, the Myc/Miz-1
complex has a different effect on TGF-β-induced senescence
(11). Similar effects have been
observed for tumor formation in medulloblastoma. Likewise,
Miz-1-deficient models show cancer repression (20). Most studies have focused on the
Miz-1/Myc complex and cell cycle inhibitors such as p21 and
p15.
p21 is an inhibitor of CDK functioning downstream of
p53 by two cellular pathways. One pathway is dependent on p53, the
other is independent. The p21 protein can inhibit the activity of
cyclin D1-CDK4 and cyclin E-CDK2, which further prevents the
phosphorylation of Rb protein, resulting in cell cycle arrest in
the G1 phase (21). The p21 protein
was shown to have the same function on the tumor necrosis factor
(TNF) and tissue plasminogen activator (r-PA) pathways (22). In tumors, p21 inhibits tumor
development (23). Low expression
of p21 and p53 mutations may play a role in the occurrence of
esophageal cancer (24). Esophageal
cancer cells show less proliferation after transfection with p21
(25). The cyclin D1 regulatory
subunit combines with CDKs to form functionally active complexes
with significant effects on cell proliferation, and overexpression
leads to uncontrolled cell growth. Various studies have shown low
expression of p21 and high expression of cyclin D1 in esophageal
cancer (26–28).
The mechanism of Miz-1 action in esophageal cancer
development is still unknown. Studies have shown that high
expression of Myc and low regulation of p21 exist in esophageal
cancer. Miz-1 has a repressive function on the expression of Myc
(29). In the present study, we
hypothesized that Miz-1 is a cancer gene acting through binding, or
in tumors with high levels of Myc, suppression of downstream genes
such as p21 and p15. The expression of Miz-1 in esophageal cancer
was determined, and its biological functions were measured after
silencing of Miz-1 by shRNA in two esophageal cancer cell lines.
Our findings confirmed that Miz-1 acts as a promoting factor and
has a vital function in esophageal cancer.
Materials and methods
Cell lines and tumor samples
A series of esophageal cancer cell lines (Eca109,
KYS150, BT9, BT, SEg, Bar-T10 and T9) were used. These cells were
obtained from Chongqing Key Laboratory of Molecular Oncology and
Epigenetics (Chongqing, China). The cell lines were maintained in
RPMI-1640 medium (HyClone Thermo, Beijing, China) supplemented with
10% fetal bovine serum (FBS; PAN Biotech, Aidenbach, Germany) and
100 U/ml of penicillin and streptomycin, and grown at 37°C in 5%
CO2. DNA and RNA samples were extracted from paired
esophageal tumor tissues and para-carcinoma tissues of distal
surgical margins. All tissues were obtained from patients who
volunteered to receive surgery due to esophageal cancer at the
Department of Surgery, The First Affiliated Hospital of Chongqing
Medical University. The present study was approved by the
Institutional Review Board of Chongqing Medical University.
cDNA and RNA extraction
Total RNA was extracted from the cell lines and
tissues using TRIzol reagent (Invitrogen, Waltham, MA, USA), and
other basic chemical reagents. Each sample was injected in 1 ml
TRIzol. The tissue samples were excised into pieces under liquid
nitrogen while using a homogenizer for 5 min, and then 200
µl of chloroform was added with mixing. The samples were
mixed for 3 min, and the supernatants were obtained after
centrifugation for 15 min at 4°C and 16,099 × g/min. The
supernatant samples to be injected (500 µl) were kept for 10
min, and then used for further centrifugation. The resulting
supernatants were discarded and 1 ml of 75% ethanol was used for
the precipitants, collected after further centrifugation for 5 min
at 4°C and 6,288 × g/min. The clear liquid was removed and diethyl
phosphorocyanidate (DEPC) water was injected for blending according
to the size of the precipitants. The RT reagent kit (Takara,
Dalian, China) was used according to the manufacturer's
specification for a 10-µl volume system using reverse
transcription of RNA into cDNA. The concentration and purity of the
cDNA were determined, and samples were stored at −80°C until
use.
Semi-quantitative RT-PCR amplification
and gel electrophoresis
Real-time reverse transcription polymerase chain
reaction (RT-PCR) samples were prepared using the premixed Taq kit
(Takara) and amplification was performed on an ABI Prism 7000
detection system (Applied Biosystems, Foster City, CA, USA)
according to the conditions recommended by the manufacturer. The
10-µl volume system contained 5 µl Taq
polymerase, 3.8 µl DEPC, 0.8 µl forward and reverse
primers and 0.4 µl cDNA. The following primers were used:
Miz-1 forward, 5′-GTGTGTGATGTGCGGTAAGG-3′ and reverse,
5′-ggACTggACgAATCTCTTgC-3′; p21 forward, 5′-CAg CTGAGGTGTGAGCAGC-3′
and reverse, 5′-GACATGGCG CCTCCTCTg-3′; cyclin D1 forward,
5′-gTgTATCgAgAg GCCAAAGG-3′ and reverse, 5′-GCAACCAGAAATGCAC
AgAC-3′; gAPDH forward, 5′-ACCACCATggAgAAgg CTGG-3′ and reverse,
5′-CTCAGTGTAGCCCAGGATGC-3′. RT-PCR was performed with 30 cycles for
Miz-1, cyclin D1 and p21, and 23 cycles for
glyceraldehyde-3-phosphate dehydrogenase (GAPDH). RT-PCR gel
electrophoresis used 2% agarose (Invitrogen, Carlsbad, CA, USA)
dissolved in 1X TAE buffer (Double Helix, Shanghai, China). Agarose
gel electrophoresis was performed at 120 mA for 25 min.
