Introduction
Laryngeal carcinoma is one of the most commonly
diagnosed head and neck cancers worldwide. Owing to advances in
both diagnostic techniques and comprehensive treatment, the
therapeutic efficacy is good in early stage disease. However, the
survival rate of advanced stage laryngeal carcinoma patients has
not significantly improved over recent years (1). Once recurrence or distant metastasis
presents, conventional therapy is almost ineffective. Therefore,
research has focused on the development of novel molecular
mechanisms underlying laryngeal carcinoma progression and on the
exploration of effective schemes for diagnosis and therapy of this
disease.
MicroRNAs (miRNAs) are endogenous non-coding small
RNAs, roughly 18–25 nucleotides in length that contribute to the
translation inhibition or degradation of their cognate target genes
through an imperfect sequence complementarity-dependent manner by
which to pair the 3′-untranslated region (3′-UTR) of a target mRNA
(2). Studies have demonstrated that
each miRNA may regulate hundreds of target genes and half of miRNAs
are located in cancer-associated genomic regions (3). In fact, miRNAs are involved in entire
signaling networks to regulate several cellular processes,
including proliferation, differentiation and apoptosis (4). Evidence has shown that a number of
miRNAs are involved in tumorigenesis and function either as
oncogenes or tumor suppressors during tumor progression (5,6).
Therefore, aberrant expression profiles of miRNAs have considerable
potential for the diagnosis, classification, clinical prognostic
information, and therapy of cancer (7–9).
Previous studies of miRNA profiles have confirmed
miR-34a as a key regulator of tumor suppression, and miR-34a is
downregulated in many solid and hematological malignancies,
including laryngeal carcinoma (10–12).
miR-34a is an important component of the p53 tumor suppressor
network, whose gene resides on chromosome 1p36.23, and is always
deregulated by gene deletion, CpG methylation or mutation of p53
(13). Ectopic expression of
miR-34a can induce apoptosis, cell cycle arrest, senescence,
differentiation, and self-renewal, by regulating several target
mRNAs, such as c-Myc, E2F, CDK4, CDK6, Bcl-2, SIRT1, c-MET and
CD44, to inhibit cancer proliferation and metastasis in various
types of cancers (14–21). Our study showed that miR-34a was
downregulated in laryngeal carcinoma tissues, and a bioinformatic
analysis predicted CCND1 as a potential target of miR-34a.
CCND1 is an established human oncogene that is
frequently overexpressed as a result of copy number variation,
mutation, or as a consequence of the deregulation of miRNAs
(22). The fundamental function of
CCND1 is to promote tumor progression through activation of the
cyclin-dependent kinases (CDKs) CDK4 and CDK6 and inactivation of
the retinoblastoma protein and release of e2F (23,24).
Various human cancers, including breast cancer, colon cancer, lung
cancer, melanoma, prostate cancer, hematopoietic malignancies and
laryngeal carcinoma, overexpress CCND1 (25). In laryngeal carcinoma,
overexpression of CCND1 is associated with unfavorable
clinicopathological features and represents an independent
significant predictor of patient prognosis (26). miR-34a was found to induce cell
cycle arrest through regulation of CCND1 in non-small cell lung
cancer (17,27). However, the potential mechanisms by
which CCND1 affects the malignant phenotype of laryngeal carcinoma
cells remain largely unknown.
In this study, we investigated the role of miR-34a
in the development and progression of laryngeal carcinoma. We
examined the expression levels of miR-34a in laryngeal carcinoma
cells and clinical samples and tested its effects on cell
proliferation, apoptosis and migration. In addition, we identified
that CCND1 was overexpressed in laryngeal carcinoma and predicted
that CCND1 was a target of miR-34a, by 3′-UTR luciferase assays and
western blot analysis. Furthermore, we validated CCND1 as the main
functional target of miR-34a through knockdown and rescue
expression of CCND1 in laryngeal carcinoma. Our study demonstrated
that miR-34a acts as a tumor suppressor by the direct targeting of
CCND1 in laryngeal carcinoma, suggesting that miR-34a is a
potential diagnostic biomarker and has therapeutic value for
laryngeal carcinoma therapy.
Materials and methods
Patient tissues
Seventy-one cases of human laryngeal squamous cell
carcinoma (LSCC) tissue specimens (tumor tissues with corresponding
normal adjacent tissues) were obtained at the time of surgical
resection from the Third Affiliated Hospital of Sun Yat-sen
University (guangzhou, Guangdong, China) between January 2011 and
September 2014. Written informed consent was obtained from all
patients for the use of their specimens before surgery. The
specimens were conserved in RNAlater (Ambion, Austin, TX, USA)
immediately after surgical resection and transferred into −80°C
freezer until use. The histological characterization and
clinicopathological staging of the LSCC tissues were determined
according to the current International Union Against Cancer (UICC)
pathological staging criteria. Use of the human tissues was
approved by the Clinical Research ethics Committee of the Third
Affiliated Hospital of Sun Yat-sen University.
Cell culture
The human laryngeal carcinoma cell line HEp-2 was
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA) and was freshly recovered from liquid nitrogen (less than
3 months). HEp-2 cells were maintained in RPMI-1640 medium
(Invitrogen, Beijing, China). Media were supplemented with 10%
fetal bovine serum (FBS; Gibco, Cappinas, Brazil). All cells were
cultured in a humidified incubator (Thermo Electron Corp., New
Castle, DE, USA) at 37°C with 5% CO2.
Bioinformatics
The potential target gene of miR-34a and its
conserved miRNA-binding site was analyzed by the algorithms of
TargetScan 5.1 (http://www.targetscan.org) and miRanda (http://www.microrna.org). According to these
algorithms, we selected the CCDN1 gene as a potential target of
miR-34a for further study.
Transient transfection of miRNA and
siRNA
The miR-34a mimics, miR-Ctrl (miRNA negative control
with non-specific), small interfering RNA duplexes targeting human
CCND1 (CCND1-siRNA, sense strand, 5′-GGAGAACAAACAGAUCAUCTTdTdT-3′
and antisense strand, 5′-GAUGAUCUGUUUGUUCUCCTCdTdT-3′) and
scrambled control siRNA (Ctrl-siRNA) (sense strand,
5′-UUCUCCGAACGUGUCACGUdTdT-3′ and antisense strand,
5′-ACGUGACACGUUCGGAGAAdTdT-3′) were purchased from RiboBio, Co.,
Ltd. (Guangzhou, China). Cells were transfected at ~60–70%
confluence using Lipofectamine 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) with miRNA mimics or siRNA
duplexes at working concentrations of 50 nM in Opti-MEM (Invitrogen
Life Technologies) according to the protocol as previously
described (w).
