Introduction
Hepatocellular carcinoma (HCC), one of the most
aggressive malignancies, is the third leading cause of
cancer-related death worldwide (1,2).
Annually, ~750,000 new cases are diagnosed and more than 75% of
cases occur in the Asia-Pacific region, and the incidence of HCC is
still increasing in the US and Europe (3–5). To
date, the treatment modality is largely reliant on surgical
resection or liver transplantation as HCC is notoriously resistant
to chemotherapy and other systemic treatment options. Moreover, the
prognosis of HCC is extremely poor due to hepatic metastasis
(6). Therefore, it is urgent to
develop an effective therapeutic method for HCC treatment.
Disturbance of the balance between proliferation and
apoptosis is a fundamental hallmark of cancer development (7,8). The
p53 tumor-suppressor protein, also known as the 'guardian of the
genome', plays a pivotal role in inducing apoptosis or cell cycle
arrest to prevent cell proliferation in response to stress such as
DNA damage, hypoxia and activation of oncogenes. Conversely,
inactivation or mutation of the p53 gene contributes to
abnormal growth due to loss of control in cell cycle regulation or
failure to trigger apoptosis, which could in turn, leads to
carcinogenesis (9–11). In the absence of stress, p53 is a
short-lived protein of extremely low levels, and is negatively
regulated by murine double minute 2 protein (MDM2) (12,13).
on the contrary, MDM2 expression is induced by p53. Hence, MDM2 and
p53 can interact to form an autoregulatory loop in which p53
activates expression of its own inhibitor (13,14).
Upon receiving the upstream stress signals, p53 is elevated to
mediate an array of apoptosis and cell cycle genes (12,15).
Commonly, p53 usually remains in its wild-type configuration in at
least 10% of human cancers displaying unusual MDM2 overexpression
(16). Furthermore, it is well
documented that inhibitors or knockdown of MDM2 can enhance the
activity and expression of p53, by which to recover its
tumor-suppressing effects (17–20).
Various elements are reported to be involved in cell
cycle transition, including cyclins, cyclin-dependent kinases
(CDKs) and cyclin-dependent kinase inhibitors (CKIs). CDKs are a
family of serine/threonine protein kinases playing pivotal roles in
such a process in combination with various endogenous cyclins
(21,22). P21, a p53 downstream gene, is known
as a CKI to stop the replication of damaged DNA (22,23).
However, it should be noted that arrested cells are not necessarily
protected from apoptosis (10). It
has been reported that p53 activates the apoptotic machinery by
regulating Bax function and mitochondrial integrity (24). In addition, restoration of p53 in
murine myeloid leukemia cell line M1 was associated with Bax
protein elevation accompanied by a simultaneous decrease in Bcl-2
(25). Furthermore, the apoptotic
function of p53 was found to be regulated by apoptosis-stimulating
proteins of the p53 (ASPP) family consisting of ASPP1, ASPP2 and
iASPP. The similar sequences at the C-terminus of the three members
which is the preferred binding site for p53 determines that ASPP1,
ASPP2 and iASPP could compete with each other to bind to p53
(26). In regards to the roles of
these proteins in apoptosis, ASPP1 and ASPP2 enhance p53-mediated
apoptosis, while iASPP inhibits the process (26–28).
The abnormal expression of ASPP members may explain the failure to
induce apoptosis of some tumors expressing wild-type p53 (26,29–31).
To date, few studies have been carried out to detect the expression
and possible roles of the ASPP family in the pathogenesis of
HCC.
All-trans retinoic acid (ATRA), a major
active metabolite of vitamin A, is reported to induce cell
differentiation of acute promyelocytic leukemia (APL) and achieve
complete remission (32,33). Subsequently, increasing studies have
been conducted to determine the efficiency of ATRA on solid tumors.
These studies revealed that it exerted anticancer effects on liver,
breast and lung cancer (34–36).
To the best of our knowledge, the activities of ATRA mainly rely on
two nuclear receptor families including retinoid receptors (RARs)
and retinoid X receptors (RXRs). RARs act as retinoid-inducible
transcriptional factors and form heterodimers with RXRs, which
regulate the expression of genes involved in cell growth,
differentiation, migration and apoptosis (37,38).
However, in clinical practice, the application of ATRA is far from
satisfactory as it may upregulate gene transcription of vascular
endothelial growth factor (VEGF), resulting in angiogenesis and
subsequent cancer growth (39). In
addition, ATRA resistance and high toxicity also hinder its
application in cancer treatment (40–42).
Although As2O3 was reported to rescue most
relapsed/refractory patients treated with ATRA, its severe
side-effects have limited its long-term use (43). Hence, it is essential to develop a
novel all-trans retinoid acid derivative with higher
sensitivity and efficiency.
4-Amino-2-trifluoromethyl-phenyl retinate (ATPR),
designed and synthesized by the College of Pharmacy, Anhui Medical
University, is a novel retinoid derivative with lower toxicity than
ATRA. our previous studies revealed that ATPR was superior to ATRA
in terms of inhibiting cellular migration as well as promoting
apoptosis and differentiation in lung, breast or gastric cancer
cell lines (44–46). However, the effects of ATPR on HepG2
cell proliferation remain unknown.
Based on the evidence described above, in the
present study, we aimed to verify the potential effects of ATPR on
the proliferation of HCC cell line HepG2 for the first time and
elucidate the involvement of p53 and the ASPP family in cell
proliferation through analysis of cell cycle progression and
apoptosis.
Materials and methods
Cell lines and reagents
Human HCC cell line HepG2 was purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). ATRA,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and dimethyl sulfoxide (DMSO) were obtained from Sigma Chemical Co.
(St. Louis, Mo, USA). ATPR was synthesized and kindly provided by
the School of Pharmacy, Anhui Medical University (China) and the
purity was 99.66%. Dulbecco's modified Eagle's medium (DMEM; low
glucose) was purchased from Gibco Life Technologies (Carlsbad, CA,
USA). Newborn calf serum was obtained from Zhejiang Tianhang
Biological Technology Co., Ltd. (China). Primary antibodies: mouse
anti-p21, -MDM2 and -β-actin were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Rabbit anti-p53, -ASPP1,
-ASPP2, -iASPP, -CDK4, mouse anti-cyclin D, -cyclin E and -CDK6
were purchased from Lianshi Biological Technology Co., Ltd.
(China). Mouse anti-Bcl-2 and rabbit anti-Bax and all secondary
antibodies were purchased from Millipore (Billerica, MA, USA).
Fluorescein (FITC)-conjugated affinitive goat anti-rabbit IgG and
4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased
from ZSGB-BIO (Beijing, China). Coulter DNA detection kit was
purchased from Beckman Coulter (Miami, FL, USA). Annexin V
apoptosis detection kit FITC was purchased from eBioscience (San
Diego, CA, USA). The BCA protein determination kit and ECL reagent
were purchased from the Beyotime Institute of Biotechnology
(China). ATRA and ATPR were dissolved in DMSO at a concentration of
20 mM, and were stored at −20°C.
Cell culture
HepG2 cells were maintained in DMEM supplemented
with 10% newborn calf serum and antibiotics containing penicillin
(100 U/ml) and streptomycin (100 U/ml) in a humidified 5%
Co2 incubator at 37°C. The medium was changed every 2 or
3 days and cells were passaged when their density reached 80–90%
confluency. To assess the cytotoxicity of ATRA and ATPR, HepG2
cells were incubated in medium with the indicated concentrations of
ATRA and ATPR. In order to exclude a potential bias of the solvent
DMSO, the DMSO group operated as the vehicle control, in which the
volume of DMSO in the culture medium equaled that contained in the
medium with 25 µM ATRA or ATPR. The cells exposed to normal
medium were used as the cell control.
Cell proliferation assessment
MTT assay was used to analyze the cell viability.
HepG2 cells (5×103 cells/well) were seeded into 96-well
plates and treated with a desired dose of ATRA or ATPR (25
µM) for different times (1–4 days) or treated with a dose
range of 5–100 µM of ATRA or ATPR for 48 h. At each time
point, 20 µl of 5 mg/ml MTT was added into each well and the
cells were incubated for an additional 4–6 h. Culture medium was
then removed and 100 µl DMSO was further added into each
well to solubilize the formazan crystals for 15 min. The optical
density (OD) value at a wavelength of 570 nm was then measured by
an absorbance microplate reader (ELX800; BioTek, Winooski, VT,
USA). Cell inhibition rate was calculated by the following formula:
Inhibition rate (%) = (OD570 of the cell control group -
OD570 of the experimental group)/OD570 of the
cell control group × 100%. The experiment was repeated at least
three times to detect the cell growth.