Construction and transfection with the
lentiviral vector shRNA
Based on the known Miz-1 gene sequence in GenBank
(Gene ID: 7709), we commissioned Shanghai Innovation Biotechnology
Co. Ltd. to provide the lentiviral vectors which contained Miz-1
shRNA. We chose shRNA in a lentiviral vector due to its stable
transfection ability, low cytotoxicity and high efficiency of
transfection and gene knockout. The following sequences were
packaged into the lentivirus vector: viral, gPH-ZBTB17-shRNA-F,
GATCCGTGTTCACTTTAAGGCTCATACTTCC TGTCAGATATGAGCCTTAAAGTGAACACTTTTTG
and gPH-ZBTB17-shRNA-R, AATTCAAAAAgTgTTCACTTT
AAGGCTCATATCTGACAGGAAGTATGAGCCTTAAAGT GAACACG. The cells
(1×105) were seeded onto 6-well plates, while injecting
20 µl (2×108 TU/ml) of the target gene or empty
lentiviral vector into each plate. The cell medium was replaced
after transfection for 12 h, and fluorescence was observed 72 h
after transfection. The transfected cells were selected with
Puromycin (Eca109, 1.2 µg/ml; KYS150, 0.8 µg/ml) on
the basis of the experiments using normal cells. After selecting
stably transfected cells, the cells were divided into three groups:
Miz-1 (transfected with shRNA for knockdown of the Miz-1 gene),
vector (transfection with empty viral vectors) and control (normal
untreated cells) groups.
Immunohistochemical staining
Immunohistochemistry was performed using the
Histostain®-Plus kit (Thermo Fisher, Waltham, MA, USA).
Sections were dewaxed by soaking in xylene for 10 min, four times
and then the concentration of ethanol was stepwise decreased from
100 to 75% for 8 min at every concentration. Endogenous peroxidase
was blocked by 3% hydrogen peroxide for 15 min. Antigen retrieval
was carried out using citrate buffer at an elevated but not a
boiling temperature for 25 min. The sections were incubated with
the primary Miz-1 antibody (1:50 dilution) overnight at 4°C,
followed by secondary antibody incubation at 37°C for 30 min.
3,3′-Diaminobenzidine (DAB) was used for staining, and hematoxylin
was used for counterstaining. Neutral gum was used to overlay the
slides. Phosphate-buffered saline (PBS) was used between each step,
for 3 min, three times. The immunohistochemical staining results
were assigned a score based on the depth of staining and the rate
of dye uptake by the stained cells. The intensity of staining was
determined as: 0, no staining; 1, weak staining; 2, moderate
staining; and 3, strong staining. Percentage of positive staining
of the cells was determined as: <5%, 0; 5–25%, 1; >25–50%, 2;
>50–75%, 3; and >75%, 4. Cytoplasmic and nuclear staining was
included in the statistical analyses. The index of staining was
determined by multiplying the score for the staining intensity by
the score for the positive rate. A score of <4 was considered as
negative staining (marked with a minus sign), and a score ≥4 was
considered as positive staining (+).
Cell proliferation assays
Log-phase cells (2×103) from three groups
were seeded onto 96-well plates. After 24 h, 10 µl of Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Santa Clara,
CA, USA) reagent was added into each plate. The optical densities
(ODs) were determined using a microplate reader (Infinite 200 PRO;
Tecan) at 450 nm after adding CCK-8 for 2 h. Samples were then
likewise measured for 5 days of culture. The experiments were
repeated independently three times. The proliferation curve of each
group was based on the absorbance at 450 nm.
Colony formation assays
Log-phase cells were seeded onto 6-well plates with
2 ml of complete medium (500 cells/well) and incubated. Plates were
initially examined by microscopy to confirm that only single cells
without clumps had been plated. Two weeks after seeding, the
colonies were fixed with 4% paraformaldehyde for 30 min and stained
with crystal violet for 30 min. Images were obtained and counting
of the cells was carried out using a scanner.
Migration and invasion assays
Each group of cells (4×105 cells) was
plated into cell culture inserts with 8-µm aperture filters
(Corning, Corning, NY, USA) coated with (invasion) or without
(migration) Matrigel® (Corning) and incubated for 24 h.
Cells migrated and invaded from the low to the high concentration
of serum. After 24 h, cells passing the filters outside the inserts
were fixed in paraformaldehyde, stained with crystal violet, and
observed by a microscope. Experiments were performed in
triplicate.
Analyses of the cell cycle and
apoptosis
Three groups of log-phase cells were collected and
centrifuged at 93 × g/min for 5 min after trypsinization. The cells
were then washed with PBS twice and fixed in ice-cold 70% ethanol
overnight at 4°C. Cells for apoptosis detection were washed with
PBS twice and mixed in PBS in 1-ml Eppendorf tubes. The samples
were assayed for cell cycle and apoptosis at Chongqing Medical
University Life Sciences Institute.
