Introduction
Human brain glioma, also known as neuroglioma,
exhibits invasive growth without an overt boundary to surrounding
tissues; it has unique cell origin, pathological structures,
biological features, and clinical symptoms (1–6).
Malignant glioma is a tumor with extremely high malignancy and
recurrence rate; low total resection rate, high postoperative
recurrence and residual lesion recurrence caused by radiation, and
chemotherapeutic tolerance constitute the main problems faced by
neurosurgeons (7–13). Considering the bottleneck in glioma
treatment, dissecting relevant molecular mechanisms and finding
tumor-related gene therapy approaches in human glioma attracts
increasing attention for improved diagnosis, treatment and
prognosis of glioma (6,14–23).
Our laboratory and others have recently screened
clinical relevant genes in glioma (24–28);
notably, we found a new glioma-expressed gene that plays an
important regulatory role in the proliferation, progression and
apoptosis of glioma cells. The gene is called ribonuclease H2,
subunit A (RNaseH2A) also known as AGS4, JUNB, RNHL, RNHIA and
RNaseHI. RNaseH2A (1148 bp), located on chromosome 19 (29,30),
is a component of ribonuclease H2 (RNAseH2) and is responsible for
its endoribonuclease activity (31–33).
RNaseH was discovered and isolated from calf thymus (34,35)
and is widely distributed in mammalian cells, yeasts, prokaryotes
and virus particles; it catalyzes in-nucleus degradation of RNA in
DNA-RNA hybrids and is involved in reverse transcriptase of
multifunctional enzymes in retroviruses, playing an important role
in various stages of viral genome transcription (36,37).
In eubacteria, RNaseH is required for several processes, including
removal of RNA primers from Okazaki fragments, transcription of
primers required for DNA polymerase I initiated DNA synthesis, and
removal of R-ring to provide conditional initiating sites required
for irregular DNA synthesis (38).
In eukaryotes, RNaseH may play similar roles (39). Recent studies have shown that
RNaseH2A mutations lead to Aicardi-Goutieres syndrome, an autosomal
recessive neurological dysfunction, which mainly causes
microcephaly, mental motor development retardation, cerebral
calcification, increased IF-α and leukocytes in cerebrospinal
fluid, fever, thrombocytopenia, and hepatitis (40–42).
RNaseH2A was proposed as a putative cancer target (31). In agreement, logistic regression
analysis revealed that expression levels of RNaseH2A, among other
genes, were positively correlated with aggressive prostate cancer
(43). However, studies assessing
RNaseH2A in relation to glioma are scarce. Therefore, in the
present study, we aimed to assess the role of RNaseH2A in glioma,
exploring the underlying mechanisms.
Our data demonstrated that RNaseH2A silencing
altered cell proliferation and clone formation and enhanced
apoptosis in the glioma cells in vitro. Meanwhile,
RNaseH2A-knockdown xenografted glioma cells were less tumorigenic
in a nude mouse model. Moreover, microarray data confirmed that
RNaseH2A regulated genes that are frequently involved in multiple
important cellular regulatory processes, including focal adhesion,
cancer, p53 signaling, and cell cycle. Our findings provide a
strong basis for finding new gene-based therapeutic approaches
against human glioma.
Materials and methods
Cell culture
The human glioblastoma (GBM) cell lines U87, U251,
A373 and A172 were maintained in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% fetal bovine serum (FBS) and 1%
penicillin-streptomycin at 37°C in 5% CO2.
RNaseH2A knockdown in GBM cells
For RNaseH2A silencing, U85 and U251 cells were
transfected with the pGCSIL-GFP plasmid containing
RNaseH2A-specific shRNA, or non-effective shRNA control fragments
(GeneChem, Montreal, QC, Canada).
Gene expression microarray analyses
Total RNA was extracted from the U87 cells
(transfected with NC or RNaseH2A shRNA) using an RNeasy Mini-kit
(Qiagen, Valencia, CA, USA), and quantified on a Nanodrop ND-1000
spectrophotometer (NanoDrop Technology). Samples with adequate RNA
quality index (>7) were used for the microarray analyses.
Genome-wide transcriptome profiles were assessed by
expression microarrays (GeneChip PrimeView human, Affymetrix) and
validated by quantitative real-time PCR. Microarray data and fully
detailed experimental procedures are published online at Gene
Expression Omnibus (GEO, NCBI) (http://www.ncbi.nlm.nih.gov/geoprofiles/).
Differentially expressed genes were identified using 1.5- or 2-fold
change as cutoff values. Gene ontology enrichment analysis was
carried out using David Functional Annotation Resources 6.7
(http://david.abcc.ncifcrf.gov/) and the
KEGG pathway analysis.
Cell proliferation, apoptosis, and cell
cycle analysis
GBM cells were seeded onto 96-well plates at
2×103 cells/well for culture. Cell proliferation was
measured using the
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay 96 h after transfection with NC or RNaseH2A shRNA. Briefly,
20 µl MTT solution (5 mg/ml) was added to each well for 4 h
at 37°C. After the medium was aspirated, 100 µl dimethyl
sulfoxide (DMSO) was added. Optical density was measured at 492 nm
on a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Viability index was calculated as experimental OD
value/control OD value. Three independent experiments were
performed in quadruplicate.
For cell cycle analysis, the cells were stained with
propidium iodide (PI) solution (50 µg/ml) and analyzed on a
FACSCalibur flow cytometer 24 h after transfection.
Apoptosis was assessed using a fluorescein
isothiocyanate (FITC) Annexin V staining kit (Life Technologies,
Grand Island, NY, USA) followed by fluorescence-activated cell
sorter (FACS) analysis according to the manufacturer's
instructions.
Colony formation assay
Following treatment, adherent cells were trypsinized
and counted to determine viability. Then, 800 viable cells were
seeded into each well of a 6-well plate (in triplicate). Cells were
allowed to adhere and grow for 10–14 days. To visualize colonies,
media were removed, and cells were fixed in paraformaldehyde for 30
min and stained with Giemsa solution (ECM 550, Chemicon). Finally,
colonies were counted; data are presented as mean colony number ±
SEM from at least three independent experiments.