Plate colony formation assay
The colony formation ability of the HepG2 cells
in vitro was measured by plate colony formation assay. HepG2
cells at logarithmic growth were plated in 6-well plates.
Twenty-four hours after plating, the cells were treated with
various concentrations (5 and 25 µM) of ATRA and ATPR, and
then harvested as single cell suspensions. Approximately 2,000
cells/well were seeded in a new 6-well plate, and the plate was
incubated for 7–10 days until the colonies were clearly visible to
the naked eye. Then, the medium in the plate was discarded and
cells were washed with phosphate-buffered saline (PBS) two times,
fixed by 4% paraformaldehyde and stained with 1% crystal violet.
The colonies (≥50 cells) were counted in five random visual fields
and photographed.
Flow cytometry for cell cycle
analysis
Flow cytometry was performed to analyze the
distribution of the cell cycle. HepG2 cells were seeded into 6-well
plates equally and incubated for 24 h in serum-free medium to
synchronize the cell cycle progression. Then, the cells were
treated with 25 µM ATRA and ATPR for 48 h. After treatment,
the adherent HepG2 cells were harvested, washed twice with ice-cold
PBS and resuspended with 100 µl PBS, and then counted and
diluted to 1×106 cells/ml. Cells were stained with 1 ml
of hypotonic propidium iodide (PI) solution (50 mg/ml PI, 0.1%
Triton X-100, 0.1% sodium citrate) in the presence of 1% RNase A
for 30 min at 37°C in the dark. Then, flow cytometry
(Becton-Dickinson, San Diego, CA, USA) was used to detect cell
cycle distribution. Finally, G0/G1, S and G2/M phase cells were
analyzed using ModFIT software.
Hoechst 33258 staining
Hoechst 33258 staining was used to visualize nuclear
changes to indicate apoptosis. HepG2 cells were cultured on sterile
circle coverslips in 6-well plates and allowed to adhere overnight
and then incubated with ATRA and ATPR (5 and 25 µM) for 48
h. Cells were sequentially washed with PBS three times, fixed with
0.5 ml/well of 4% paraformaldehyde for 10 min, and washed again
with PBS two times, finally followed by the addition of 0.1 ml of a
DNA-specific fluorescent dye, Hoechst 33258 for 5 min. After being
washed with PBS, the slides were mounted with aqueous-based
anti-fade mounting agent and then examined and photographed under
an upright fluorescence microscope.
Flow cytometric analysis of
apoptosis
Flow cytometry was used to further confirm the
occurrence of apoptosis. After treatment for 48 h, the cells were
collected, washed twice with PBS, and resuspended in 100 µl
of Annexin V binding buffer at 1×106 cells/ml. Then,
cells were stained with 5 µl of Annexin V-FITC and incubated
for 15 min in the dark. Next, 400 µl of Annexin V binding
buffer and 5 µl of PI were added, and the cells were
immediately analyzed by flow cytometry.
Immunofluorescence assay
The cellular fluorescent density of p53 was observed
by immunofluorescence assay. HepG2 cells were cultured in 12-well
plates with sterile circle coverslips and allowed to adhere
overnight, followed by the treatments (ATRA and ATPR, 25 µM)
for 48 h. Cells were washed with PBS three times, fixed with 4%
paraformaldehyde for 20 min at room temperature and permeabilized
with 0.1% Triton X-100. To minimize the non-specific binding of the
antibody, cells were blocked with 5% bovine serum albumin (BSA) for
2 h at room temperature, then incubated with p53 primary antibody
(1:50 dilution in 5% BSA) overnight at 4°C, washed and incubated
with FITC-conjugated secondary antibody (1:100 dilution in PBS) for
2 h at room temperature away from light. DAPI was used to stain
nuclei for 5 min and finally the coverslips with cells were mounted
with aqueous-based anti-fade mounting medium. Images of the stained
cells were visualized by a fluorescence microscope (Leica DMI4000
B).
Western blot analysis
HepG2 cells exposed to ATRA and ATPR (5 and 25
µM) for 48 h were washed with cold PBS three times and lysed
in lysis buffer (Tris-HCl, pH 7.14, 150 mM NaCl, 1 mM EDTA, 1%
Triton X-100, 0.1% SDS, 1 mM leupeptin, 1 mM PMSF) on ice for 30
min. The protein extracts were quantified with a BCA kit and
resolved in SDS sample buffer. Equal amounts of protein were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidene
fluoride (PVDF) membranes. The membranes were blocked with blocking
buffer [TBST buffer (20 mM Tris-HCl pH 7.6, 150 mM NaCl and 0.05%
Tween-20), 5% skimmed milk] for 2 h at room temperature and
incubated with diluted primary antibodies overnight at 4°C with the
indicated primary antibody against p53 (1:500), MDM2 (1:250), ASPP1
(1:500), ASPP2 (1:500), iASPP (1:500), p21 (1:250), cyclin D
(1:1,000), cyclin E (1:1,000), CDK4 (1:1,000), CDK6 (1:1,000),
Bcl-2 (1:500), Bax (1:1,000) and β-actin (1:1,000) respectively,
then exerted to incubation with the correspondingly diluted
horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h
at room temperature. The protein bands were visualized with
enhanced chemiluminescence (ECL) reagent. Finally, blots were
exposed to Kodak film. Negatives were scanned using a
ScanPrisa1240oUT (Acer, China). The data were quantified from three
independent experiments using Quantity one software.
Real-time quantitative PCR (qRT-PCR)
Total RNA was extracted using the TRIzol procedure
(Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized
from 4 µg RNA using the reverse transcription system
(Takara, China). The resulting 4 µg cDNA was amplified by
UltraSYBR Mixture (CWBiotech, China) using a Real-Time PCR system
(Thermo, USA) with the following primer sets: p53,
5′-gttcc-gagagctgaatgagg-3 (forward) and 5′-ttatggcgggaggtagactg-3′
(reverse); GAPDH, 5′-aggtcggagagtcaacggatttg-3′ (forward) and
5′-cctggaagatggtgatgggat-3′ (reverse). All primers were synthesized
by Sangon Biotech Co., Ltd. (Shanghai, China). GAPDH served as a
reference gene. Real-time PCR cycle program was as follows: an
initial denaturation at 95°C for 30 sec, followed by 40 cycles of a
5-sec extension step at 95°C, and annealing for 30 sec at 60°C.
Results are shown as the mean normalized values of cDNA levels
among each group to the reference gene.
Statistical analysis
All data are expressed as mean ± standard deviation
(SD). SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) was used
for all statistical analyses. Data from multiple groups were
analyzed by one-way ANOVA, followed by Dunnett's or LSD t-tests. A
value of P<0.05 was considered statistically significant.