Western blot analyses
Cells with stable transfection or wild-type were
collected by a cell wiper and washed with PBS twice. Cell proteins
were extracted by one mixture of 99% RIPA lysis buffer (RIPA) and
1% phenylmethanesulfonyl fluoride (PMSF) (Beytime Biotechnology,
Shanghai, China) according to the cell mass weight after
centrifugation at 4°C for 40 min, then centrifuged at 4°C, at
12,000 rpm for 15 min. After centrifugation, loading buffer was
added at a volume of 25% of the liquid supernatant and samples were
boiled for 5 min. A total of 40 µg of protein was separated
by sodium dodecyl sulphate-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred onto a polyvinylidene fluoride
(PVDF) membrane using the laboratory manual instructions. Membranes
were blocked in 5% skimmed milk powder for 2 h. The primary
antibodies used were: Miz-1 (Santa Cruz Biotechnology, Santa Cruz,
CA, USA), p21, cleaved caspase 3, polyADP ribose polymerase (PARP)
(all from Cell Signaling Technology, Danvers, MA, USA), cyclin D1
(Epitomics, Burlingame, CA, USA) and GAPDH used as a control (Cell
Signaling Technology). Membranes were incubated with the primary
antibodies at 4°C overnight, washed with Tris-buffered saline and
Tween-20 (TBST) twice, and immersed in the corresponding secondary
antibodies for 2 h. Proteins were visualized by BeyoECL Plus
(Beyotime, Chengdu, China) in the dark.
Statistical analyses
All statistical analyses were performed with SPSS
17.0 software (SPSS, Inc., Chicago, IL, USA). Data are expressed as
the average ± standard deviation (SD). Student's t-tests were used
for comparisons between any two means and single factor analyses of
variance (ANOVA) were used for comparisons among more than two
groups. The Chi-square test was used for two constituent ratios in
two different groups. For all values, p<0.05 was considered to
indicate a statistically significant result. All graphs were
created by GraphPad Prism (GraphPad, La Jolla, CA, USA).
Results
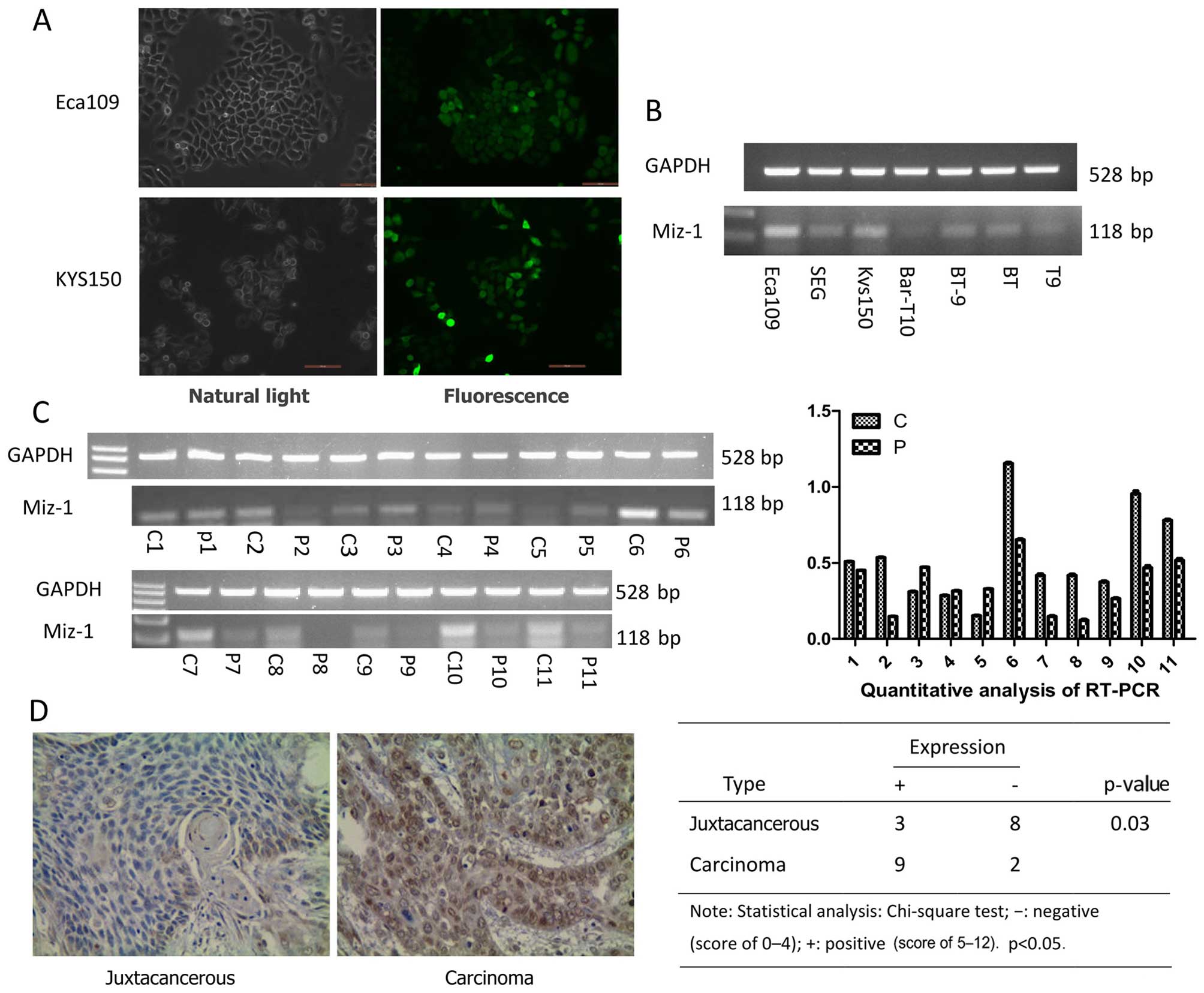
Miz-1 is highly expressed in esophageal
cancer and cell lines
We collected 22 esophageal tumor tissues and paired
distal surgical margin samples from normal esophageal tissues to
examine the expression of Miz-1. Half of the samples were used for
RT-PCR and the other half for immunohistochemical staining. We
found that expression of Miz-1 was upregulated in 8 tumor tissue
samples compared with that in the normal esophageal tissues, as
determined by RT-PCR (Fig. 1C).