RNA extraction, reverse transcription and
quantitative real-time PCR (qPCR)
The total RNA extraction, reverse transcription and
qPCR procedure were executed as previously described (20). In brief, total RNA from the cultured
cells and surgical LSCC tissues were extracted by
TRIzol® reagent (Invitrogen Life Technologies). To
quantitate the expression level of miR-34a, reverse transcription
was carried out by a specific stem-loop real-time PCR miRNA kit
(Ribobio, Co., Ltd.). After reverse transcription at 42°C for 60
min followed by denaturation at 70°C for 10 min, qPCR was carried
out using the Platinum SYBR Green qPCR SuperMix-UDG system
(Invitrogen Life Technologies) on an ABI 7900HT Real-Time PCR
system (Applied Biosystems Life Technologies, Foster City, CA,
USA). Data were analyzed by ABI SDS version 2.3 (Applied Biosystems
Life Technologies). All procedures were performed according to the
protocols of the manufacturer. Each sample was analyzed in
triplicate and normalized to internal controls, 5S ribosomal RNA,
and the fold changes were calculated according to the relative
quantification method (RQ = 2−ΔΔCT). The results are
expressed as fold-change in the expression levels in the cells or
tissues.
The primers of miR-34a and 5S rRNA used for
stem-loop real-time PCR are listed as follows: miR-34a stem-loop
RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAAGGGCAG-3′;
miR-34a forward, 5′-GCGGCCAATCAGCAAGTATACT-3′; miR-34a reverse,
5′-GTGCAGGTCCGAGGT-3′; 5S rRNA stem-loop RT,
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATCGCACTGGATACGACCAGGCG-3′; 5S rRNA
forward, 5′-CTGGTTAGTACTTGGACGGGAGAC-3′; 5S rRNA reverse,
5′-GTGCAGGGTCCGAGGT-3′.
MTT assay
The cell growth and proliferation of the HEp-2 cells
transfected with miRNAs or siRNAs were assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Sigma-Aldrich, St. Louis, MO, USA). The cells were plated in
96-well plates at 5×103 cells/well and transfected with
miRNAs or siRNAs. After incubation at the appropriate time points,
the culture medium was replaced by 100 µl of fresh DMEM. And
25 µl of MTT (5 g/l in phosphate-buffered saline) was added
to each well, to obtain a working concentration of MTT, 1 g/l.
Then, incubation was carried out for an additional 4 h, the medium
was replaced with dimethyl sulfoxide (DMSO; Sigma-Aldrich), and the
absorbance was determined using a SpectraMax M5 microplate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA) at 570 nm. The cell
viability was normalized to the cells without transfection of the
miRNA or siRNA. Three independent experiments (triplicate in each)
were analyzed.
Apoptosis assays
To determine cell apoptosis in vitro, we used
the Annexin V-fluorescein isothiocyanate apoptosis detection kit
(KeyGen, Nanjing, China) according to the manufacturer's
instructions. Briefly, after a 48-h transfection, the cells were
collected by trypsinization and washed with PBS, and resuspended in
binding buffer at a concentration of 1×106 cells/ml.
Next, the cells were incubated with 2 µl of propidium iodide
and 2 µl of Annexin V-fluorescein isothiocyanate for 15 min,
washed and resuspended in 500 µl PBS. Flow cytometric
analysis was carried out by a FACSCalibur (Becton-Dickinson, San
Jose, CA, USA).
Transwell invasion assay
To evaluate cell migration in vitro,
Transwell chambers (8 µm; BD Biosciences, San Jose, CA, USA)
were used according to the manufacturer's instructions. Briefly,
the transfected cells were collected, re-suspended
(5×104 cells/well) in 200 µl serum-free medium,
and added to the top chamber. In the bottom chamber, culture medium
containing 10% FBS served as a chemoattractant. After incubation
for 24 h at 37°C, the cells that did not migration through the
pores were carefully removed by cotton swabs. Then the invasive
cells were fixed and stained with 0.5% crystal violet
(Sigma-Aldrich) for 30 min, counted with an inverted microscope
(Olympus IX71; Olympus Corp., Tokyo, Japan), and the relative
number of migratory cells was calculated from five random fields
from digital images (×200). The data are expressed as the means ±
SD of three independent experiments.
Plasmid
The 3′-UTR sequences of human CCND1 containing the
putative miR-34a binding sites were obtained from healthy and
disease-free volunteer gDNA using PCR amplification and cloned into
the pGL3 luciferase reporter plasmid (Promega, Corp., Madison, WI,
USA), which was termed as wild-type 3′-UTR (wt 3′-UTR). The point
mutations were created by the Quick-Change site-Directed
Mutagenesis kit (Stratagene, La Jolla, CA, USA), according to the
manufacturer's instructions. The resultant product served as the
mutated 3′-UTR (mut 3′-UTR). Both inserted fragment sequences were
confirmed by DNA sequencing.
To overexpress CCND1, the cDNA of CCND1 containing
the putative miR-34a binding sites was cloned into the pcDNA3.1
vector (Invitrogen Life Technologies), which was termed as
wild-type 3′-UTR-CCND1 (wt 3′-UTR-CCND1). The mut 3′-UTR-CCND1 was
obtained as described above. Cells were co-transfected with 50 nM
of miRNA mimics and 500 ng of plasmid in a 6-well plate for the
rescue experiment.
Luciferase assays
For the luciferase reporter assay, HEp-2 cells
(6×104) were seeded in triplicate in 24-well plates 24 h
before transfection. Vectors, as described above, and the control
vector pRL-TK (Promega, Corp.) coding for Renilla luciferase
(for normalization) were co-transfected with miRNA in triplicate by
Lipofectamine 2000. The cells were harvested and lysed 48 h after
transfection, and the luciferase activity was measured using the
Dual-Glo luciferase assay kit (Promega, Corp.). The firefly
luciferase values were normalized to Renilla, and the
relative ratios of firefly to Renilla activity are shown.