Results
ATPR significantly inhibits HepG2 cell
proliferation in vitro
The effects of ATRA and ATPR on the proliferation of
HepG2 cells were determined by MTT assay, plate colony formation
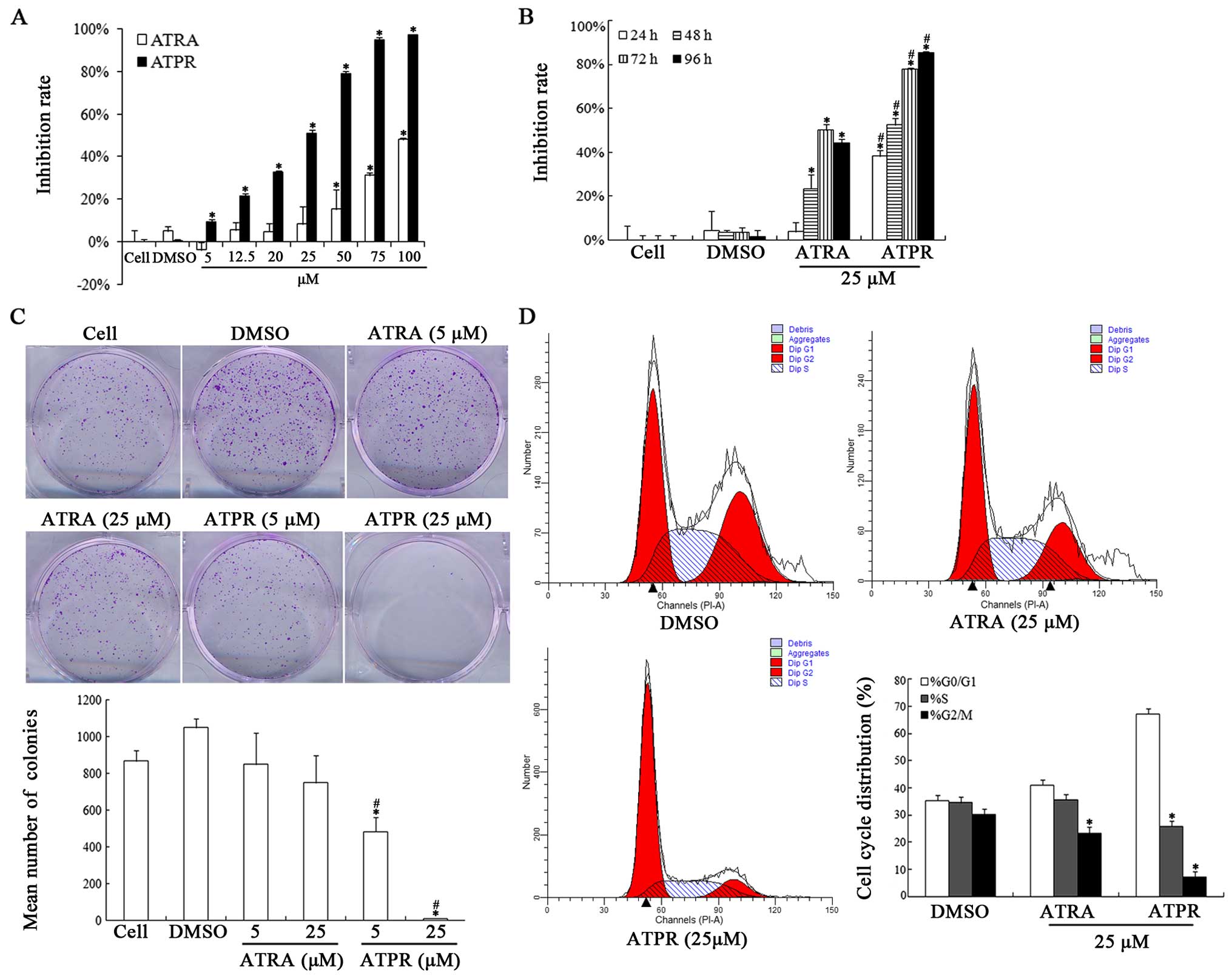
and flow cytometric analysis, respectively. As shown in Fig. 1A and B, ATPR significantly inhibited
the cell viability of the HepG2 cells in a dose- and time-dependent
manner. The inhibition rate of ATPR was much higher than that of
ATRA at the same dose. According to the data, the IC50
value of ATPR was 25.723 µM. Finally, two concentrations (5
and 25 µM) were chosen for the following assays. In
addition, plate colony formation assay was carried out to evaluate
the colony formation of the HepG2 cells in vitro. The
results indicated the ATPR induced a decrease in the density and
size of the colonies in a dose-dependent manner (Fig. 1C). Taken together, it is reasonable
to conclude that ATPR induced marked inhibition of HepG2 cell
proliferation.
Cell cycle analysis was used to further confirm the
growth inhibitory effects of ATPR on HepG2 cells. As shown in
Fig. 1D ATPR (25 µM) caused
a significant accumulation of G0/G1 phase cells. The percentage of
cells arrested in the G0/G1 phase increased from 35.37±1.59 to
67.11±2.54% and cells in the S phase decreased from 34.52±2 to
25.73±1.53% after ATPR interference (25 µM) compared with
those of the vehicle group. No significant difference was observed
in these percentages in the ATRA group (25 µM) compared with
the vehicle group (Fig. 1D).
Therefore, we speculated that ATPR may suppress HepG2 proliferation
by arresting cells in the G0/G1 phase.
ATPR induces apoptosis in HepG2
cells
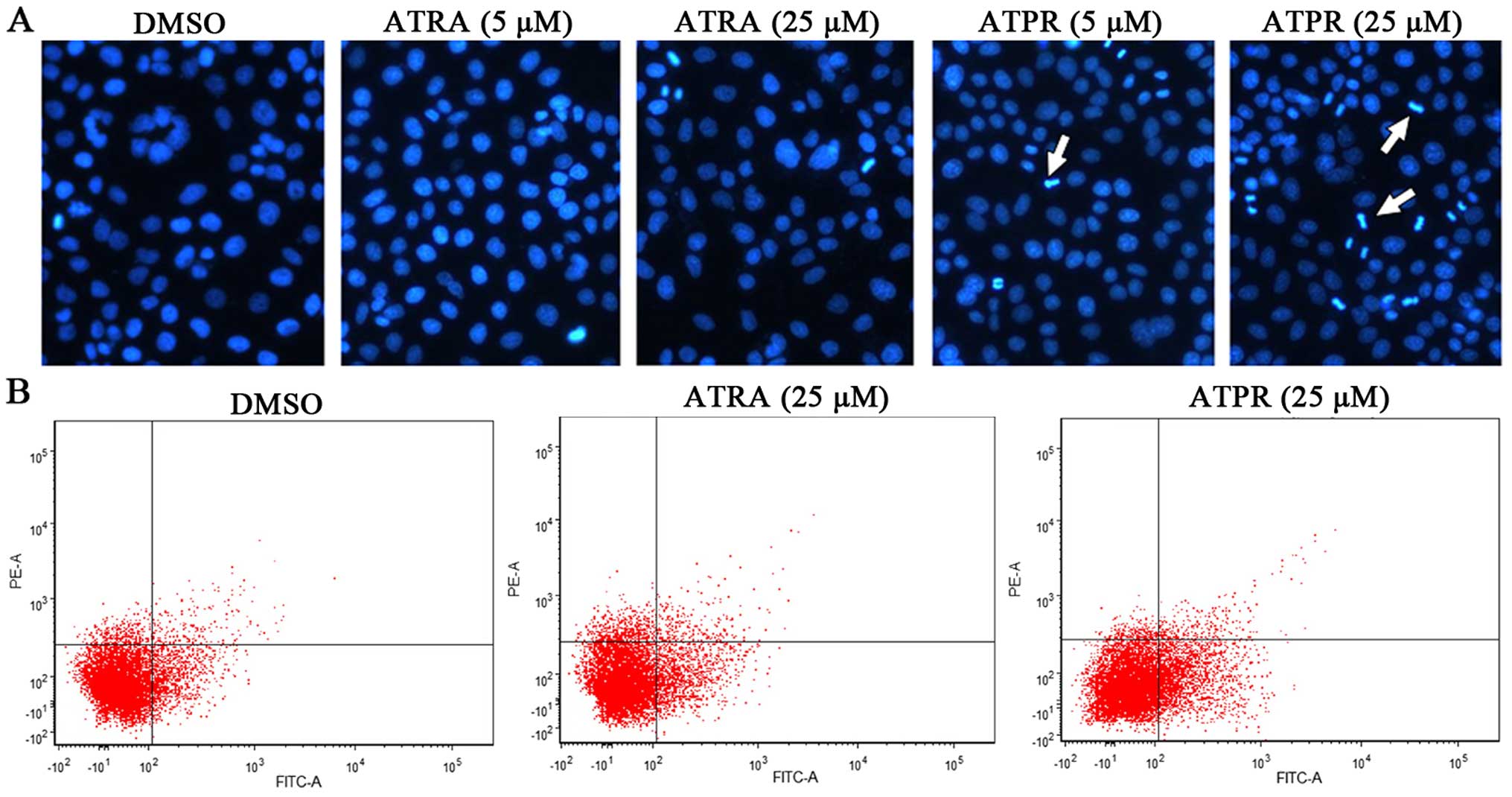
The apoptotic morphology of the cells was assessed
with Hoechst staining. The nuclei were homogeneous in the control
group, while in the ATPR groups (5 and 25 µM), the nuclei
were condensed with enhanced fraction, and appeared as obvious
apoptotic bodies (Fig. 2A).