Similar results were found using immunohistochemical staining. Nine
of 11 samples were positive (Fig.
1D) in the group of tumor tissues; however, three samples were
positive in the control samples. We also detected Miz-1 expression
in 7 esophageal cancer cell lines, by RT-PCR. The results showed
that Miz-1 was expressed at high levels in the esophageal cancer
cell lines (Eca109 and KYS150) (Fig.
1B). The results suggest that Miz-1 is an oncogene in
esophageal cancer.
Transfection verification
The green fluorescence-containing shRNA lentivirus
showed the Miz-1 (transfected with shRNA to knockout the Miz-1
gene) and the vector (transfected with the empty viral vector) cell
groups by use of fluorescence microscopy. After stable
transfection, Miz-1 mRNA levels were measured by RT-PCR and the
protein expression of Miz-1 was measured by western blot analyses.
We confirmed that cells were successfully transfected with the
lentivirus by the presence of green fluorescence (Fig. 1A). We also further confirmed Miz-1
knockdown in the cells by RT-PCR and western blot analyses
(Fig. 4A and B).
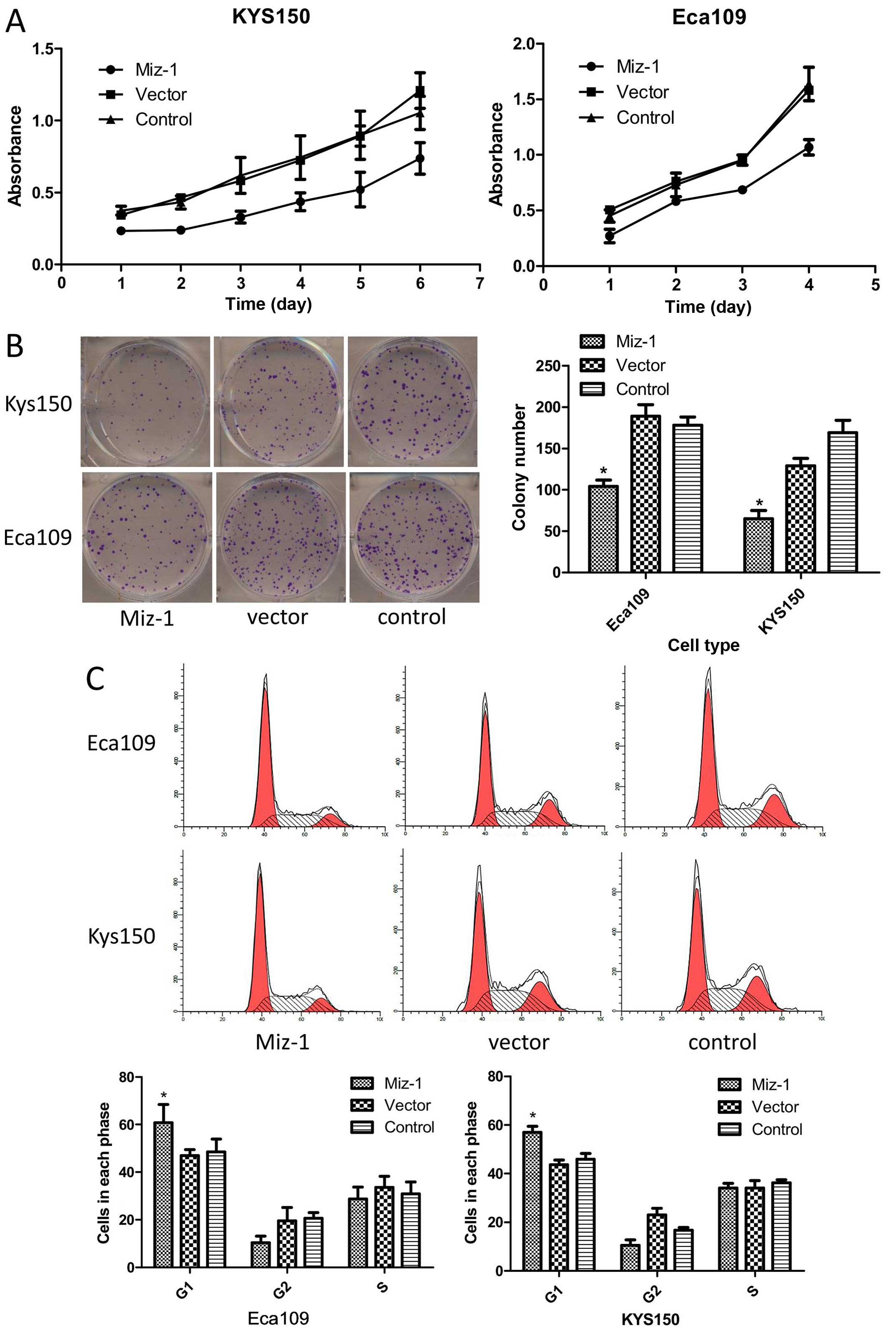
Miz-1 promotes cell clonogenicity and
proliferation
After we confirmed that Miz-1 is differentially
expression in tumor tissues vs. normal tissues, we determined
whether the cell functions were altered after knockdown of Miz-1.