RT-PCR and quantitative real-time
PCR
RNA extraction was performed with TRIzol (Invitrogen
Life Technologies, Carlsbad, CA, USA); cDNA was synthesized using
RT-Phusion kit (Thermo Scientific, Waltham, MA, USA). Gene-specific
mRNA levels were quantified by standard RT-PCR or quantitative PCR
(qPCR), using the ΔΔCt method, as previously described (44). Primer sequences are listed in
Table I.
 | Table IList of primers. |
Table I
List of primers.
| Gene | Sequence |
|---|
| RNaseH2A-F |
AAGACCCTATTGGAGAGCGAG |
| RNaseH2A-R |
AGTTCAGGTTGTATTTGACCCG |
| GAPDH-F |
TGACTTCAACAGCGACACCCA |
| GAPDH-R |
CACCCTGTTGCTGTAGCCAAA |
| MDM2-F |
GAATCATCGGACTCAGGTACATC |
| MDM2-R |
TCTGTCTCACTAATTGCTCTCCT |
| SMAD3-F |
GACTACAGCCATTCCATCC |
| SMAD3-R |
CAGGTCCAAGTTATTATGTGC |
| FGF2-F |
ATCAAAGGAGTGTGTGCTAACC |
| FGF2-R |
ACTGCCCAGTTCGTTTCAGTG |
| SMAD2-F |
AGAGGGAAACAAGAACAGG |
| SMAD2-R |
ATGCTCTGGCGTCTACTG |
| IGF1R-F |
TGCGTGAGAGGATTGAGTTTC |
| IGF1R-R |
CTTATTGGCGTTGAGGTATGC |
| IL6-F |
CAAATTCGGTACATCCTCG |
| IL6-R |
CTCTGGCTTGTTCCTCACTA |
| IL8-F |
TGGCAGCCTTCCTGATTT |
| IL8-R |
AACCCTCTGCACCCAGTT |
| FAS-F |
ACACTCACCAGCAACACCAA |
| FAS-R |
CTTCCTTTCTCTTCACCCAAACA |
| BIRC5-F |
ACCGCATCTCTACATTCAAG |
| BIRC5-R |
CAAGTCTGGCTCGTTCTC |
| TGFBR2-F |
GTGCCAACAACATCAACC |
| TGFBR2-R |
GACTGCCACTGTCTCAAACT |
Western blot analysis
After treatment, the cells were harvested and lysed
in RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1%
Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) that contained
a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA)
for 30 min at 4°C. Forty micrograms of protein from each lysate
were fractionated by 10% SDS-PAGE and transferred to polyvinylidene
difluoride membranes (Millipore, Bedford, MA, USA). After blocking
with 5% nonfat milk in PBS-Tween-20 for 1 h at room temperature,
the membranes were blotted with the appropriate primary antibody.
Primary antibodies against RNaseH2A (1:1,000; Proteintech), IL-6
(1:2,000; Abcam), FAS (1:1,000; Abcam), IGTA2 (1:10,000; Abcam),
GAPDH (1:20,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA) were incubated at 4°C overnight. After washing four times with
TBST, the membranes were incubated with a horseradish peroxidase
(HRP)-conjugated anti-rabbit or anti-mouse secondary antibody
(Santa Cruz Biotechnology, Inc.) for 2 h. The proteins were
visualized using enhanced chemiluminescence (ECL; Beyotime
Institute of Biotechnology, Nantong, China).
In vivo GBM xenograft studies
All mouse care and experiments were carried out with
approval of the Institutional Animal Care and Use Committee at the
Beijing Tian Tan Hospital, Capital Medical University. Subcutaneous
xenograft models were performed by injecting U87 cells transfected
with NC or RNaseH2A shRNA as previously described (45).
Statistical analysis
Results are representative of 3 independent
experiments. Data are expressed as the mean ± standard deviation
(SD). Student's t-test was employed to compare groups. P<0.05
was considered statistically significant. SPSS 10.0 software (SPSS,
Chicago, IL, USA) was used for statistical analyses.
Results
The RNaseH2A gene is expressed in GBM
cells
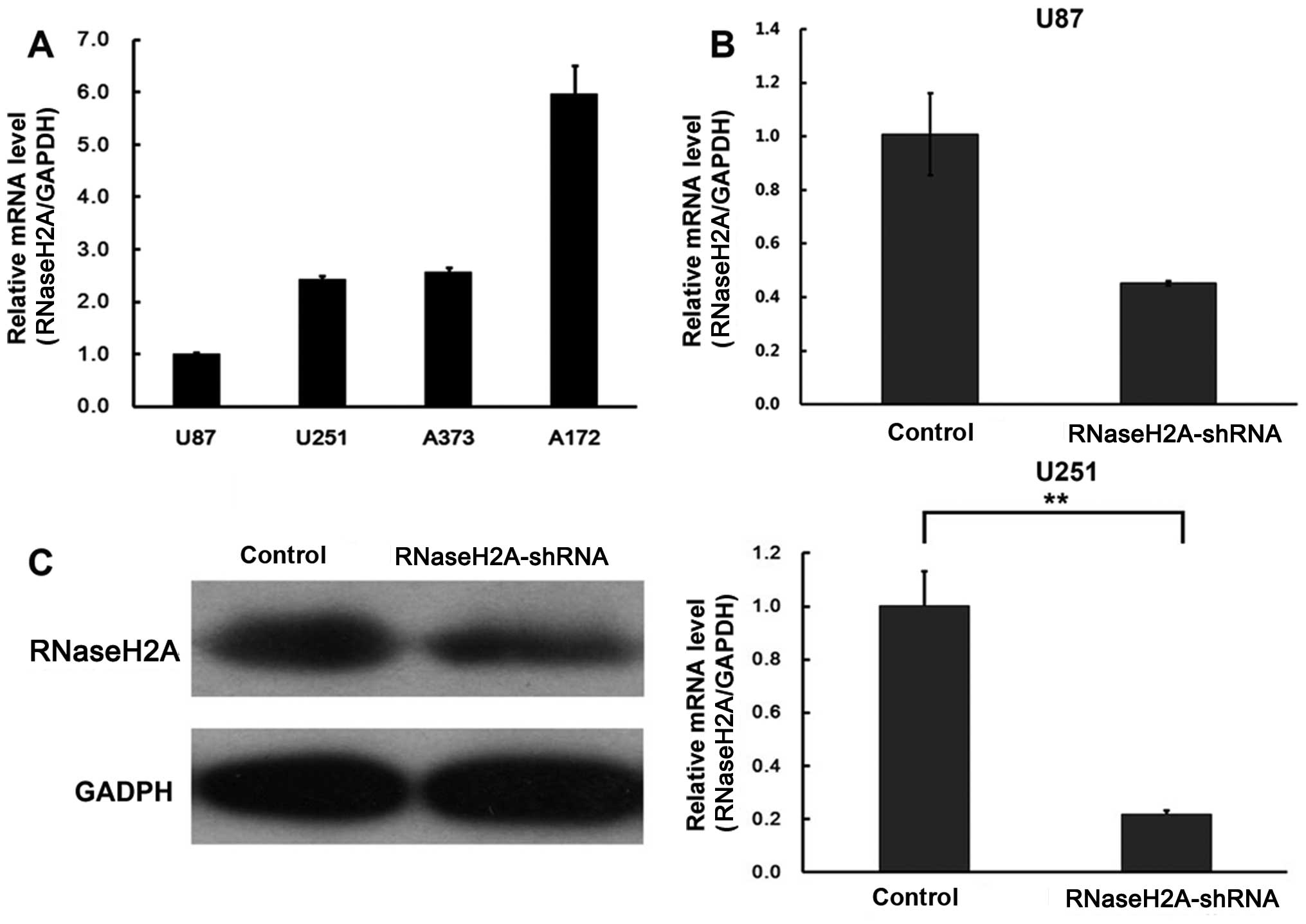
Four GBM cell lines, including U87, U251, A373 and
A172 were assessed for RNaseH2A expression by RT-PCR. RNaseH2A was
detected in all of the cell lines (Fig.