Moreover, HepG2 cells were double-stained with Annexin V and PI and
analyzed through flow cytometry. The fourth- and the first-quadrant
cells represented early apoptotic cells (Annexin
V+/PI−) and late apoptotic cells (Annexin
V+/PI+), respectively. As shown in Fig. 2B, the apoptosis rate in the ATPR
group (25 µM) was significantly increased when compared with
the vehicle group. These results demonstrated that HepG2 cells
underwent apoptosis following ATPR treatment.
ATPR increases expression of p53 in the
HepG2 cells
To illuminate the participation of p53 in
ATPR-inhibited proliferation, the level of p53 was examined. In the
immunofluorescence assay, p53 was labeled with green fluorescent
protein and the fluorescence was evenly distributed in the nuclei
in the control group (Fig. 3A).
After treatment with ATPR (25 µM), the fluorescent density
of p53 was higher in the nuclei compared with that noted in the
control and ATRA (25 µM) groups (Fig. 3A). Meanwhile, western blotting and
qRT-PCR were applied to detect the protein and mRNA expression of
p53. As shown in Fig. 3B, p53
protein expression was significantly activated in a dose-dependent
manner after exposure to ATRA and ATPR for 48 h, particularly in
the ATPR groups. qRT-PCR analysis displayed that the mRNA level of
p53 was markedly elevated in the ATRA (25 µM) and ATPR (5
and 25 µM) groups (Fig. 3C).
In contrast, no statistical significance was noted in regards to
the mRNA level in the ATRA group (5 µM) compared to the
vehicle group. These outcomes demonstrated that ATPR increased p53
accumulation in the HepG2 cells.
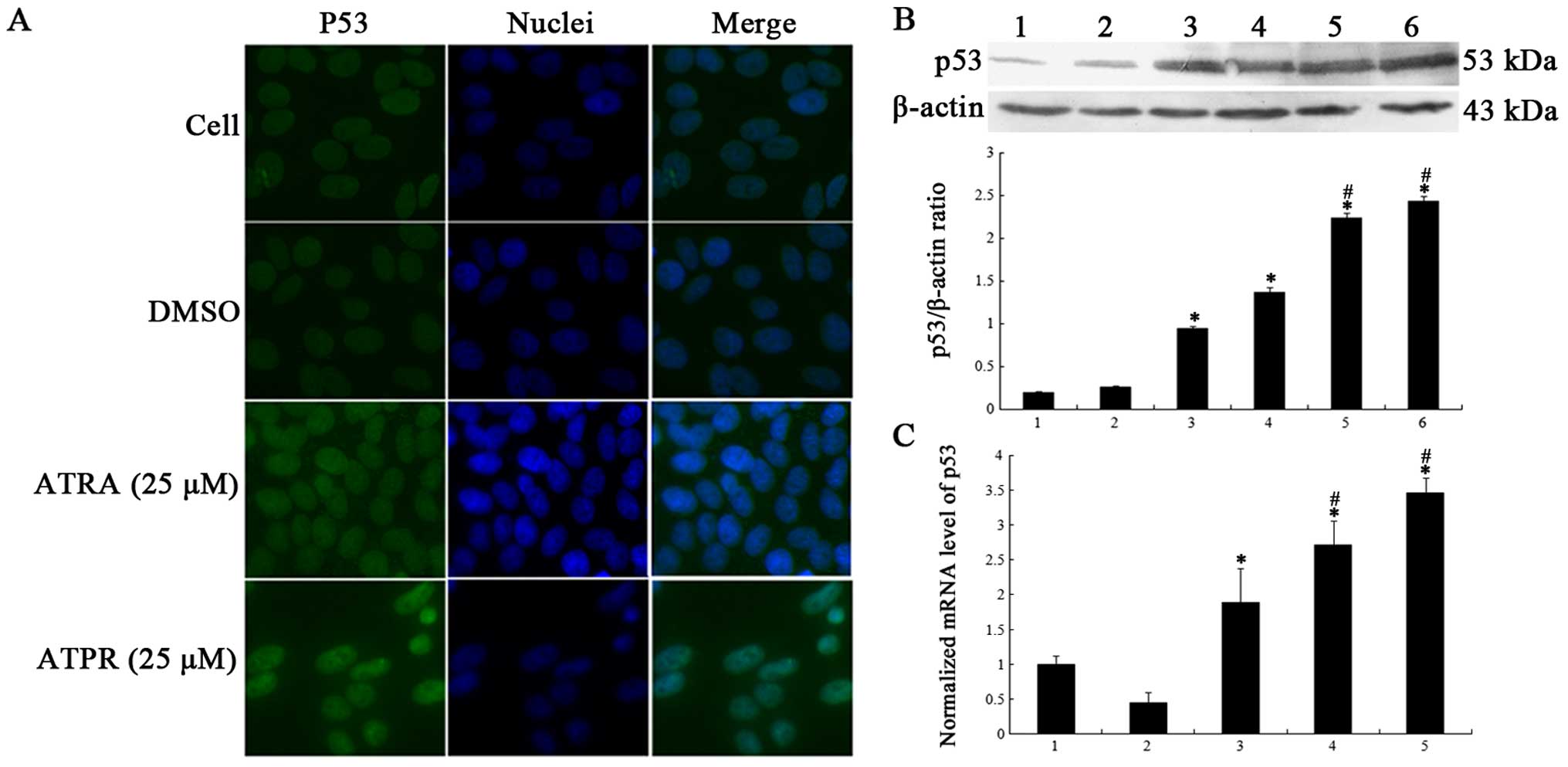 | Figure 3Effects of ATPR on p53 expression in
HepG2 cells. Cells were treated with 5 or 25 µM ATRA and
ATPR for 48 h. (A) ATPR elevated p53 accumulation in the nuclei.
Immunofluorescence microscopy showed that p53 was accumulated in
the nuclei in the ATPR-treated group. (B) Western blot analyses
revealed that ATPR increased more protein expression of p53 than
ATRA. Lane 1, cells; lane 2, DMSO; lane 3, 5 µM ATRA; lane
4, 25 µM ATRA; lane 5, 5 µM ATPR; and lane 6, 25
µM ATPR. (C) ATPR markedly upregulated the mRNA levels of
p53 in the HepG2 cells. Lane 1, DMSO; lane 2, 5 µM ATRA;
lane 3, 25 µM ATRA; lane 4, 5 µM ATPR; and lane 5, 25
µM ATPR. All numerical data are presented as mean ± SD.