The colony formation and CCK-8 assays were used to determine the
cell proliferation status. The results showed that the colony
formation numbers (104 in Eca109 and 65 in KYS150) in the group
with silenced Miz-1 was markedly reduced compared to that in the
control group (p<0.05) (Fig.
2B). Similarly, the CCK-8 assay showed inhibition of cell
growth in the two cell lines after Miz-1 was silenced (p<0.05)
(Fig. 2A).
Miz-1 reduces cell cycle arrest and
apoptosis
We further investigated the effect of Miz-1 on
esophageal cancer cell apoptosis and the cell cycle using flow
cytometry. The Miz-1 cell group with knockdown of Miz-1 had
increased numbers of Eca109 (60.8%) and KYS150 (57%) cells in the
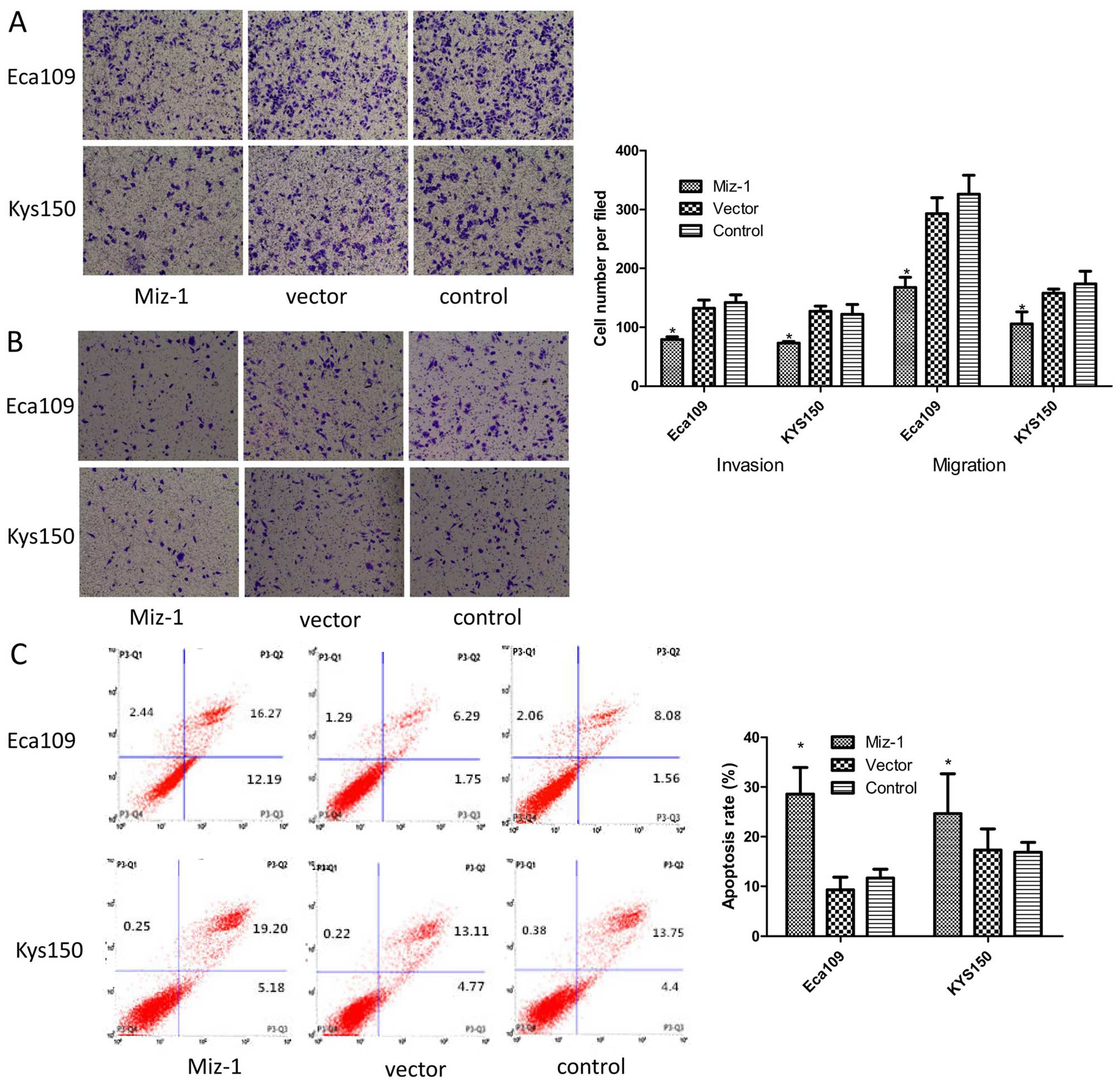
g0–g1 phase compared with the control cells (p<0.05) (Fig. 2C). Furthermore, knockdown of Miz-1
induced cell apoptosis from 9.3 to 28.6% in the Eca109 cells and
from 16.8 to 24.7% in the KYS150 cells (p<0.05) (Fig. 3C). These results suggest that the
cell cycle and apoptosis were also affected by Miz-1. Moreover,
knockdown of Miz-1 induced apoptosis.