1A).
RNaseH2A silencing suppresses glioma cell
proliferation in vitro and in vivo
Two cell lines (U87 and U251) were selected for RNA
interference experiments, to assess RNaseH2A function in glioma
cells. As shown in Fig. 1B and C,
RNaseH2A was successfully silenced in both cell lines. RNaseH2A
mRNA expression was reduced to a greater extent in the U251 cell
line.
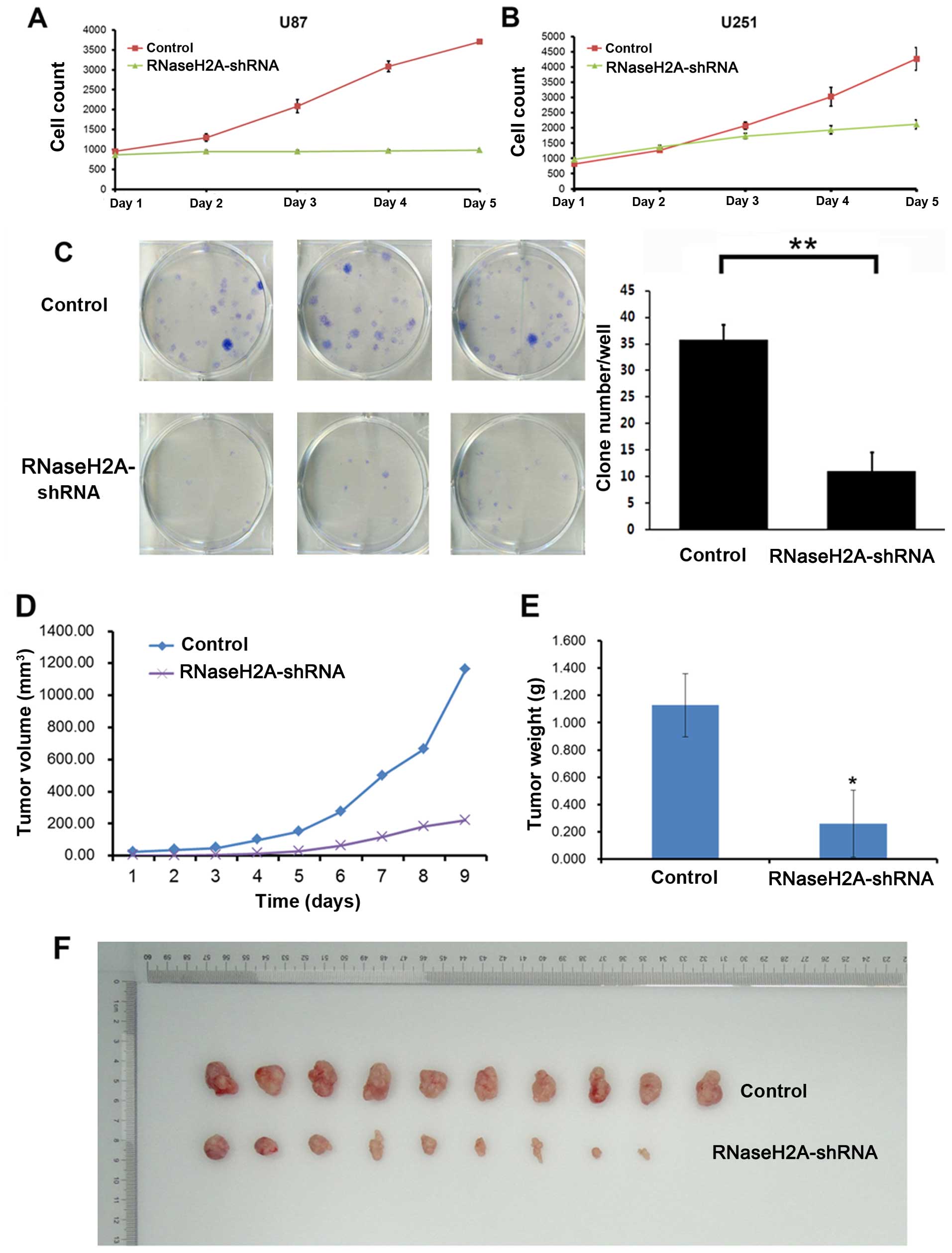
Cell proliferation analysis was performed in the two
glioma cell lines (U87 and U251) transfected with RNaseH2A-shRNA.
Both glioma cell lines showed significantly reduced viability after
RNaseH2A silencing (Fig. 2A and B).
Five days after shRNA transfection, cell viability was reduced by
70.7 and 57.8% in the U87 and U251 cells, respectively. In
accordance, colony formation was also significantly decreased
(P<0.05) in the U87 cells after RNaseH2A knockdown, compared
with the control values (Fig. 2C).