Three independent experiments were carried out, and the data are
represented as the mean ± SD.
Western blot analysis
According to standard western blot analysis
procedures, briefly, after culturing for 72 h, the transfected
cells were harvested on ice using RIPA lysis and extraction buffer
[25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium
deoxycholate, 0.1% SDS, protease inhibitor cocktail (Pierce,
Rockford, IL, USA)]. Then, 10 µg protein of each sample was
separated by sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) on 10% polyacrylamide gels, and then
transferred to polyvinlidene fluoride membranes (PVDF; Millipore,
Billerica, MA, USA). After blocking for 1 h at room temperature and
incubation overnight at 4°C in Tris-buffered saline containing
0.05% Tween (TBST) with 5% milk, the membranes were incubated with
mouse monoclonal antibody against human CCND1 (Cell Signaling
Technology, Danvers, MA, USA) or tubulin (Sigma-Aldrich) as a
protein loading control, followed by horseradish peroxidase
(HRP)-conjugated goat-anti-mouse IgG (Abcam), and the bands were
detected using the Supersignal West Pico ECL chemiluminescence kit
(Pierce) and Kodak X-ray film (Eastman Kodak Co, Rochester, NY,
USA).
Statistical analysis
SPSS 13.0 software was used for statistical
analysis. All experiments were performed independently three times,
and all samples were in triplicate. The data are expressed as the
mean ± SEM unless otherwise mentioned; two-tailed Student's t-test
was used for statistical analysis when only two groups were tested.
A one-way analysis of variance was used to compare multiple groups.
Spearman's correlation analysis was used to determine the
correlation between miR-34a and CCND1 expression. P-values of
<0.05 were considered to indicate statistical significance.
Results and Discussion
miR-34a is downregulated in human LSCC
specimens and is inversely associated with advanced stage and lymph
node metastasis in the patients
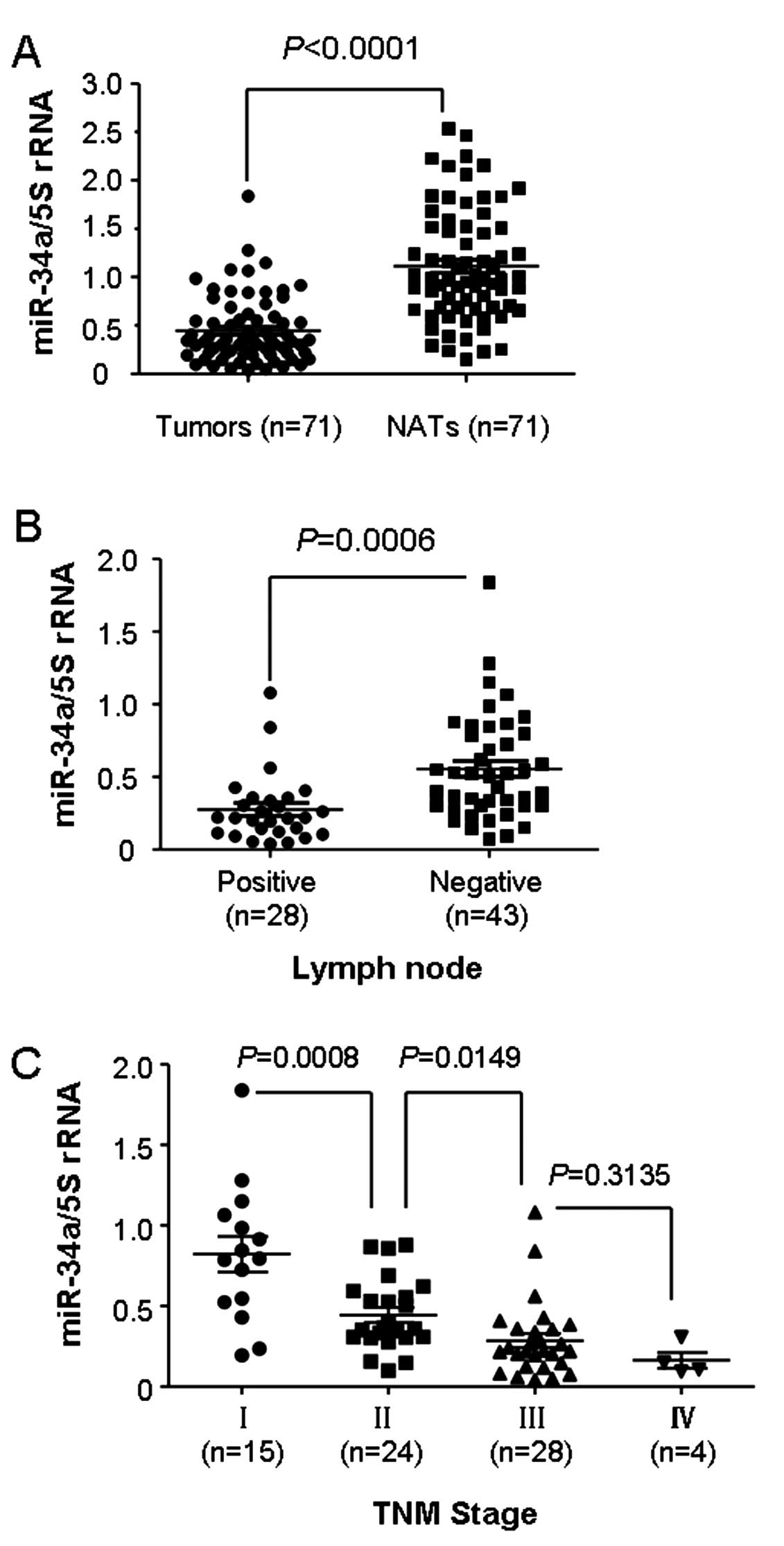
To investigate the expression of miR-34a and its
significance in LSCC, we first examined the expression levels of
miR-34a in 71 pairs of primary LSCC and their corresponding normal
adjacent tissues by stem-loop RT-PCR. The correlation between the
miR-34a expression levels and the clinicopathological
characteristics of the LSCC patients, including gender, age,
primary location, lymph node status, T classification and clinical
stage are summarized in Table I.