Miz-1 induces invasiveness and
migration
Cellular motility through invasiveness and migration
experiments was further determined. The cells transfected with the
Miz-1 shRNA had a low capability for invasiveness and migration,
using both Eca109 and KYS150 cells (Fig. 3A and B). In the absence of a matrix,
the number of Eca109 cells with Miz-1 knockdown (168 cells) passing
through the Transwell was less than the control cells (293
knockdown and 326 control cells) in five microscopic fields
(p<0.05). Similar results were also observed for the
invasiveness assays using the KYS150 cell line (p<0.05).
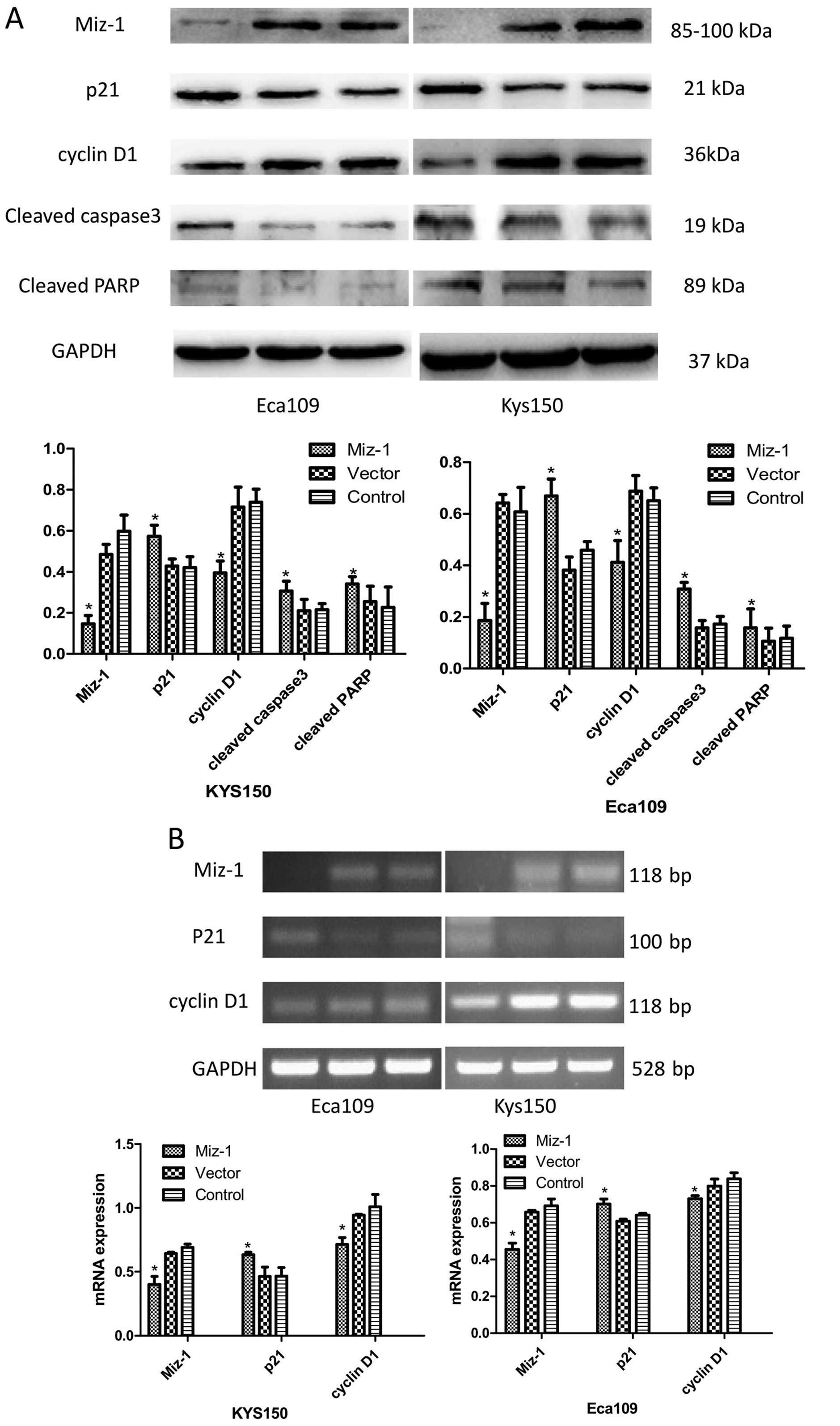
Miz-1 decreases the expression of p21,
caspase 3, PARP and p21-arrested cyclin D1
We also examined Miz-1 as a potential downstream
target gene using RT-PCR and western blot analyses. We hypothesized
that Miz-1 could be a link between the c-Myc oncogene and the
downstream target genes p21 and cyclin D1. The results suggested
that without Miz-1, p21 was expressed at high levels and reduced
downstream cyclin D1, leading to cell cycle arrest (Fig. 4A and B). Moreover, knockdown of
Miz-1 induced apoptotic factors caspase 3 and PARP (Fig. 4A and B) (p<0.05).
Discussion
Esophageal cancer is one of the most common cancers.
It originates in the esophageal mucosal epithelium. Cell cycle
dysregulation is one fundamental aspect of cancer incidence and
development. The proliferation of cancer cells is attributed to
unchecked cell cycling (32),
leading to the lack of cell death and perhaps to cell pleomorphism.