*p<0.05 vs. the control group. #p<0.05
vs. the ATRA groups. |
ATPR alters the expression of MDM2 and
ASPP family proteins in the HepG2 cells
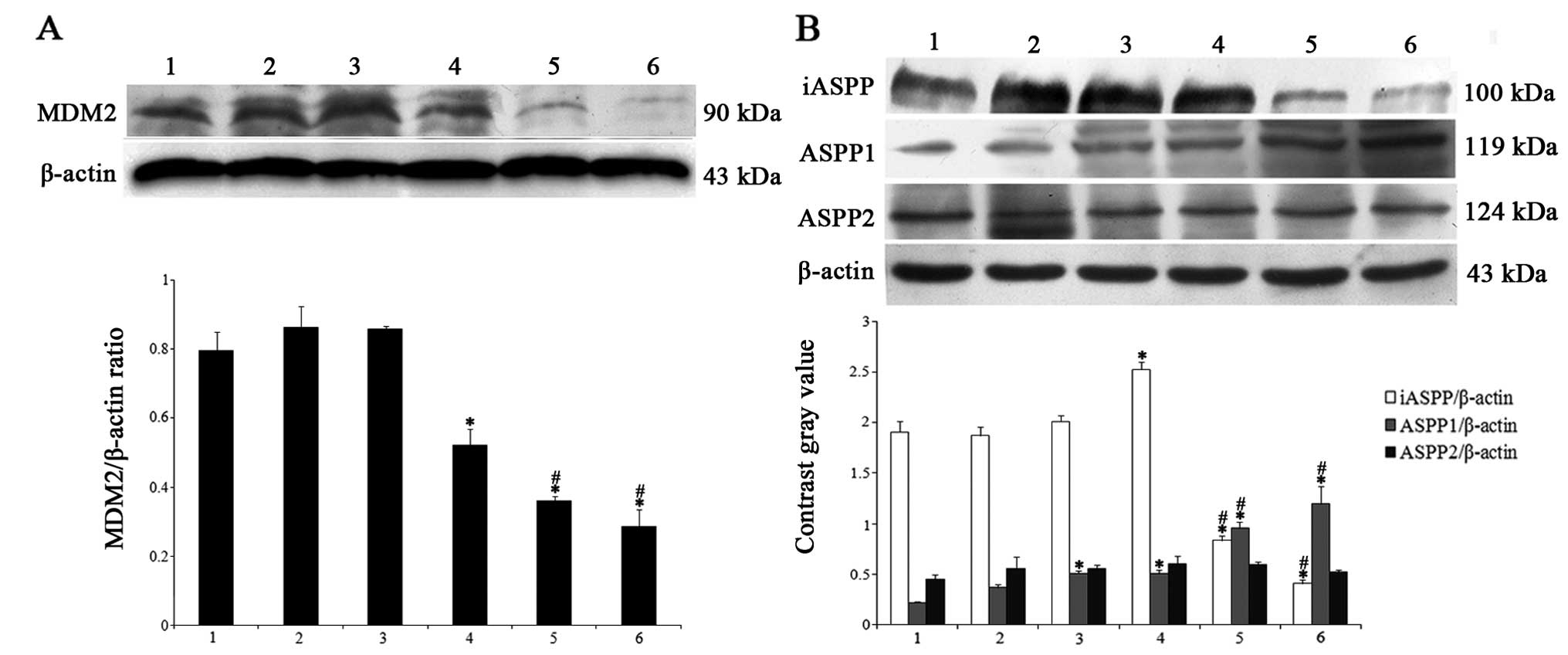
We next determined the expression of MDM2, one of
the main factors causing p53 degradation, and the ASPP family,
specific regulators of p53, in HepG2 cells. As shown in Fig. 4A, ATPR inhibited MDM2 expression in
a dose-dependent manner compared with control group. In addition,
ATPR induced elevation of ASPP1 expression and downregulation of
iASPP expression in the HepG2 cells. Nevertheless, no statistical
difference was noted in regards to the ASPP2 expression in the
HepG2 cells subject to ATPR compared with the control group
(Fig. 4B). Therefore, MDM2, ASPP1
and iASPP proteins may participate in the mechanisms mediating
ATPR-inhibited proliferation.
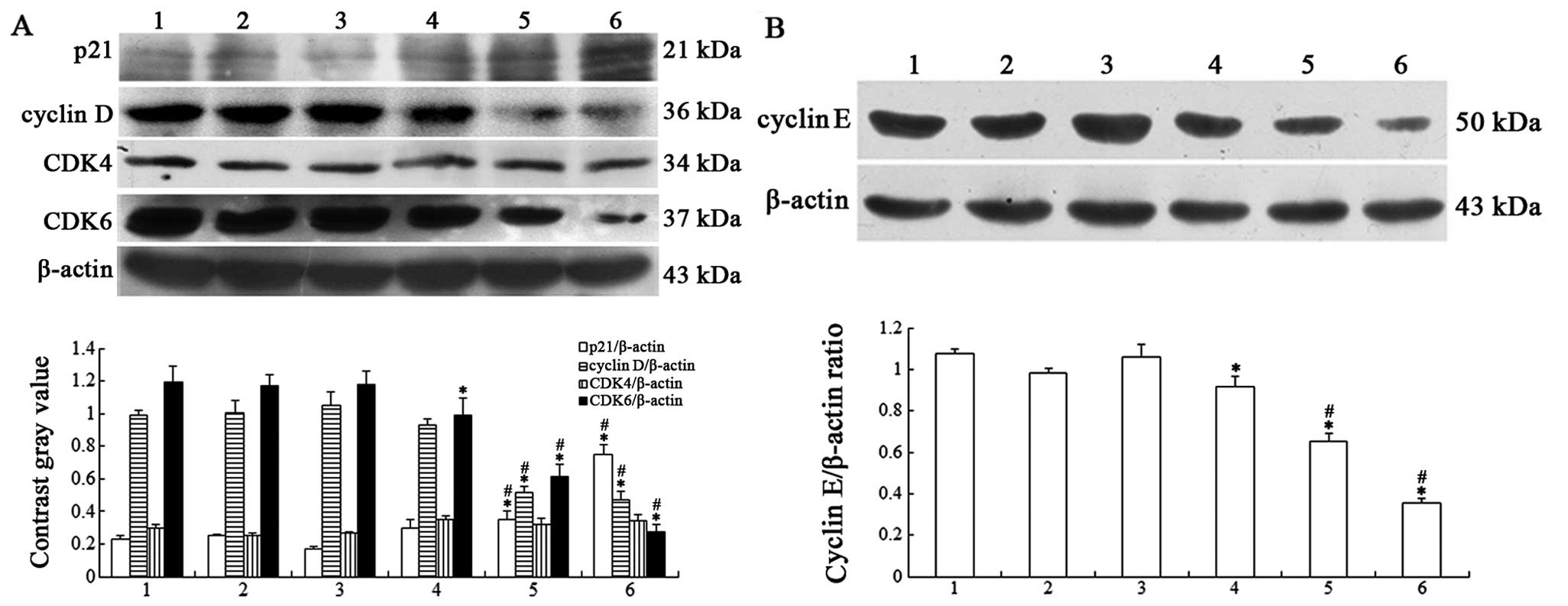
Effects of ATPR on cell cycle-related
proteins in HepG2 cells
To further examine the mechanisms of G0/G1 cell
cycle arrest induced by ATPR in HepG2 cells, western blotting was
conducted to determine the expression of cycle-related proteins
p21, cyclin E and D, CDK4 and CDK6 that are critical for G1-S
progression. The results revealed that the expression of p21 was
increased, while that of cyclin E and D, and CDK6 was decreased in
the ATPR groups (Fig. 5A and B). No
statistical difference was observed in the expression of CDK4 in
any treatment groups compared with the control group. This
indicated that induction of G0/G1 phase arrest by ATPR may be
associated with upregulation of p21 and downregulation of cyclin E
and D, and CDK6.