The data demonstrated no obvious correlations between miR-34a
expression and gender, age and primary location. However, as shown
in Fig. 1A, the comparative
analysis indicated that the median expression level of miR-34a was
lower in the LSCC tissues compared with the median level in the
paired normal adjacent tissues (median, 0.3432 and 0.9756,
respectively; P<0.0001). In regards to lymph node status, we
separated 71 LSCC patients into two groups: lymphatic node
metastasis-positive or -negative. Notably, miR-34a expression was
significantly lower in the lymphatic node metastasis-positive group
than that in the negative group (Table
I and Fig. 1b; P=0.0006).
Furthermore, the expression of miR-34a was lower in the LSCC cases
with advanced TNM stage (stage III and IV), compared to that in
early stage LSCC patients (stage I and II; Table I and Fig. 1C). Collectively, these data
suggested that miR-34a was downregulated in LSCC and a decreased
expression of miR-34a could be attributed to the progression and
metastasis of LSCC.
 | Table IRelationship between miR-34a
expression and clinicopathological parameters of the laryngeal
cancer cases. |
Table I
Relationship between miR-34a
expression and clinicopathological parameters of the laryngeal
cancer cases.
| Clinicopathological
parameters | No. of cases | Median expression of
miR-34a | P-value |
|---|
| Gender |
| Male | 56 | 0.3432±0.0533 | 0.6160 |
| Female | 15 | 0.3487±0.0616 | |
| Age (years) |
| ≤60 | 33 | 0.3854±0.0631 | 0.5328 |
| >60 | 38 | 0.3225±0.0535 | |
| Primary
location |
| Supraglottic | 29 | 0.3024±0.0567 | 0.2869 |
| Glottic | 42 | 0.3579±0.0566 | |
| Lymph node
metastasis |
| Negative | 43 | 0.5028±0.0551 | 0.0006 |
| Positive | 28 | 0.2202±0.0441 | |
|
Differentiation |
| Well | 45 | 0.3990±0.0562 | 0.0079 |
| Moderate and
low | 26 | 0.2317±0.0430 | |
| T
classification |
| T1+T2 | 44 | 0.4029±0.0576 | 0.0380 |
| T3+T4 | 27 | 0.2641±0.0460 | |
| Clinical stage |
| I+II | 39 | 0.5250±0.0578 | 0.0001 |
| III+IV | 32 | 0.2202±0.0392 | |
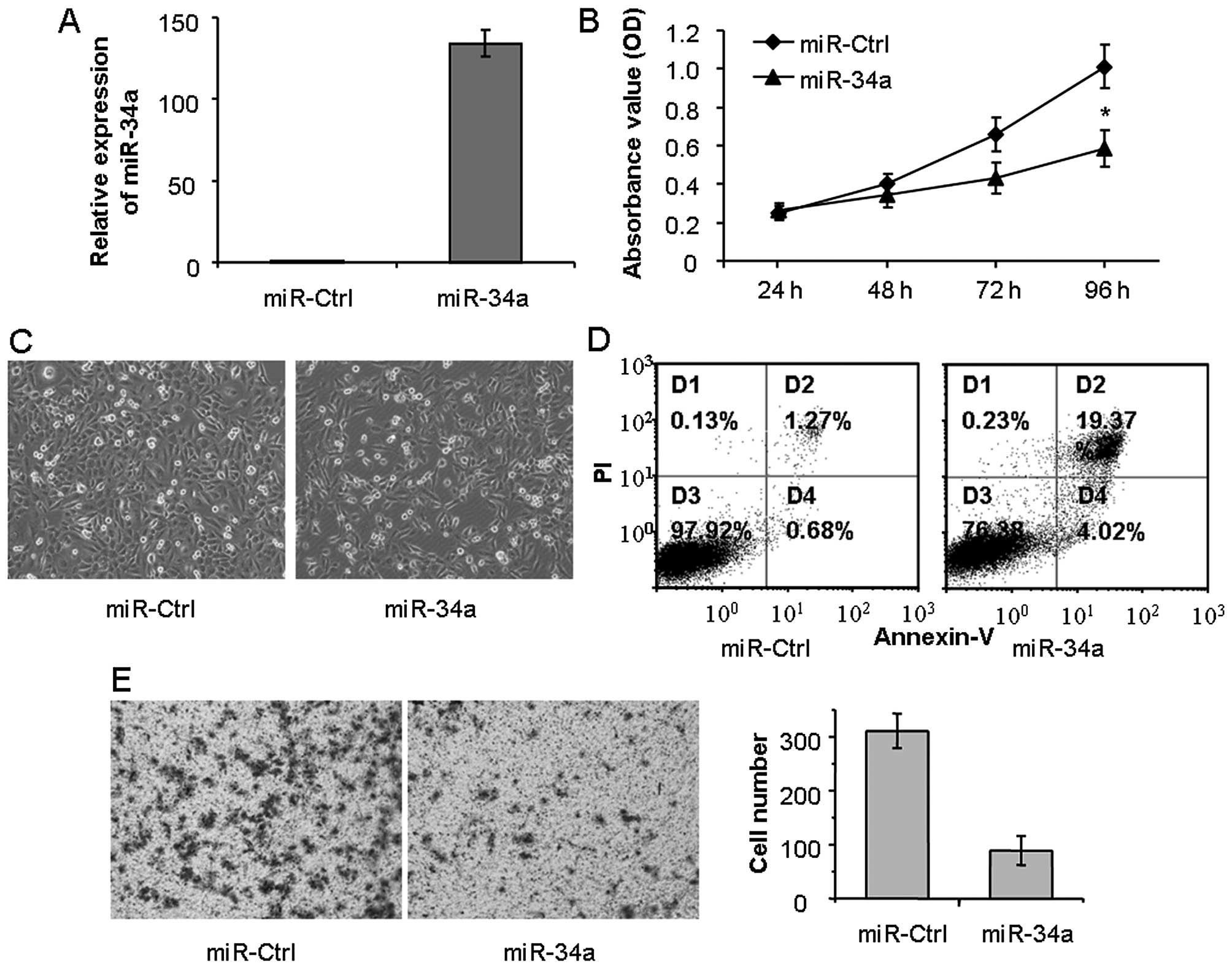
miR-34a inhibits the proliferation,
migration and induces apoptosis in the HEp-2 cells
To explore the function of miR-34a on LSCC cell
proliferation, we transfected LSCC HEp-2 cells with miR-34a mimics.