The p21 is one inhibitor of CDK that monitors abnormal
proliferation in cells. In esophageal cancer, there is a low level
of p21, and cell cycle dysregulation. The cause may be a mutation
of p21 itself, or the loss of regulation of upstream c-Myc
oncogenes. Several studies have shown high levels of c-Myc in
esophageal cancer. We hypothesized that this cell cycle signaling
pathway may be critical for esophageal cancer development.
To determine the pathway of oncogenesis, we showed
that Miz-1 one zinc finger protein interaction between Myc was
necessary for inhibition of p21 by Myc. To further confirm these
findings, we used two stable esophageal cancer cell lines with
Miz-1 silencing via specific shRNA packaged in a lentiviral vector.
Fluorescence microscopy images confirmed the success of the
transfection. The RT-PCR and western blot analyses verified the
Miz-1 downregulation in the transfected cell lines. Using the same
methodology, we measured the expression levels of both p21 and
cyclin D1.
The most obvious changes in the cell cycle and
proliferation were observed in the Miz-1-silenced cells. Likewise,
cyclin D1 restored the inhibition of p21. Flow cytometric results
suggested that the cells with knockdown of Miz-1 remained in G0-G1
phase, even in the presence of cyclin D1, a factor that maintains
cells in the G1-S phase. Since the cell cycle was arrested in the
G1 phase, the cells were smaller and had less proliferation. Our
studies revealed that the cell group with downregulated Miz-1 had
less growth and proliferation compared with the other cell groups
as determined by the CCK-8 and clone formation assays.
Decreasing Miz-1 not only had an effect on the cell
cycle and proliferation, but also influenced apoptosis and cell
migration. The migration and invasion assays showed that the cells
moved more slowly after knockdown of the target gene. The data also
suggested that Miz-1 participated in apoptotic signaling pathways
through altered expression of PARP and caspase 3. The results of
the cell apoptosis experiments suggested that cells without Miz-1
had a higher rate of apoptosis compared with the other two cell
groups.
Our findings are consistent with the hypothesis that
Miz-1 is a key regulator in the signaling pathway of cell cycle
arrest induced by CDKN1a and cyclin D1. Moreover, we found that
Miz-1 also participated in apoptosis through caspase 3 and
PARP.
Overall, our results strongly suggest that Miz-1 is
highly expressed in esophageal cancer tissues compared with
juxtacancerous tissues (at least 5 cm distance from the tumor).
This suggested that esophageal cancer may develop when the levels
of Miz-1 are elevated. When we knocked down Miz-1, we found changes
in cell proliferation, in the cell cycle distribution, apoptosis,
migration and invasion. We therefore propose a signaling pathway by
which Miz-1 suppresses p21, a normal cancer suppressor gene that
regulates the cell cycle. One possible mechanism is that Miz-1 acts
with some oncogene such as Myc. Miz-1 could also function
independently; however, its mechanism remains unknown.
We chose to measure two typical apoptosis-related
proteins in the classic signaling pathways. The alteration of
caspase 3 and PARP demonstrated that Miz-1 participates in
apoptotic signaling pathways. The migration and invasion of cells
are attributed to the functions of integrins or the influences of
cell viability and cell cycle arrest. To confirm these results,
further coimmunoprecipitation assays and/or dual-luciferase
reporter systems should be used to confirm a relationship between
these two target genes.
Our findings are consistent with the hypothesis that
Miz-1 has a different expression pattern in tumors vs. juxta
cancerous tissues and may act as a key regulator of oncogenesis by
affecting various cell functions, including the cell cycle and
apoptosis. These findings indicate that Miz-1 may be an effective
and novel target for esophageal cancer therapy.
Acknowledgments
The present study was supported by the Key
Scientific Research Project of Chongqing Municipal Bureau of Health
(grant no. 2012-1-015).
References
|
1
|
Dang CV: c-Myc target genes involved in
cell growth, apoptosis, and metabolism. Mol Cell Biol. 19:1–11.
1999. View Article : Google Scholar
|
|
2
|
Wu Y and Li Z: Oncogene c-myc and
malignant tumor. Clin Res. 25:1698–1700. 2008.
|
|
3
|
Kotake T, Saiki S, Kinouchi T, Shiku H and
Nakayama E: Detection of the c-myc gene product in urinary bladder
cancer. Jpn J Cancer Res. 81:1198–1201. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Masters JR, Vesey SG, Munn CF, Evan GI and
Watson JV: c-myc oncoprotein levels in bladder cancer. Urol Res.
16:341–344. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levens D: Disentangling the MYC web. Proc
Natl Acad Sci USA. 99:5757–5759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li LH, Nerlov C, Prendergast G, Macgregor
D and Ziff EB: c-Myc represses transcription in vivo by a novel
mechanism dependent on the initiator element and Myc box II. EMBO
J. 13:4070–4079. 1994.PubMed/NCBI
|
|
7
|
Peukert K, Staller P, Schneider A,
Carmichael G, Hänel F and Eilers M: An alternative pathway for gene
regulation by Myc. EMBO J. 16:5672–5686. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Phan RT, Saito M, Basso K, Niu H and
Dalla-Favera R: BCL6 interacts with the transcription factor Miz-1
to suppress the cyclin-dependent kinase inhibitor p21 and cell
cycle arrest in germinal center B cells. Nat Immunol. 6:1054–1060.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Basu S, Liu Q, Qiu Y and Dong F: gfi-1
represses CDKN2B encoding p15INK4B through interaction
with Miz-1. Proc Natl Acad Sci USA. 106:1433–1438. 2009. View Article : Google Scholar
|
|
10
|
Lu J, Chen M, Ren XR, Wang J, Lyerly HK,
Barak L and Chen W: Regulation of Hedgehog signaling by
Myc-interacting zinc finger protein 1, Miz1. PLoS One.