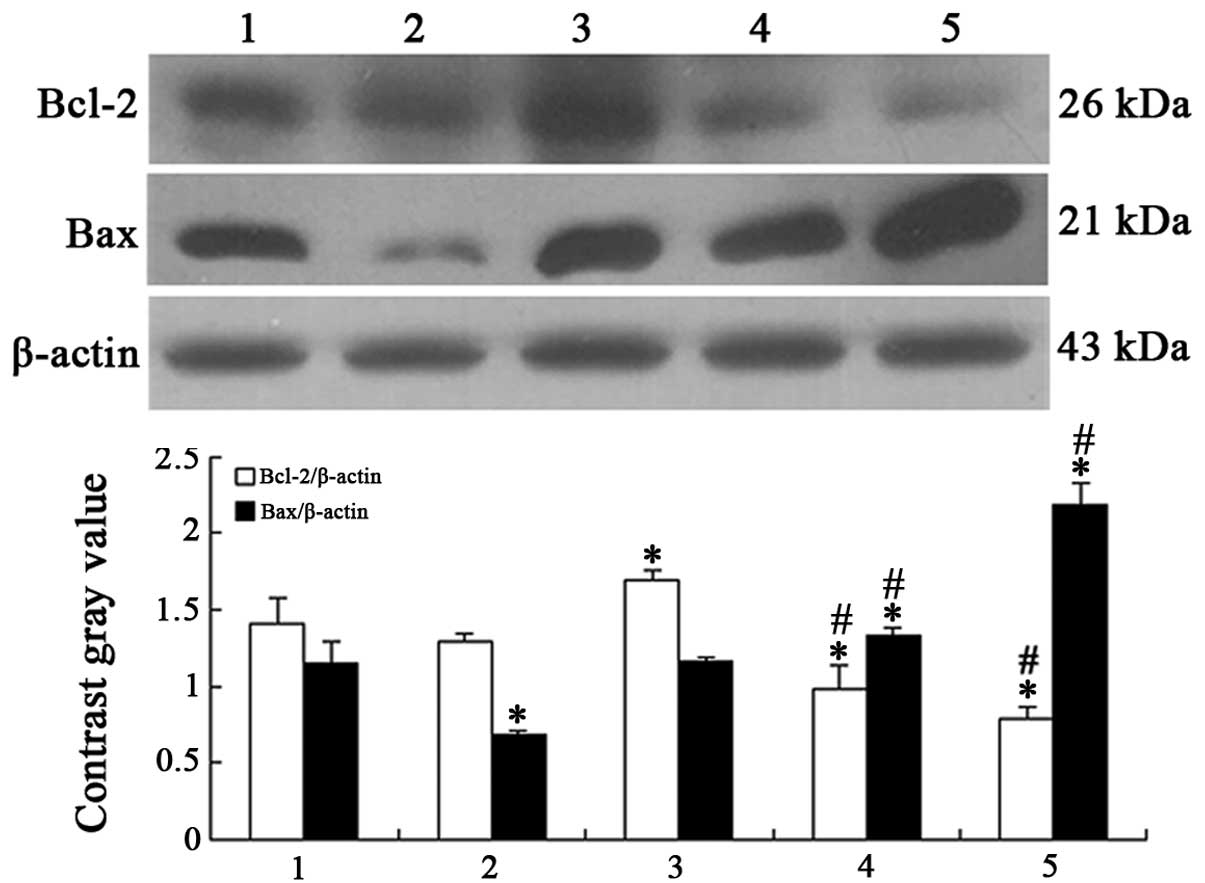
Effects of ATPR on expression of Bcl-2
family proteins in the HepG2 cells
To evaluate the involvement of the Bcl-2 family in
ATPR-induced apoptosis in HepG2 cells, the expression of Bcl-2 and
Bax was measured. As shown in Fig.
6, Bax expression was enhanced, while the expression of Bcl-2
was reduced in a dose-dependent manner, which clearly demonstrated
that ATPR may contribute to the apoptosis in HepG2 cells.
Discussion
Generally, aberrant proliferation of cancer cells
mainly relies on the activation of oncogenes and inactivation of
tumor-suppressor genes (47). p53,
commonly regarded as a cell transcription factor, shows
anti-carcinogenic activities by inducing cell cycle arrest and
apoptosis in malignant cells. In contrast, malfunction of p53 has
been observed in up to 50% of HCC for carcinogenesis and
chemotherapeutic resistance (48).
In the present study, we demonstrated that ATPR more effectively
inhibited the proliferation of HepG2 cells than ATRA by inducing
G0/G1 arrest and apoptosis by increasing expression of p53 and
ASPP1, and reducing iASPP expression, which may provide a promising
option to restore functional p53 expression in HCC therapy.
Currently, numerous studies have been carried out to
explore the mechanisms of anticancer drugs through therapeutic
targeting of p53-mediated apoptosis in cancers (24,49–51).
For example, Mu et al reported that oroxylin A promotes
apoptosis in cancer cells by modulating the expression of p53
(49). In addition, chrysin and
cisplatin promote the apoptosis of HepG2 cells by upregulating p53
(24). Yet, certain factors may be
responsible for resisting ATRA-induced apoptosis in HCC (52). Therefore, in the present study, we
investigated the effects of a novel ATRA derivative, ATPR, on the
proliferation, functional recovery of p53 and its downstream
targets in HepG2 cells. our results indicated that ATPR inhibited
the proliferation of HepG2 cells in a dose- and time-dependent
manner compared to ATRA. To address the preliminary mechanism,
immunofluorescence assay, western blot and qRT-PCR analyses were
performed to detect p53 expression in the HepG2 cells in the
presence of ATRA or ATPR. The accumulation of fluorescence in the
nuclei, the protein and mRNA expression of p53 were obviously
elevated in the ATPR groups, whereas, in the ATRA groups, no
significant changes were observed in these aspects. Based on this,
it is reasonable to speculate that p53 may be involved in
ATPR-inhibited proliferation in HepG2 cells, while the high ATRA
resistance of HepG2 cells may be related to the loss of regulation
in p53 expression. In future studies, knockdown or overexpression
of the p53 gene could be used to better understand the role
of p53 in the ATPR-mediated inhibition of proliferation in the
HepG2 cells.
As is known, MDM2 negatively regulates p53 by
binding to the N-terminus transactivation domain, and forming the
MDM2-p53 complex to export p53 from the nucleus towards proteasomal
degradation (53). Accordingly,
several strategies have been explored to disrupt the p53-MDM2
interaction for the functional recovery of p53, including the use
of small peptides, antisense oligonucleotides, and inhibitors of
MDM2 (54). Based on this, MDM2
inhibition may represent an appealing therapeutic strategy for the
treatment of cancer. Intriguingly, in the present study, ATPR
induced a decrease in MDM2 in the HepG2 cells. It blocked the entry
of MDM2 into the nucleus, and thereby allowed p53 to escape from
the negative feedback loop. Further experiments on their
interactions are needed.
ASPP family proteins have been reported to be
involved in regulating the function of p53 (26,27).
In cancer cells, upregulation of the iASPP protein and
downregulation of ASPP1 and ASPP2 were found, and may play
important roles in the pathogenesis of cancer (31,55).
Similarly, in the present study, the levels of ASPP1 and ASPP2 were
low, while iASPP expression was comparatively high in the HepG2
cells, which may have weakened p53-induced apoptosis. Notably, the
expression of ASPP1 and iASPP was reversed after interference of
ATPR in the HepG2 cells, however, the expression of ASPP2 showed no
change after ATPR interference. Thus, it was plausible to
demonstrate that ATPR could recover the function of p53 by inducing
the elevation of ASPP1 and the decrease of iASPP. In line with our
results, some studies have reported that downregulation of iASPP
inhibited the proliferation and promoted apoptosis in cells
expressing wild-type p53 (31,56,57).
Although studies have reported that ASPP2 promoted apoptosis in
cancers (55,58,59),
ASPP2 was not involved in the apoptosis process after ATPR
treatment, indicating that iASPP and ASPP1 compete with ASPP2 to
bind to p53. Taken together, ATPR induced a decline in MDM2 and
iASPP and an elevation in ASPP1 in the HepG2 cells, which may
partly enhance the function of p53, leading to an inhibitory effect
on proliferation.
Upon activation of p53, downstream target genes were
found to be upregulated, including Bax, p53-upregulated modulator
of apoptosis (PUMA), and cell cycle regulator p21 (60). The dysfunction of the cell cycle
causes malignant cell proliferation (61). The early G1 stage of the cell cycle
is coordinated by cyclin D/CDK4 and cyclin D/CDK6 complexes that
result in phosphorylation and inactivation of the retinoblastoma
(Rb) protein. Subsequently, this process leads to the derepression
and activation of E2F target genes such as cyclin E gene
facilitating G1 and S phase progression (62). p21 serves as a specific inhibitor of
the cyclin/CDK complexes and its overexpression can inactivate them
to promote cell cycle arrest in the G0/G1 or G2/M phase. Growth
arrest through overexpression of p21 is the normal response to p53
activation in cancer cells (60).
In the present study, the expression of p21 was increased by the
elevation of ATPR. Unlike previous drugs (e.g. melatonin and
oridonin) causing HepG2 cell cycle arrest in the G2/M phase through
increasing expression of p53 and p21 (63,64),
ATPR markedly arrested HepG2 cells in the G0/G1 phase, and induced
a marked reduction in G1 and S transition-related proteins,
including cyclin D and E, and CDK6. However, CDK4 expression was
constant in each group. Collectively, our results demonstrated that
ATPR may induce G0/G1 arrest through increasing the expression of
p21 and decreasing the expression of cyclin D and E, and CDK6 via
accumulation of p53. The results are in agreement with previous
studies in which MDM2-mediated p53 activation promoted cell cycle
arrest by increasing p21 expression (65,66).