The effectiveness of miR-34a ectopic overexpression in the HEp-2
cells was tested by quantitative real-time PCR. Compared with the
negative control (miR-Ctrl), the mimics significantly elevated the
expression of miR-34a (Fig. 2A). As
shown in the MTT assay, restoration of miR-34a inhibited the
proliferation of HEp-2 cells, compared with the miR-Ctrl (Fig. 2B). Then, we observed the morphologic
images of the HEp-2 cells transfected with the miR-34a mimics. The
cells exhibited significantly reduced cell numbers, compared with
the miR-Ctrl-transfected cells (100 vs. ~40% confluence,
respectively). These data were concordant with the MTT assay.
Meanwhile, the cells treated with miR-34a displayed morphological
changes characterized by extension of the cytoplasmic portion and
rounding of the cell body, while the rest of the spindled cells
showed conspicuous shrinkage and extensive detachment (Fig. 2C).
It is well known that miR-34a is a key regulator of
tumor suppression, which can induce cancer cell apoptosis. In order
to explore the biological effect of miR-34a in HEp-2 cells, flow
cytometric analysis of Annexin V-FITC/PI staining was used to
detected apoptotics cells. As shown in Fig. 2D, compared with miR-Ctrl,
transfection with miR-34a mimics obviously increased the number of
apoptotic cells (1.95 vs. 23.39%; P<0.05). Therefore, ectopic
expression of miR-34a may reduce cell growth of LSCC by inhibiting
cell proliferation and inducing cell apoptosis. In addition, we
investigated the role of miR-34a in cell migration using Transwell
assay. As shown in Fig. 2E, ectopic
expression of miR-34a obviously reduced the cell migration,
compared with the miR-Ctrl. Taken together, miR-34a exhibited a
tumor suppressor function, and inhibited LSCC cell proliferation
and migration.
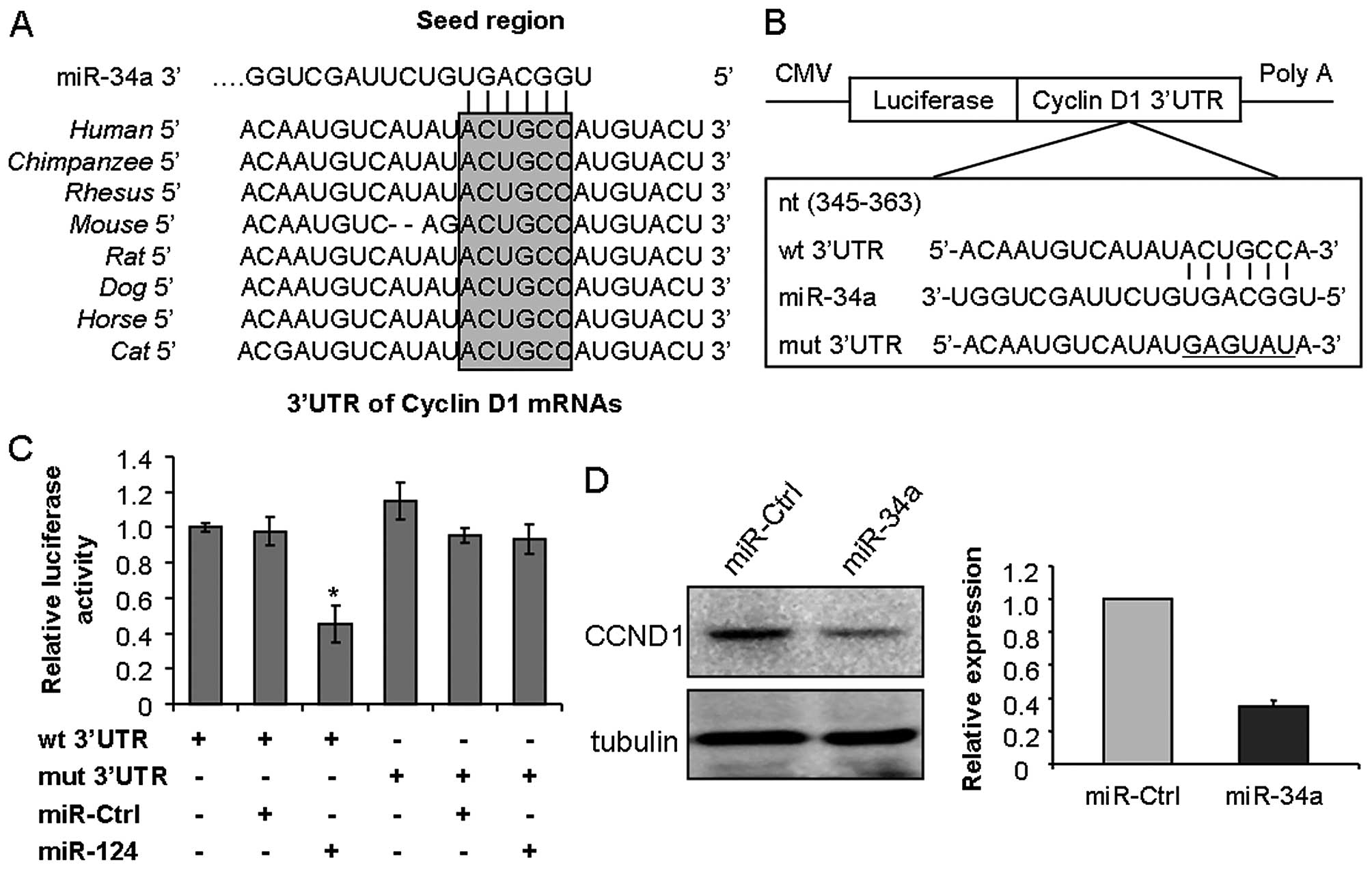
miR-34a downregulates CCND1 expression by
directly targeting its 3′-UTR
To explore the molecular mechanism underlying the
ability of miR-34a to inhibit cancer cell proliferation and
migration, bioinformatic analysis was used to identify the putative
protein-coding gene targets of miR-34a, especially for those
oncogenes which involved in proliferation, migration and potential
therapeutic targets of cancer. According to this principle, CCND1
was chosen as one of the potential targets of miR-34a for further
experimental validation, which was strictly consistent among
different species and whose 3′-UTR of mRNA has one potential
complementary site for the seed region of miR-34a (Fig. 3A). To test whether CCND1 is a direct
target of miR-34a, the target fragment sequence of CCND1 3′-UTR (wt
3′-UTR) and the corresponding mutant counterpart sequence (mut
3′-UTR) were cloned into a luciferase reporter vector (Fig. 3B). The constructed reporter vectors
as described were co-transfected with miR-34a mimics or miR-Ctrl
into the HEp-2 cells. As expected, miR-34a reduced the luciferase
activity of the CCND1 wt 3′-UTR by ~50%, while the luciferase
activity was not obviously changed in the 3′-UTR with mutant
binding site construct. Meanwhile, miR-Ctrl did not attenuate the
luciferase activity of either the wt or mut 3′-UTR construct
(Fig. 3C). In addition, western
blotting results showed that ectopic expression of miR-34a
significantly reduced CCND1 expression to ~40% in the HEp-2 cells
(Fig. 3D). These data indicated
that miR-34a suppressed CCND1 expression dependent on the specific
seed binding sites of the CCND1 3′-UTR.