8:e633532013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Inoue S, Hao Z, Elia AJ, Cescon D, Zhou L,
Silvester J, Snow B, Harris IS, Sasaki M, Li WY, et al:
Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by
preventing c-Myc/Miz1-mediated down-regulation of p21 and p15.
genes Dev. 27:1101–1114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Riggelen J, Müller J, Otto T, Beuger
V, Yetil A, Choi PS, Kosan C, Möröy T, Felsher DW and Eilers M: The
interaction between Myc and Miz1 is required to antagonize
TGFbeta-dependent autocrine signaling during lymphoma formation and
maintenance. genes Dev. 24:1281–1294. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gebhardt A, Kosan C, Herkert B, Möröy T,
Lutz W, Eilers M and Elsässer HP: Miz1 is required for hair
follicle structure and hair morphogenesis. J Cell Sci.
120:2586–2593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Seoane J, Le HV and Massagué J: Myc
suppression of the p21Cip1 Cdk inhibitor influences the
outcome of the p53 response to DNA damage. Nature. 419:729–734.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu S, Cetinkaya C, Munoz-Alonso MJ, von
der Lehr N, Bahram F, Beuger V, Eilers M, Leon J and Larsson LG:
Myc represses differentiation-induced p21CIP1 expression via
Miz-1-dependent interaction with the p21 core promoter. Oncogene.
22:351–360. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adhikary S, Peukert K, Karsunky H, Beuger
V, Lutz W, Elsässer HP, Möröy T and Eilers M: Miz1 is required for
early embryonic development during gastrulation. Mol Cell Biol.
23:7648–7657. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gebhardt A, Frye M, Herold S, Benitah SA,
Braun K, Samans B, Watt FM, Elsässer HP and Eilers M: Myc regulates
keratinocyte adhesion and differentiation via complex formation
with Miz1. J Cell Biol. 172:139–149. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saba I, Kosan C, Vassen L and Möröy T:
IL-7R-dependent survival and differentiation of early T-lineage
progenitors is regulated by the BTB/POZ domain transcription factor
Miz-1. Blood. 117:3370–3381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hönnemann J, Sanz-Moreno A, Wolf E, Eilers
M and Elsässer HP: Miz1 is a critical repressor of cdkn1a during
skin tumorigenesis. PLoS One. 7:e348852012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Yan J, Jiang S, Wen J, Chen L, Zhao
Y and Lin A: Site-specific ubiquitination is required for relieving
the transcription factor Miz1-mediated suppression on TNF-α-induced
JNK activation and inflammation. Proc Natl Acad Sci USA.
109:191–196. 2012. View Article : Google Scholar
|
|
21
|
Li Z and Li B: Correlation of gene
polymorphism of p21 and p27 with tumor. Int J gene. 29:317–320.
2006.
|
|
22
|
Krauss Gerhard; Sun C, Liu JS, et al:
Biochemistry of Signal Transduction and Regulation. Chemical
Industry Press; pp. 338–377. 2005
|
|
23
|
Palazzo JP, Mercer WE, Kovatich AJ and
McHugh M: Immunohistochemical localization of
p21WAF1/CIP1 in normal, hyperplastic, and neoplastic
uterine tissues. Hum Pathol. 28:60–66. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Lv H, Jing L and Qin J:
Expression of p21cip1/WAF, p53 and the infection of
HPV-16 in the esophageal cancer. Shi Yong Ai Zheng Za Zhi She.
19:488–490. 2004.In Chinese.
|
|
25
|
Tanaka Y, Fujii T, Yamana H, Kato S,
Morimatsu M and Shirouzu K: Experimental gene therapy using
p21Waf1 gene for esophageal squamous cell carcinoma by
gene gun technology. Int J Mol Med. 14:545–551. 2004.PubMed/NCBI
|
|
26
|
Liu J, Hu Y, Hu W, Xie X, Ela Bella A, Fu
J and Rao D: Expression and prognostic relevance of
p21WAF1 in stage III esophageal squamous cell carcinoma.
Dis Esophagus. 25:67–71. 2012. View Article : Google Scholar
|
|
27
|
Zhang H, Liu Y and Hu M: Expression and
significance of p53, p21, p16, cyclin D and CDK, in esophageal
squamous cell carcinoma. Immunological J. 4:312–315. 2003.
|
|
28
|
Möröy T, Saba I and Kosan C: The role of
the transcription factor Miz-1 in lymphocyte development and
lymphomagenesis-binding Myc makes the difference. Semin Immunol.
23:379–387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Montgomery EA: Oesophageal cancer. (World
Cancer Report 2014). Stewart BW and Wild CP: International Agency
for Research on Cancer; Lyon: pp. 374–382. 2014
|
|
30
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Gobal cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen W, Zheng R, Zhang S, Zhao P, Zeng H
and Zou X: Report of cancer incidence and mortality in China, 2010.
Ann Transl Med. 2:612014.PubMed/NCBI
|
|
32
|
Deshpande A, Sicinski P and Hinds PW:
Cyclins and cdks in development and cancer: A perspective.
Oncogene. 24:2909–2915. 2005. View Article : Google Scholar : PubMed/NCBI
|