Furthermore, xenografted tumor growth in the RNaseH2A-shRNA group
was markedly reduced compared with that of the normal control group
in vivo (Fig. 2D–F). Indeed,
34 days after U87 cell injection, tumor volumes were 219.29±246.43
and 1160.26±222.61 mm3 in the RNaseH2A-shRNA and normal
control groups, respectively; tumor weights of 1.127±0.232 and
0.261±0.245 g were obtained from the control and RNaseH2A-silenced
cells, respectively (P<0.01). These findings demonstrated that
RNaseH2A was required for glioma cell proliferation, both in
vitro and in vivo.
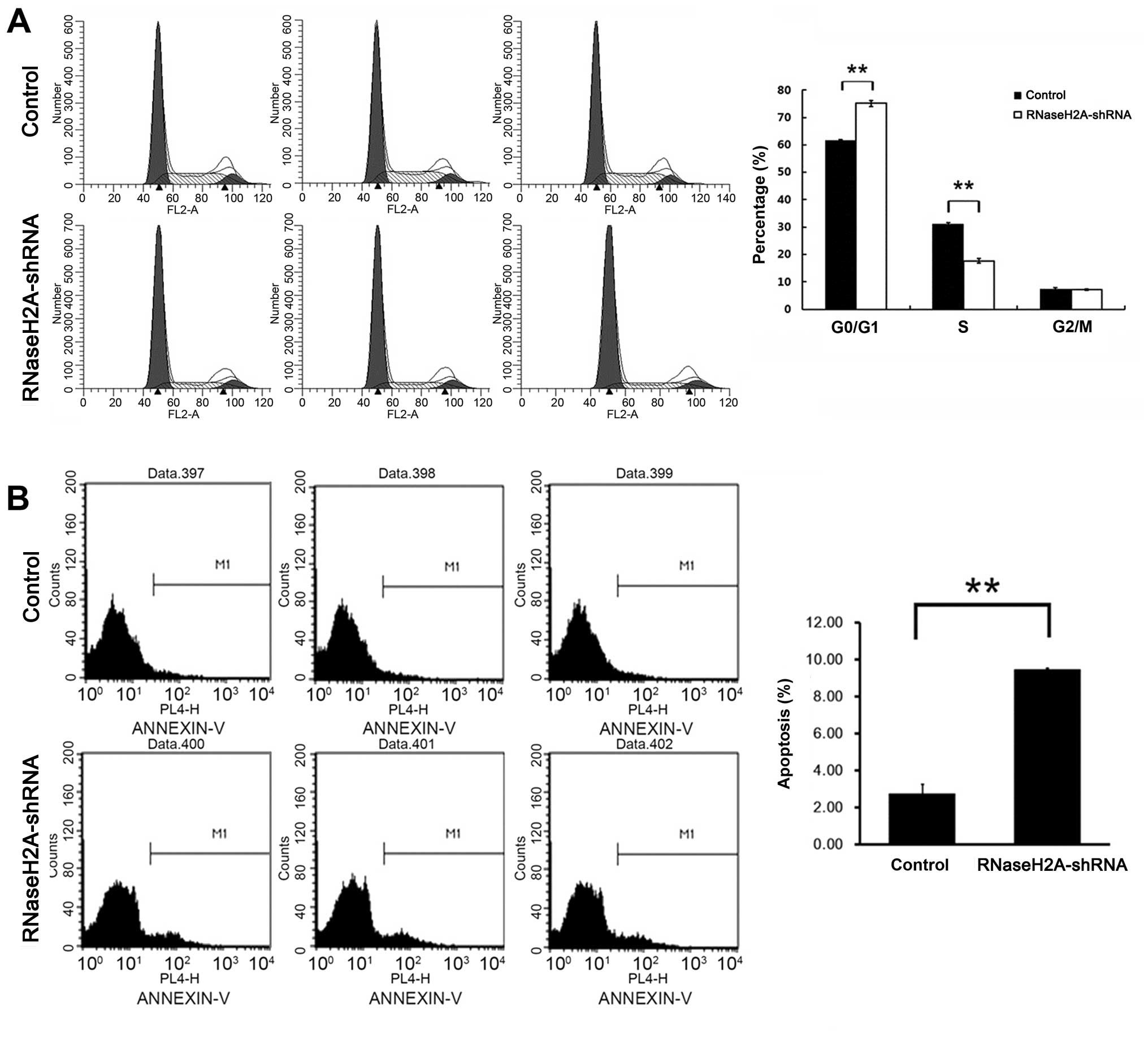
RNaseH2A silencing causes cell cycle
arrest in the G0/G1 phase and apoptosis in glioma cells
To explore the mechanism underlying the observed
impaired growth of glioma cells after RNaseH2A silencing, cell
cycle distribution was assessed by flow cytometry. RNaseH2A
silencing resulted in an increased percentage of U87 cells in the
G0/G1 phase, with 75.14±1.004 and 61.51±0.43% obtained
respectively, in the RNaseH2A knockdown and control groups,
respectively (P<0.01, Fig.
3A).
The effect of RNaseH2A on apoptosis in glioma cells
(Fig. 3B) was also investigated. As
shown in Fig. 3B, RNaseH2A
knockdown induced cell apoptosis by 4.5-fold compared to the normal
control cells.