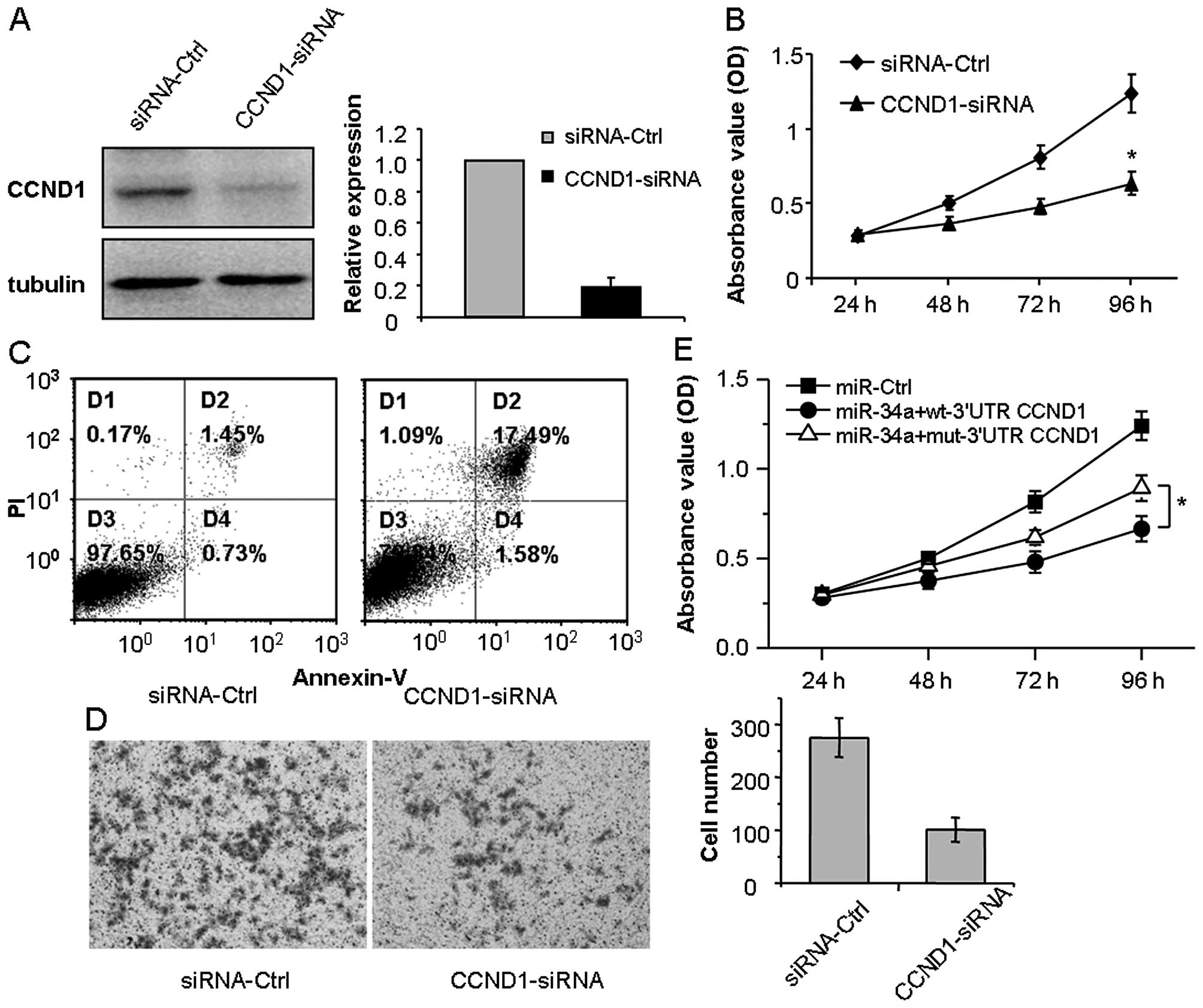
CCND1 is involved in the effects of
miR-34a on proliferation and migration
To evaluate the role of CCND1 in LSCC, HEp-2 cells
were transfected with CCND1-specific siRNAs (CCND1-siRNA) or
negative control (siRNA-Ctrl). After 48 h, proteins were collected
and western blotting assay showed that CCND1 protein in the cells
transfected with 50 nM CCND1-siRNA was significantly reduced
(Fig. 4A). The MTT assays
demonstrated that the knockdown of CCND1 suppressed the
proliferation of HEp-2 cells (Fig.
4b; p<0.05). Furthermore, flow cytometric analysis indicated
that the knockdown of CCND1 induced the apoptosis of HEp-2 cells
(Fig. 4C; P<0.05). In addition,
Transwell assays revealed that knockdown of CCND1 inhibited the
cell migration of HEp-2 cells (Fig.
4D; P<0.05). Take together, the effects of the knockdown of
CCND1 in HEp-2 cells had effects similar to those of miR-34a.