In addition, ATPR-induced apoptosis in HepG2 cells was observed by
Hoechst staining featured by nuclear condensation and fragmentation
and flow cytometry. It has been reported that the Bcl-2/Bax ratio
could reflect the release of cytochrome c and the initiation
of apoptosis (67). Several studies
have revealed that accumulation and activation of p53 induced
apoptosis by regulating the expression of Bax. In addition, Bcl-2
blocked apoptosis by binding to Bax to prevent the release of the
pro-apoptotic proteins (24,49,51).
Similarly, in the present study, the subsequent elevation in Bax
and the reduction in Bcl-2 may be associated with accumulation of
p53 and modulation of the ASPP family in HepG2 cells treated with
ATPR. These downstream molecular events perhaps support the
critical roles of p53 in the apoptosis of HepG2 cells induced by
ATPR. However, the detailed mechanism of p53 in apoptosis
progression should be further studied.
In summary, ATPR was superior to ATRA in inhibiting
the proliferation of HepG2 cells in vitro. Upregulation of
p53 and ASPP1 as well as downregulation of iASPP may be involved in
the ATPR-mediated inhibition of proliferation by inducing G0/G1
cell cycle arrest and apoptosis. Therefore, ATPR may be a novel and
efficient solution to restore the function of p53 in HCC therapy.
In the future, other cell lines with different phenotypes (e.g.
mutated or null p53) should be used to further confirm the
roles of p53 and the ASPP family in cancer cell proliferation.
Acknowledgments
The present study was financially supported by the
National Natural Science Foundation of China (no. 81272399), and
the youth Projects of Wannan Medical College (no. WK201403).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
But DY, Lai CL and Yuen MF: Natural
history of hepatitis-related hepatocellular carcinoma. World J
Gastroenterol. 14:1652–1656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deuffic S, Poynard T, Buffat L and
Valleron AJ: Trends in primary liver cancer. Lancet. 351:214–215.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
El-Serag HB, Davila JA, Petersen NJ and
McGlynn KA; EI-Serag HB: The continuing increase in the incidence
of hepatocellular carcinoma in the United States: An update. Ann
Intern Med. 139:817–823. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Avila MA, Berasain C, Sangro B and Prieto
J: New therapies for hepatocellular carcinoma. Oncogene.
25:3866–3884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feitelson MA, Arzumanyan A, Kulathinal RJ,
Blain SW, Holcombe RF, Mahajna J, Marino M, Martinez-Chantar ML,
Nawroth R, Sanchez-Garcia I, et al: Sustained proliferation in
cancer: Mechanisms and novel therapeutic targets. Semin Cancer
Biol. 35(Suppl): S25–S54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi KW, Suh H, Jang S, Kim D and Lee CH:
HY253, a novel decahydrofluorene analog, induces apoptosis via
intrinsic pathway and cell cycle arrest in liver cancer HepG2
cells. J Microbiol Biotechnol. 25:413–417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vousden KH and Prives C: Blinded by the
light: The growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haupt S, Berger M, Goldberg Z and Haupt Y:
Apoptosis - the p53 network. J Cell Sci. 116:4077–4085. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meek DW: The p53 response to DNA damage.
DNA Repair. 3:1049–1056. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J and Kurokawa M: Regulation of MDM2
stability after DNA damage. J Cell Physiol. 230:2318–2327. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Michael D and Oren M: The p53-Mdm2 module
and the ubiquitin system. Semin Cancer Biol. 13:49–58. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giaccia AJ and Kastan MB: The complexity
of p53 modulation: Emerging patterns from divergent signals. Genes
Dev. 12:2973–2983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reifenberger G, Liu L, Ichimura K, Schmidt
EE and Collins VP: Amplification and overexpression of the MDM2
gene in a subset of human malignant gliomas without p53 mutations.
Cancer Res. 53:2736–2739. 1993.PubMed/NCBI
|
|
17
|
Zhao K, Zhou Y, Qiao C, Ni T, Li Z, Wang
X, Guo Q, Lu N and Wei L: Oroxylin A promotes PTEN-mediated
negative regulation of MDM2 transcription via SIRT3-mediated
deacetylation to stabilize p53 and inhibit glycolysis in wt-p53
cancer cells. J Hematol oncol. 8:412015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lauria A, Tutone M, Ippolito M, Pantano L
and Almerico AM: Molecular modeling approaches in the discovery of
new drugs for anti-cancer therapy: The investigation of p53-MDM2
interaction and its inhibition by small molecules. Curr Med Chem.
17:3142–3154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang W and Hu Y: Small molecule agents
targeting the p53-MDM2 pathway for cancer therapy. Med Res Rev.
32:1159–1196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montes de Oca Luna R, Wagner DS and Lozano
G: Rescue of early embryonic lethality in mdm2-deficient mice by
deletion of p53. Nature. 378:203–206. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
De Falco M and De Luca A: Cell cycle as a
target of antineoplastic drugs. Curr Pharm Des. 16:1417–1426. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Okayama H: Cell cycle control by anchorage
signaling. Cell Signal. 24:1599–1609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barroso-González J and Thomas G: Endosome
traffic machinery meets the p53–p21 axis. Mol Cell oncol.
2:e9750752015. View Article : Google Scholar
|
|
24
|
Li X, Huang JM, Wang JN, Xiong XK, Yang XF
and Zou F: Combination of chrysin and cisplatin promotes the
apoptosis of Hep G2 cells by up-regulating p53. Chem Biol Interact.
232:12–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sullivan A and Lu X: ASPP: A new family of
oncogenes and tumour suppressor genes. Br J Cancer. 96:196–200.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ahn J, Byeon IJ, Byeon CH and Gronenborn
AM: Insight into the structural basis of pro- and antiapoptotic p53
modulation by ASPP proteins. J Biol Chem. 284:13812–13822. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bergamaschi D, Samuels Y, Sullivan A,
Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, Smith P, Gasco
M, et al: iASPP preferentially binds p53 proline-rich region and
modulates apoptotic function of codon 72-polymorphic p53. Nat
Genet. 38:1133–1141. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agirre X, Román-Gómez J, Jiménez-Velasco
A, Garate L, Montiel-Duarte C, Navarro G, Vázquez I, Zalacain M,
Calasanz MJ, Heiniger A, et al: ASPP1, a common activator of TP53,
is inactivated by aberrant methylation of its promoter in acute
lymphoblastic leukemia. Oncogene. 25:1862–1870. 2006. View Article : Google Scholar
|
|
30
|
Song B, Bian Q, Zhang YJ, Shao CH, Li G,
Liu AA, Jing W, Liu R, Zhou YQ, Jin G, et al: Downregulation of
ASPP2 in pancreatic cancer cells contributes to increased
resistance to gemcitabine through autophagy activation. Mol Cancer.
14:1772015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen J, Xie F, Zhang L and Jiang WG: iASPP
is over-expressed in human non-small cell lung cancer and regulates
the proliferation of lung cancer cells through a p53 associated
pathway. BMC Cancer. 10:6942010. View Article : Google Scholar
|
|
32
|
Kwok SK, Park MK, Cho ML, Oh HJ, Park EM,
Lee DG, Lee J, Kim HY and Park SH: Retinoic acid attenuates
rheumatoid inflammation in mice. J Immunol. 189:1062–1071. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Siddikuzzaman GC, Guruvayoorappan C and
Berlin Grace VM: All trans retinoic acid and cancer.
Immunopharmacol Immunotoxicol. 33:241–249. 2011. View Article : Google Scholar
|
|
34
|
Ginestier C, Wicinski J, Cervera N,
Monville F, Finetti P, Bertucci F, Wicha MS, Birnbaum D and
Charafe-Jauffret E: Retinoid signaling regulates breast cancer stem
cell differentiation. Cell Cycle. 8:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee JH, Yoon JH, Yu SJ, Chung GE, Jung EU,
Kim HY, Kim BH, Choi DH, Myung SJ, Kim YJ, et al: Retinoic acid and
its binding protein modulate apoptotic signals in hypoxic
hepatocellular carcinoma cells. Cancer Lett. 295:229–235. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang HN, Zeng B, Zhang Y, Daskoulidou N,
Fan H, Qu JM and Xu SZ: Involvement of TRPC channels in lung cancer
cell differentiation and the correlation analysis in human
non-small cell lung cancer. PLoS one. 8:e676372013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gauchotte G, Lacomme S, Brochin L,
Tournier B, Cahn V, Monhoven N, Piard F, Klein M, Martinet N,
Rochette-Egly C, et al: Retinoid acid receptor expression is
helpful to distinguish between adenoma and well-differentiated
carcinoma in the thyroid. Virchows Arch. 462:619–632. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tang XH and Gudas LJ: Retinoids, retinoic
acid receptors, and cancer. Annu Rev Pathol. 6:345–364. 2011.