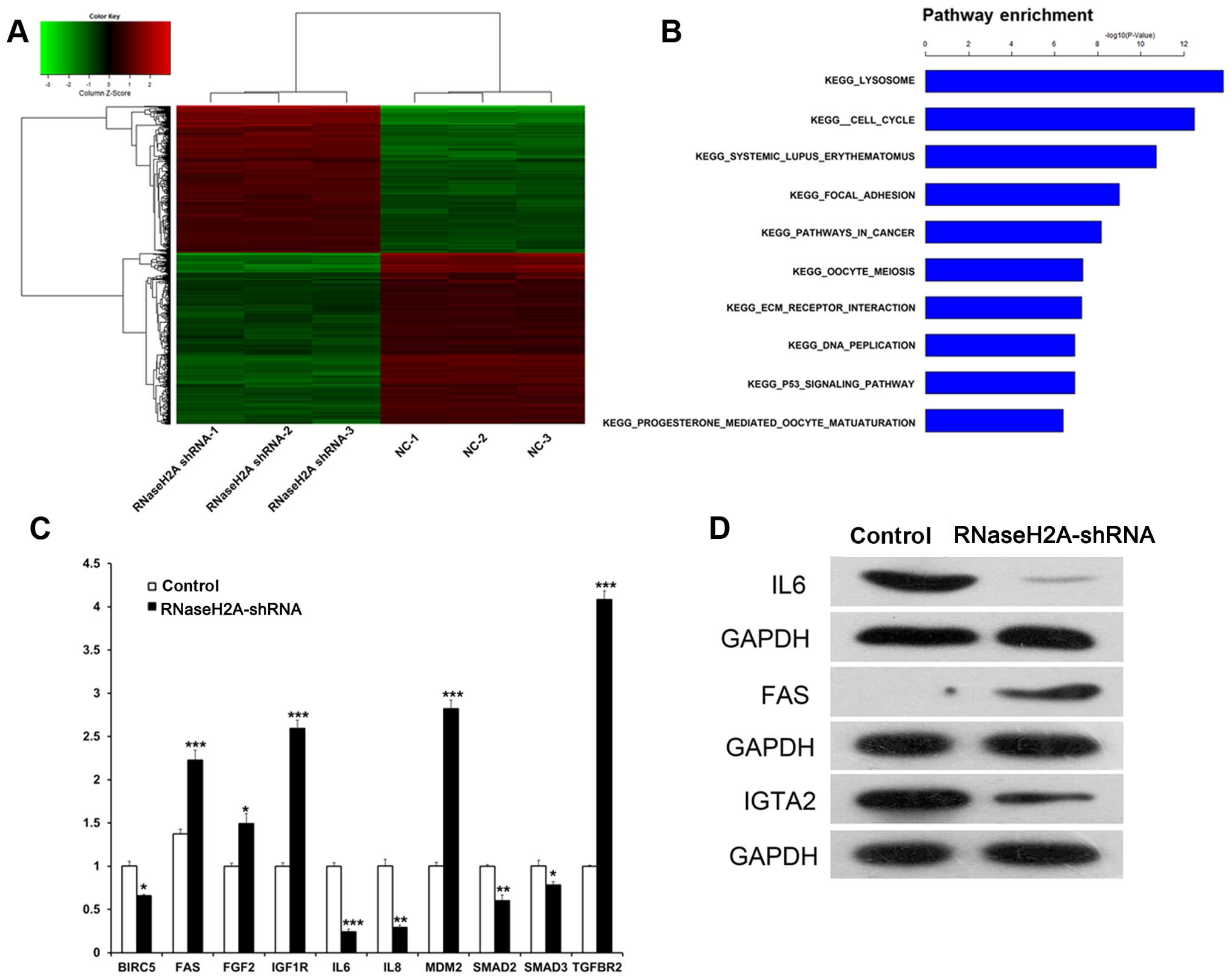
RNaseH2A silencing induces differential
expression of genes involved in multiple cell signaling
pathways
To further explore the biological significance of
RNaseH2A in glioma cells, gene expression profiles of U87 cells
transfected with or without RNaseH2A shRNA were assessed. As shown
in the heat map (Fig. 4A), gene
expression profiles in the NC and RNaseH2A shRNA groups were
remarkably different. A total of 821 upregulated and 941
downregulated genes were found in the RNaseH2A shRNA-transfected
U87 cells. GO and KEGG pathway analysis revealed that the
differentially expressed genes are frequently involved in multiple
important cellular regulatory processes, including focal adhesion,
cancer, p53 signaling, and cell cycle (Fig. 4B). Therefore, our data suggest that
RNaseH2A shRNA affects various genes involved in important
biological processes.
To validate the microarray data, qRT-PCR and western
blot analysis were carried out. Protein and gene expression levels
were consistent with the microarray data. For instance, IL-6 mRNA
levels were decreased significantly in the RNaseH2A shRNA group
compared to the control group, while FAS mRNA amounts were
increased significantly in the RNaseH2A shRNA group compared to the
control group (P<0.001); western blotting yielded similar
results. These findings indicated that IL-6 and FAS might be
suitable RNaseH2A target genes (Fig. 4C
and D).
Discussion
RNaseH2A was expressed in all glioma cells assessed.
Notably, RNaseH2A knockdown by shRNA silencing resulted in reduced
proliferation and viability of the tumor cells, which were blocked
in the G0/G1 phase. In addition, RNaseH2A-knockdown cells showed
increased apoptosis in vitro. In agreement, clone formation
(in vitro) and tumor growth (in vivo) were
significantly decreased after RNaseH2A silencing. Gene-chip based
transcriptome assay indicated that RNaseH2A plays an important role
in several signaling pathways involved in cell proliferation,
including the IL-6 and FAS pathways.
Gene dysregulation is a hallmark of tumor genesis
and progression (46–51), with post-transcriptional regulation
of messenger RNA constituting an important step. Ribonucleases
(RNases) catalyze RNA breakdown, thus influencing mRNA turnover and
gene expression; their dysfunction is linked to various types of
tumors. For instance, failure to recruit PARN, a poly A
ribonuclease, has been observed in malignant glioma (52,53).
In addition, reduction and/or depletion of XRN1, a 5′–3′
exonuclease which initiates mRNA decay, is implicated in primary
osteogenic sarcoma and its derived cell lines (54). Furthermore, the gene encoding
truncated RNase L is positively correlated with hereditary prostate
cancer (55), while moderated
reduction in enzyme activity is related to a higher risk of
prostate and colorectal cancers (56) as well as pancreatic carcinoma
(57–59). IRE1, a transmembrane
endoribonuclease found in the endoplasmic reticulum (60), can act as a tumor suppressor,
deciding the fate of cancer cells (61). RNases from the miRNA pathway,
including Drosha, Dicer, and Ago2, are also implicated in tumor
biology. For instance, elevated expression of Drosha was found in
esophageal cancers (62), with its
suppression leading to decreased cancer cell proliferation; in
addition, elevated mRNA levels and genomic copy numbers of Drosha
were found in clinical cervical squamous cell carcinoma samples and
derived cell lines (63). Several
studies have reported overexpression of Dicer in multiple cancers,
including salivary gland, lung, prostate, and ovary carcinomas, as
well as Burkitt's lymphoma; Ago2-overexpression was also found in
these cancers (64–68).
RNaseH2A is a component of the heterotrimeric type
II ribonuclease H enzyme (RNAseH2), which provides the main
ribonuclease H activity (31–33).