To determine whether or not CCND1 is the direct
functional regulator of the miR-34a effect in HEp-2 cells, we
co-transfected miR-34a mimics accompanied by wt/mut 3′-UTR-CCND1
plasmid which contained wild-type or mutant 3′-UTR-CCND1 cDNA into
HEp-2 cells. MTT results indicated that mut 3′-UTR-CCND1 slightly
attenuated the miR-34a-mediated effects in the HEp-2 cells, to
regain the proliferation, compared with the miR-Ctrl (Fig. 4E; P<0.05). These results suggest
that CCND1 plays an important role in the regulation of miR-34a in
HEp-2 cells.
miR-34a and CCND1 are inversely
correlated in LSCC tissues
Finally, we examined whether miR-34a and
downregulation-mediated CCND1 in clinical LSCC were correlation. We
measured the mRNA levels of CCND1 in 23 cases of LSCC specimens and
their corresponding normal adjacent tissues by real-time PCR. These
cases included 5 cases of clinical stage I, 8 cases of stage II, 8
cases of stage III, and 2 cases of stage IV LSCC. The results
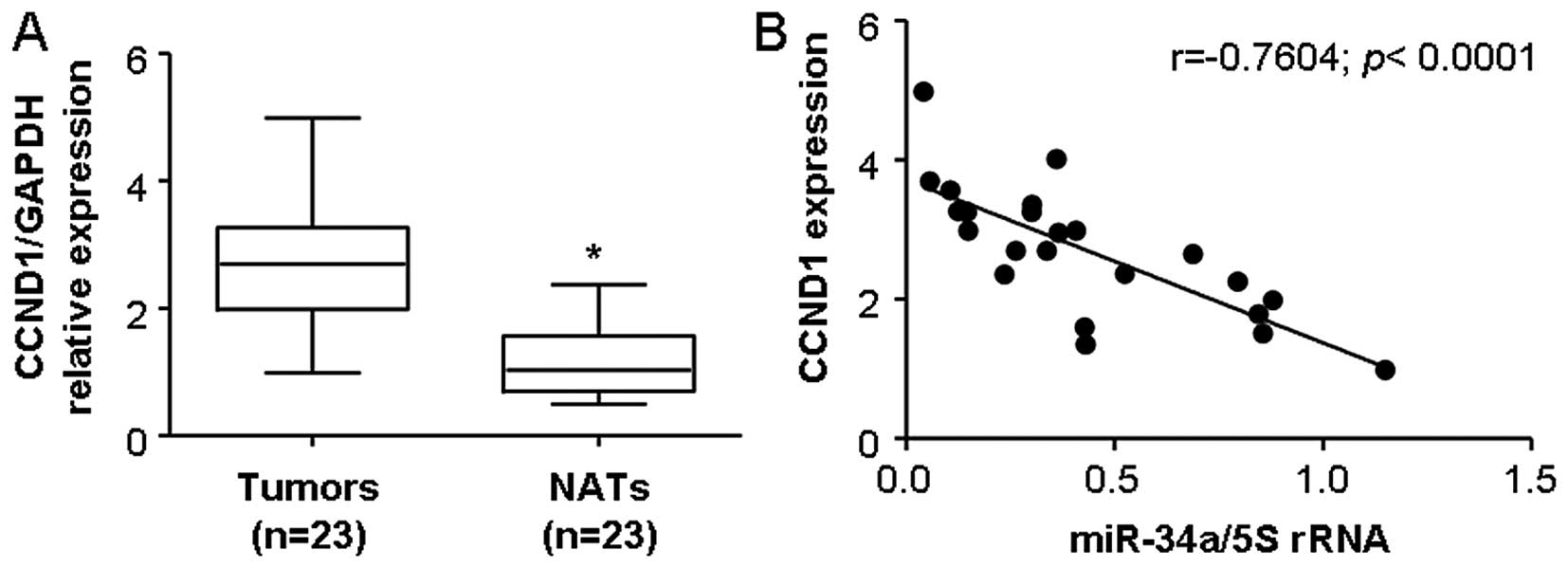
indicated that the average expression level of CCND1 was
significantly higher in LSCC specimens than that in the
corresponding normal adjacent tissues (Fig. 5A; P<0.05). We further correlated
CCND1 with the miR-34a expression levels in the same LSCC
specimens. As shown in Fig. 5b, a
significant inverse correlation was observed when CCND1 expression
levels were plotted against miR-34a expression levels (2-tailed
Spearman's correlation, r=−07604; P <0.0001).
Cancer is a group of diseases involving uncontrolled
growth of abnormal cells, which is caused not only by the change in
a series of important proteins but also by a global dysregulation
in the miRNA profile (9). miRNAs
are conserved small non-coding RNAs that participate in the
regulation of a series of fundamental biological processes, such as
development, proliferation, apoptosis, differentiation, survival
and cell death. In view of the pivotal function of miRNAs, their
alteration naturally promotes numerous cell physiological and
pathological processes, and are eventually involved in the etiology
of many human diseases, such as autoimmune, cardiovascular disease,
diabetes, and cancer. In this study, we found that miR-34a was
frequently downregulated in LSCC tissues, and its expression level
was inversely correlated with lymph node metastasis and advanced
clinical stage. Then, restoration of miR-34a suppressed cancer cell
proliferation, migration and induced the apoptosis in HEp-2 cells
in vitro. Furthermore, we identified CCND1, a putative
therapeutic target oncogene (25),
as a direct and functional target of miR-34a by binding to the
specific 3′-UTR of CCND1. Our study indicated that miR-34a serves
as a novel proliferation and metastasis suppressor in LSCC.