View Article : Google Scholar
|
|
39
|
Saito A, Sugawara A, Uruno A, Kudo M,
Kagechika H, Sato Y, Owada Y, Kondo H, Sato M, Kurabayashi M, et
al: All-trans retinoic acid induces in vitro angiogenesis via
retinoic acid receptor: Possible involvement of paracrine effects
of endogenous vascular endothelial growth factor signaling.
Endocrinology. 148:1412–1423. 2007. View Article : Google Scholar
|
|
40
|
Freemantle SJ, Spinella MJ and Dmitrovsky
E: Retinoids in cancer therapy and chemoprevention: Promise meets
resistance. Oncogene. 22:7305–7315. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Su M, Alonso S, Jones JW, Yu J, Kane MA,
Jones RJ and Ghiaur G: All-trans retinoic acid activity in acute
myeloid leukemia: Role of cytochrome P450 enzyme expression by the
microenvironment. PLoS one. 10:e01277902015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou DC, Hallam SJ, Lee SJ, Klein RS,
Wiernik PH, Tallman MS and Gallagher RE: Constitutive expression of
cellular retinoic acid binding protein II and lack of correlation
with sensitivity to all-trans retinoic acid in acute promyelocytic
leukemia cells. Cancer Res. 58:5770–5776. 1998.PubMed/NCBI
|
|
43
|
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM,
Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al: Use of arsenic
trioxide (As2O3) in the treatment of acute
promyelocytic leukemia (APL): II. Clinical efficacy and
pharmacokinetics in relapsed patients. Blood. 89:3354–3360.
1997.PubMed/NCBI
|
|
44
|
Wang H, Gui SY, Chen FH, Zhou Q and Wang
Y: New insights into 4-amino-2-tri-fluoromethyl-phenyl ester
inhibition of cell growth and migration in the A549 lung
adenocarcinoma cell line. Asian Pac J Cancer Prev. 14:7265–7270.
2013. View Article : Google Scholar
|
|
45
|
Wang N, Ge JF, Pan CX, Peng XQ, Chen HH,
Wang XQ, Tang J, Hu W and Chen FH: Anti-tumor effect of
4-amino-2-trifluoromethyl-phenyl retinate on human breast cancer
MCF-7 cells via up-regulation of retinoid receptor-induced gene-1.
Biomed Pharmacother. 67:687–692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hu A, Yang Y, Zhang S, Zhou Q, Wei W and
Wang Y: 4-Amino-2-trifluoromethyl-phenyl retinate inhibits the
migration of BGC-823 human gastric cancer cells by downregulating
the phosphorylation level of MLC II. Oncol Rep. 32:1473–1480.
2014.PubMed/NCBI
|
|
47
|
Su C: Survivin in survival of
hepatocellular carcinoma. Cancer Lett. Jun 25–2015.Epub ahead of
print. piiS0304–3835. (15): 00409–7. View Article : Google Scholar
|
|
48
|
Chaparro M, González Moreno L,
Trapero-Marugán M, Medina J and Moreno-Otero R: Review article:
Pharmacological therapy for hepatocellular carcinoma with sorafenib
and other oral agents. Aliment Pharmacol Ther. 28:1269–1277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mu R, Qi Q, Gu H, Wang J, Yang Y, Rong J,
Liu W, Lu N, You Q and Guo Q: Involvement of p53 in oroxylin
A-induced apoptosis in cancer cells. Mol Carcinog. 48:1159–1169.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fujioka S, Schmidt C, Sclabas GM, Li Z,
Pelicano H, Peng B, Yao A, Niu J, Zhang W, Evans DB, et al:
Stabilization of p53 is a novel mechanism for proapoptotic function
of NF-kappaB. J Biol Chem. 279:27549–27559. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ren J, Cheng H, Xin WQ, Chen X and Hu K:
Induction of apoptosis by 7-piperazinethylchrysin in HCT-116 human
colon cancer cells. Oncol Rep. 28:1719–1726. 2012.PubMed/NCBI
|
|
52
|
Zhu MY, Guo JL, Xia H, Li W, Lu Y, Dong X,
Chen Y, Fu SG, Xie XJ and Li FS: The anti-apoptotic effect of
cytoplasmic alpha-fetoprotein in hepatoma cells induced by
all-trans retinoic acid involves activation of the PI3K/AKT
signaling pathway. Zhonghua Gan Zang Bing Za Zhi. 22:837–842.
2014.In Chinese. PubMed/NCBI
|
|
53
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang Y, Ludwig RL, Jensen JP, Pierre SA,
Medaglia MV, Davydov IV, Safiran YJ, Oberoi P, Kenten JH, Phillips
AC, et al: Small molecule inhibitors of HDM2 ubiquitin ligase
activity stabilize and activate p53 in cells. Cancer Cell.
7:547–559. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang Y, Godin-Heymann N, Dan Wang X,
Bergamaschi D, Llanos S and Lu X: ASPP1 and ASPP2 bind active RAS,
potentiate RAS signalling and enhance p53 activity in cancer cells.
Cell Death Differ. 20:525–534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu ZJ, Cai Y, Hou L, Gao X, Xin HM, Lu X,
Zhong S, Gu SZ and Chen J: Effect of RNA interference of iASPP on
the apoptosis in MCF-7 breast cancer cells. Cancer Invest.
26:878–882. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liu H, Wang M, Diao S, Rao Q, Zhang X,
Xing H and Wang J: siRNA-mediated down-regulation of iASPP promotes
apoptosis induced by etoposide and daunorubicin in leukemia cells
expressing wild-type p53. Leuk Res. 33:1243–1248. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang B, Qiao L, Shi Y, Feng X, Chen D and
Guo H: ASPP2 inhibits oxaliplatin-induced autophagy and promotes
apoptosis of colon cancer cells. Xi Bao yu Fen Zi Mian yi Xue Za
Zhi. 31:898–904. 2015.In Chinese. PubMed/NCBI
|
|
59
|
Hou Q, Zhao H, Gong W, Zhu Z, Han Y, Chen
D and Guo H: Phosphorylation status of ASPP2 modulates p53
apoptotic function in oxaliplatin-induced apoptosis of colorectal
cancer HCT116 cells. Zhonghua Zhong Liu Za Zhi. 36:418–423. 2014.In
Chinese. PubMed/NCBI
|
|
60
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lange CA and Yee D: Killing the second
messenger: Targeting loss of cell cycle control in
endocrine-resistant breast cancer. Endocr Relat Cancer. 18:C19–C24.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Moeller SJ and Sheaff RJ: G1 phase:
Components, conundrums, context. Results Probl Cell Differ.
42:1–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Martín-Renedo J, Mauriz JL, Jorquera F,
Ruiz-Andrés O, González P and González-Gallego J: Melatonin induces
cell cycle arrest and apoptosis in hepatocarcinoma HepG2 cell line.
J Pineal Res. 45:532–540. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang H, Ye Y, Chui JH, Zhu GY, Li YW, Fong
DW and Yu ZL: Oridonin induces G2/M cell cycle arrest and apoptosis
through MAPK and p53 signaling pathways in HepG2 cells. oncol Rep.
24:647–651. 2010.PubMed/NCBI
|
|
65
|
Vousden KH: Switching from life to death:
The Miz-ing link between Myc and p53. Cancer Cell. 2:351–352. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Proietti S, Cucina A, Dobrowolny G,
D'Anselmi F, Dinicola S, Masiello MG, Pasqualato A, Palombo A,
Morini V, Reiter RJ, et al: Melatonin down-regulates MDM2 gene
expression and enhances p53 acetylation in MCF-7 cells. J Pineal
Res. 57:120–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tan ML, Ooi JP, Ismail N, Moad AI and
Muhammad TS: Programmed cell death pathways and current antitumor
targets. Pharm Res. 26:1547–1560. 2009. View Article : Google Scholar : PubMed/NCBI
|