Consistent with our findings, a previous study indicated that
RNaseH2A is a putative anticancer drug target in transformed stem
cells (31). However, how RNaseH2A
is involved in tumor genesis and progression remains unclear. In
this study, gene silencing and transcriptome profiling were
combined to assess downstream signaling pathways mediating the
effect of RNaseH2A. Multiple genes involved in important cellular
processes, including focal adhesion, cancer, p53 signaling, and
cell cycle, were differentially expressed after RNaseH2A silencing.
Further analysis demonstrated that IL-6 was downregulated after
RNaseH2A knockdown. GBM cells produced IL-6 in vitro and
in vivo (69,70), and Goswami et al reported
that IL-6 mediated autocrine growth promotion in the human GBM
multiforme cell line U87MG (71);
in addition, targeting IL-6Rα or IL-6 expression in GSCs impaired
cell growth and increased the survival of mice bearing intracranial
human glioma xenografts (71), in
agreement with our findings of decreased xenograft growth after
RNaseH2A silencing. We also found that FAS expression was
upregulated after RNaseH2A silencing, which is consistent with the
enhanced apoptosis described above after RNaseH2A silencing. FAS
receptor is a death receptor on the surface of cells that leads to
programmed cell death (72).
Activation of FAS was shown to initiate apoptosis in different
glioma cell lines (73).
Fas-ligand, which is expressed in GBM cell lines and primary
astrocytic brain tumors (74), can
lead to >90% inhibition of clonal tumor cell growth in high
grade gliomas ex vivo (73).
Thus, silencing of RNaseH2A may inhibit proliferation and promote
apoptosis in glioma cells, via downregulation of IL-6 and
upregulation of FAS simultaneously.
In summary, we unveiled a possible mechanism by
which RNaseH2A contributes to glioma cell proliferation. Indeed,
RNaseH2A silencing resulted in growth arrest and apoptosis in
glioma cells, possibly through IL-6 and FAS regulation. These
findings indicate that RNaseH2A upregulation may contribute to
gliomagenesis and progression, via modulation of factors involved
in cell growth and apoptosis. Therefore, RNaseH2A should be
considered as a potential molecular target for glioma diagnosis and
treatment.
Acknowledgments
This study was supported by the National Key
Technologies R&D Program of the Ministry of Science and
Technology of China (no. 2013BAI09B03)
References
|
1
|
Adamczyk LA, Williams H, Frankow A, Ellis
HP, Haynes HR, Perks C, Holly JM and Kurian KM: Current
understanding of circulating tumor cells–potential value in
malignancies of the central nervous system. Front Neurol.
6:1742015. View Article : Google Scholar
|
|
2
|
Aparicio-Blanco J and Torres-Suárez AI:
Glioblastoma multiforme and lipid nanocapsules: a review. J Biomed
Nanotechnol. 11:1283–1311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cuddapah VA, Robel S, Watkins S and
Sontheimer H: A neurocentric perspective on glioma invasion. Nat
Rev Neurosci. 15:455–465. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Errico A: CNS cancer: new options for
glioblastoma. Nat Rev Clin Oncol. 11:1242014.PubMed/NCBI
|
|
5
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Westphal M and Lamszus K: Circulating
biomarkers for gliomas. Nat Rev Neurol. 11:556–566. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang ZZ, Shields LB, Sun DA, Zhang YP,
Hunt MA and Shields CB: The art of intraoperative glioma
identification. Front Oncol. 5:1752015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Samdani AF, Torre-Healy A, Khalessi A,
McGirt M, Jallo GI and Carson B: Intraventricular ganglioglioma: a
short illustrated review. Acta Neurochir (Wien). 151:635–640. 2009.
View Article : Google Scholar
|
|
9
|
Gautschi OP, van Leyen K, Cadosch D,
Hildebrandt G and Fournier JY: Fluorescence guided resection of
malignant brain tumors-breakthrough in the surgery of brain tumors.
Praxis Bern (1994). 98:643–647. 2009.In German. View Article : Google Scholar
|
|
10
|
Signorelli F, Guyotat J, Elisevich K and
Barbagallo GM: Review of current microsurgical management of
insular gliomas. Acta Neurochir (Wien). 152:19–26. 2010. View Article : Google Scholar
|
|
11
|
Bello L, Fava E, Carrabba G, Papagno C and
Gaini SM: Present day's standards in microsurgery of low-grade
gliomas. Adv Tech Stand Neurosurg. 35:113–157. 2010.PubMed/NCBI
|
|
12
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lacroix M, Abi-Said D, Fourney DR,
Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch
SJ, Holland E, et al: A multivariate analysis of 416 patients with
glioblastoma multiforme: prognosis, extent of resection, and
survival. J Neurosurg. 95:190–198. 2001. View Article : Google Scholar
|
|
14
|
Ohgaki H and Kleihues P: Genetic
alterations and signaling pathways in the evolution of gliomas.
Cancer Sci. 100:2235–2241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marumoto T and Saya H: Molecular biology
of glioma. Adv Exp Med Biol. 746:2–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patel M, Vogelbaum MA, Barnett GH, Jalali
R and Ahluwalia MS: Molecular targeted therapy in recurrent
glioblastoma: current challenges and future directions. Expert Opin
Investig Drugs. 21:1247–1266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Spasic M, Chow F, Tu C, Nagasawa DT and
Yang I: Molecular characteristics and pathways of Avastin for the
treatment of glioblastoma multiforme. Neurosurg Clin N Am.
23:417–427. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu JJ and Wong ET: Personalized medicine
for glioblastoma: current challenges and future opportunities. Curr
Mol Med. 13:358–367. 2013.PubMed/NCBI
|
|
19
|
Goodenberger ML and Jenkins RB: Genetics
of adult glioma. Cancer Genet. 205:613–621. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Assi H, Candolfi M, Baker G, Mineharu Y,
Lowenstein PR and Castro MG: Gene therapy for brain tumors: basic
developments and clinical implementation. Neurosci Lett. 527:71–77.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carén H, Pollard SM and Beck S: The good,
the bad and the ugly: epigenetic mechanisms in glioblastoma. Mol
Aspects Med. 34:849–862. 2013. View Article : Google Scholar :
|
|
22
|
Rizzo D, Ruggiero A, Martini M, Rizzo V,
Maurizi P and Riccardi R: Molecular biology in pediatric high-grade
glioma: impact on prognosis and treatment. BioMed Res Int.
2015:2151352015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mischel PS and Cloughesy TF: Targeted
molecular therapy of GBM. Brain Pathol. 13:52–61. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Chen LH, Wan H, Yang R, Wang Z,
Feng J, Yang S, Jones S, Wang S, Zhou W, et al: Exome sequencing
identifies somatic gain-of-function PPM1D mutations in brainstem
gliomas. Nat Genet. 46:726–730. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wan W, Xu X, Jia G, Li W, Wang J, Ren T,
Wu Z, Zhang J, Zhang L and Lu Y: Differential expression of p42.3
in low- and high-grade gliomas. World J Surg Oncol. 12:1852014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Melton C, Reuter JA, Spacek DV and Snyder
M: Recurrent somatic mutations in regulatory regions of human
cancer genomes. Nat Genet. 47:710–716. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peters I, Tezval H, Kramer MW, Wolters M,
Grünwald V, Kuczyk MA and Serth J: Implications of TCGA network
data on 2nd generation immunotherapy concepts based on PD-L1 and
PD-1 target structures. Aktuelle Urol. 46:481–485. 2015.In German.
PubMed/NCBI
|
|
28
|
Chen X, Shi K, Wang Y, Song M, Zhou W, Tu
H and Lin Z: Clinical value of integrated-signature miRNAs in
colorectal cancer: miRNA expression profiling analysis and
experimental validation. Oncotarget. 6:37544–37556. 2015.PubMed/NCBI
|
|
29
|
Moelling K and Broecker F: The reverse
transcriptase-RNase H: from viruses to antiviral defense. Ann NY
Acad Sci. 1341:126–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Natiq A, Elalaoui SC, Miesch S, Bonnet C,
Jonveaux P, Amzazi S and Sefiani A: A new case of de novo
19p13.2p13.12 deletion in a girl with overgrowth and severe
developmental delay. Mol Cytogenet. 7:402014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Flanagan JM, Funes JM, Henderson S, Wild
L, Carey N and Boshoff C: Genomics screen in transformed stem cells
reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug
targets. Mol Cancer Ther. 8:249–260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng S and Cao Z: Is the role of human
RNase H2 restricted to its enzyme activity? Prog Biophys Mol Biol.
Nov 19–2015.Epub ahead of print. PubMed/NCBI
|
|
33
|
Reijns MA, Bubeck D, Gibson LC, Graham SC,
Baillie GS, Jones EY and Jackson AP: The structure of the human
RNase H2 complex defines key interaction interfaces relevant to
enzyme function and human disease. J Biol Chem. 286:10530–10539.
2011. View Article : Google Scholar :
|
|
34
|
Hausen P and Stein H: Ribonuclease H. An
enzyme degrading the RNA moiety of DNA-RNA hybrids. Eur J Biochem.
14:278–283. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stein H and Hausen P: Enzyme from calf
thymus degrading the RNA moiety of DNA-RNA hybrids: effect on
DNA-dependent RNA polymerase. Science. 166:393–395. 1969.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Coté ML and Roth MJ: Murine leukemia virus
reverse transcriptase: structural comparison with HIV-1 reverse
transcriptase. Virus Res. 134:186–202. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mizuno M, Yasukawa K and Inouye K: Insight
into the mechanism of the stabilization of moloney murine leukaemia
virus reverse transcriptase by eliminating RNase H activity. Biosci
Biotechnol Biochem. 74:440–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schultz SJ and Champoux JJ: RNase H
activity: structure, specificity, and function in reverse
transcription. Virus Res. 134:86–103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rice GI, Forte GM, Szynkiewicz M, Chase
DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM,
Balottin U, et al: Assessment of interferon-related biomarkers in
Aicardi-Goutières syndrome associated with mutations in TREX1,
RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control
study. Lancet Neurol. 12:1159–1169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Orcesi S, La Piana R and Fazzi E:
Aicardi-Goutieres syndrome. Br Med Bull. 89:183–201. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Crow YJ: Aicardi-Goutières syndrome. Handb
Clin Neurol. 113:1629–1635. 2013. View Article : Google Scholar
|
|
42
|
Crow YJ and Manel N: Aicardi-Goutières
syndrome and the type I interferonopathies. Nat Rev Immunol.
15:429–440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Williams KA, Lee M, Hu Y, Andreas J, Patel
SJ, Zhang S, Chines P, Elkahloun A, Chandrasekharappa S, Gutkind
JS, et al: A systems genetics approach identifies CXCL14, ITGAX,
and LPCAT2 as novel aggressive prostate cancer susceptibility
genes. PLoS Genet. 10:e10048092014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT Method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
45
|
Dai B, Wan W, Zhang P, Zhang Y, Pan C,
Meng G, Xiao X, Wu Z, Jia W, Zhang J, et al: SET and MYND
domain-containing protein 3 is overexpressed in human glioma and
contributes to tumorigenicity. Oncol Rep. 34:2722–2730.
2015.PubMed/NCBI
|
|
46
|
Yun K, Fidler AE, Eccles MR and Reeve AE:
Insulin-like growth factor II and WT1 transcript localization in
human fetal kidney and Wilms' tumor. Cancer Res. 53:5166–5171.
1993.PubMed/NCBI
|
|
47
|
Pritchard-Jones RO, Dunn DB, Qiu Y, Varey
AH, Orlando A, Rigby H, Harper SJ and Bates DO: Expression of
VEGFxxxb, the inhibitory isoforms of VEGF, in malignant
melanoma. Br J Cancer. 97:223–230. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ismail PM, Lu T and Sawadogo M: Loss of
USF transcriptional activity in breast cancer cell lines. Oncogene.
18:5582–5591. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Macé K, Aguilar F, Wang JS, Vautravers P,
Gómez-Lechón M, Gonzalez FJ, Groopman J, Harris CC and Pfeifer AM:
Aflatoxin B1-induced DNA adduct formation and p53
mutations in CYP450-expressing human liver cell lines.
Carcinogenesis. 18:1291–1297. 1997. View Article : Google Scholar
|
|
50
|
Cox C, Bignell G, Greenman C, Stabenau A,
Warren W, Stephens P, Davies H, Watt S, Teague J, Edkins S, et al:
A survey of homozygous deletions in human cancer genomes. Proc Natl
Acad Sci USA. 102:4542–4547. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Doyle GA, Bourdeau-Heller JM, Coulthard S,
Meisner LF and Ross J: Amplification in human breast cancer of a
gene encoding a c-myc mRNA-binding protein. Cancer Res.
60:2756–2759. 2000.PubMed/NCBI
|
|
52
|
Suswam E, Li Y, Zhang X, Gillespie GY, Li
X, Shacka JJ, Lu L, Zheng L and King PH: Tristetraprolin
down-regulates interleukin-8 and vascular endothelial growth factor
in malignant glioma cells. Cancer Res. 68:674–682. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lai WS, Kennington EA and Blackshear PJ:
Tristetraprolin and its family members can promote the cell-free
deadenylation of AU-rich element-containing mRNAs by poly(A)
ribonuclease. Mol Cell Biol. 23:3798–3812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang K, Dion N, Fuchs B, Damron T,
Gitelis S, Irwin R, O'Connor M, Schwartz H, Scully SP, Rock MG, et
al: The human homolog of yeast SEP1 is a novel candidate tumor
suppressor gene in osteogenic sarcoma. Gene. 298:121–127. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Rökman A, Ikonen T, Seppälä EH, Nupponen
N, Autio V, Mononen N, Bailey-Wilson J, Trent J, Carpten J,
Matikainen MP, et al: Germline alterations of the RNASEL gene, a
candidate HPC1 gene at 1q25, in patients and families with prostate
cancer. Am J Hum Genet. 70:1299–1304. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Krüger S, Silber AS, Engel C, Görgens H,
Mangold E, Pagenstecher C, Holinski-Feder E, von Knebel Doeberitz
M, Moeslein G, Dietmaier W, et al German Hereditary Non-Polyposis
Colorectal Cancer Consortium: Arg462Gln sequence variation in the
prostate-cancer-susceptibility gene RNASEL and age of onset of
hereditary non-polyposis colorectal cancer: a case-control study.
Lancet Oncol. 6:566–572. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bartsch DK, Fendrich V, Slater EP,
Sina-Frey M, Rieder H, Greenhalf W, Chaloupka B, Hahn SA,
Neoptolemos JP and Kress R: RNASEL germline variants are associated
with pancreatic cancer. Int J Cancer. 117:718–722. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shook SJ, Beuten J, Torkko KC,
Johnson-Pais TL, Troyer DA, Thompson IM and Leach RJ: Association
of RNASEL variants with prostate cancer risk in Hispanic Caucasians
and African Americans. Clin Cancer Res. 13:5959–5964. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rennert H, Zeigler-Johnson CM, Addya K,
Finley MJ, Walker AH, Spangler E, Leonard DG, Wein A, Malkowicz SB
and Rebbeck TR: Association of susceptibility alleles in
ELAC2/HPC2, RNASEL/HPC1, and MSR1 with prostate cancer severity in
European American and African American men. Cancer Epidemiol
Biomarkers Prev. 14:949–957. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sidrauski C and Walter P: The
transmembrane kinase Ire1p is a site-specific endonuclease that
initiates mRNA splicing in the unfolded protein response. Cell.
90:1031–1039. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Davies MP, Barraclough DL, Stewart C,
Joyce KA, Eccles RM, Barraclough R, Rudland PS and Sibson DR:
Expression and splicing of the unfolded protein response gene XBP-1
are significantly associated with clinical outcome of
endocrine-treated breast cancer. Int J Cancer. 123:85–88. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sugito N, Ishiguro H, Kuwabara Y, Kimura
M, Mitsui A, Kurehara H, Ando T, Mori R, Takashima N, Ogawa R, et
al: RNASEN regulates cell proliferation and affects survival in
esophageal cancer patients. Clin Cancer Res. 12:7322–7328. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Muralidhar B, Goldstein LD, Ng G, Winder
DM, Palmer RD, Gooding EL, Barbosa-Morais NL, Mukherjee G, Thorne
NP, Roberts I, et al: Global microRNA profiles in cervical squamous
cell carcinoma depend on Drosha expression levels. J Pathol.
212:368–377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kaul D and Sikand K: Defective
RNA-mediated c-myc gene silencing pathway in Burkitt's lymphoma.
Biochem Biophys Res Commun. 313:552–554. 2004. View Article : Google Scholar
|
|
65
|
Flavin RJ, Smyth PC, Finn SP, Laios A,
O'Toole SA, Barrett C, Ring M, Denning KM, Li J, Aherne ST, et al:
Altered eIF6 and Dicer expression is associated with
clinicopathological features in ovarian serous carcinoma patients.
Mod Pathol. 21:676–684. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chiosea S, Jelezcova E, Chandran U,
Acquafondata M, McHale T, Sobol RW and Dhir R: Up-regulation of
dicer, a component of the MicroRNA machinery, in prostate
adenocarcinoma. Am J Pathol. 169:1812–1820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chiosea S, Jelezcova E, Chandran U, Luo J,
Mantha G, Sobol RW and Dacic S: Overexpression of Dicer in
precursor lesions of lung adenocarcinoma. Cancer Res. 67:2345–2350.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chiosea SI, Barnes EL, Lai SY, Egloff AM,
Sargent RL, Hunt JL and Seethala RR: Mucoepidermoid carcinoma of
upper aerodigestive tract: clinicopathologic study of 78 cases with
immunohistochemical analysis of Dicer expression. Virchows Arch.
452:629–635. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang H, Lathia JD, Wu Q, Wang J, Li Z,
Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, et
al: Targeting interleukin 6 signaling suppresses glioma stem cell
survival and tumor growth. Stem Cells. 27:2393–2404. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chang CY, Li MC, Liao SL, Huang YL, Shen
CC and Pan HC: Prognostic and clinical implication of IL-6
expression in glioblastoma multiforme. J Clin Neurosci. 12:930–933.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Goswami S, Gupta A and Sharma SK:
Interleukin-6-mediated autocrine growth promotion in human
glioblastoma multiforme cell line U87MG. J Neurochem. 71:1837–1845.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Green DR and Llambi F: Cell Death
Signaling. Cold Spring Harb Perspect Biol. 7:2015.pii: a006080.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Frei K, Ambar B, Adachi N, Yonekawa Y and
Fontana A: Ex vivo malignant glioma cells are sensitive to Fas
(CD95/APO-1) ligand-mediated apoptosis. J Neuroimmunol. 87:105–113.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gratas C, Tohma Y, Van Meir EG, Klein M,
Tenan M, Ishii N, Tachibana O, Kleihues P and Ohgaki H: Fas ligand
expression in glioblastoma cell lines and primary astrocytic brain
tumors. Brain Pathol. 7:863–869. 1997. View Article : Google Scholar : PubMed/NCBI
|