As a tumor suppressor microRNA, miR-34a is one of
the most characterized miRNAs which has attracted much attention in
various cancers. Reduced expression of miR-34a, triggers
dysregulation of cell homeostasis, which could be a selective
advantage for cancer cells. Indeed, miR-34a is a direct p53 target,
and ectopic expression can recapitulate some tumor suppressor
biological functions of p53 such as apoptosis, cell cycle arrest,
inhibition of cancer stem cells viability and metastasis formation
(28). Shen et al showed
that miR-34a was downregulated in LSCC using real-time PCR analysis
and its levels were negatively correlated with histological
differentiation and were positively correlated with survival rate
and might be a potential biomarker of progression and prognosis for
LSCC. Ectopic expression of miR-34a significantly inhibited cell
proliferation by arresting cells at the G0/G1 phase through
targeting survivin in HEp-2 cells (10). However, the miR-34a expression
levels in clinical tissues and its definite mechanism in LSCCare
not completely understood. In the present study, we demonstrated
that miR-34a was downregulated in LSCC tissues and the reduced
miR-34a expression level was closely correlated to lymphatic node
metastasis and advanced TNM stage of LSCC, suggesting that a
reduced expression level of miR-34a is associated with LSCC
progression. Taken together, these data suggest that miR-34a
expression is frequently downregulated in LSCC, and is responsible
for the formation and progression of LSCC. However, the exact
function of miR-34a in LSCC is not completely elucidated.
Cell proliferation and migration are required for
the process of tumorigenesis and progression. miR-34a as an
important tumor suppressor miRNA participates in the regulation of
multiple steps of the process of cells (13). In the present study, to determine
the function of miR-34a in LSCC, we used laryngeal carcinoma cell
line HEp-2 as a model, and ectopic expression of miR-34a
significantly suppressed HEp-2 cell proliferation and migration
(Fig. 2). Our results indicate that
miR-34a is a potential therapeutic target for LSCC.
As an important oncogene, more than 100 proteins
have been identified to interact with CCND1 in human cancer
(29). These proteins are involved
in cell cycle control, transcriptional regulation, DNA repair, RNA
metabolism, protein folding, cell structure and cell organization.
Therefore, the dyregulation of CCND1 affects multiple cellular
processes, which could have oncogenic consequences. The
overexpression of CCND1 has been shown in 80% of head and neck
squamous cell carcinoma (30). A
translocation that juxtaposes CCND1 with the immunoglobulin heavy
chain locus, CCND1 copy number amplification, mutations in the
3′-UTR cause stabilization of the CCND1 mRNA, and as a consequence
causes oncogenic activation of mitogenic signaling pathways leading
to CCND1 overexpression (25).
CCND1 overexpression is associated with poor prognosis and
increased metastasis. In this study, we identified CCND1 as a
target of miR-34a in LSCC, and overexpression of CCND1 was
inversely correlated with miR-34a in LSCC clinical tissues.
Meanwhile, we found that knockdown of CCND1 caused inhibition of
proliferation and migration in HEp-2 cells, and restoration of
CCND1 partially abrogated the miR-34a-mediated effects in HEp-2
cells. These results suggest that miR-34a serves as a tumor
suppressor miRNA by regulating CCND1 to suppress the proliferation
and metastasis of LSCC.
Taken together, our study demonstrated that miR-34a
as an important tumor suppressor miRNA is downregulated in LSCC.
Ectopic expression of miR-34a inhibited HEp-2 cell proliferation
and migration by directly targeting CCND1. These results suggest
that the downregulation of miR-34a in LSCC contributes to LSCC
tumorigenesis and progression and that miR-34a may be novel
diagnostic biomarker and a potential therapeutic target for
LSCC.
Acknowledgments
This study was supported by grants from the Science
and Technology Planning Project of Guangdong Province, China (grant
no. 2013B021800088 to J.Y., no. 2014A020212477 to LS.L.), and the
Natural Science Foundation of Guangdong Province, China (grant no.
2014A030313057 to J.Y., no. 2015A030313035 to LS.L.), and the
Specialized Research Fund for the Doctoral program of Higher
education (grant no. 20130171120069 to LS.L.).
References
|
1
|
Forastiere AA, Zhang Q, Weber RS, Maor MH,
Goepfert H, Pajak TF, Morrison W, Glisson B, Trotti A, Ridge JA, et
al: Long-term results of RTOG 91-11: a comparison of three
nonsurgical treatment strategies to preserve the larynx in patients
with locally advanced larynx cancer. J Clin Oncol. 31:845–852.
2013. View Article : Google Scholar :
|
|
2
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen CZ: MicroRNAs as oncogenes and tumor
suppressors. N Engl J Med. 353:1768–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Luo J, Wang B, Wang D, Xie X, Yuan
L, Guo J, Xi S, Gao J, Lin X, et al: Microrna-124 targets
flotillin-1 to regulate proliferation and migration in breast
cancer. Mol Cancer. 12:1632013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Andorfer CA, Necela BM, Thompson EA and
Perez EA: MicroRNA signatures: clinical biomarkers for the
diagnosis and treatment of breast cancer. Trends Mol Med.
17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Banwait JK and Bastola DR: Contribution of
bioinformatics prediction in microRNA-based cancer therapeutics.
Adv Drug Deliv Rev. 81:94–103. 2015. View Article : Google Scholar
|
|
9
|
Berindan-Neagoe I, Monroig PC, Pasculli B
and Calin GA: MicroRNAome genome: a treasure for cancer diagnosis
and therapy. CA Cancer J Clin. 64:311–336. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shen Z, Zhan G, Ye D, Ren Y, Cheng L, Wu Z
and Guo J: MicroRNA-34a affects the occurrence of laryngeal
squamous cell carcinoma by targeting the antiapoptotic gene
survivin. Med Oncol. 29:2473–2480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar B, Yadav A, Lang J, Teknos TN and
Kumar P: Dysregulation of microRNA-34a expression in head and neck
squamous cell carcinoma promotes tumor growth and tumor
angiogenesis. PLoS One. 7:e376012012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Agostini M and Knight RA: miR-34: From
bench to bedside. Oncotarget. 5:872–881. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar
|
|
14
|
Tazawa H, Tsuchiya N, Izumiya M and
Nakagama H: Tumor-suppressive miR-34a induces senescence-like
growth arrest through modulation of the E2F pathway in human colon
cancer cells. Proc Natl Acad Sci USA. 104:15472–15477. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Winton DJ: miR-34a sets the 'sweet spot'
for notch in colorectal cancer stem cells. Cell Stem Cell.
12:499–501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu C, Kelnar K, Liu B, Chen X,
Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, et
al: The microRNA miR-34a inhibits prostate cancer stem cells and
metastasis by directly repressing CD44. Nat Med. 17:211–215. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bandi N and Vassella E: miR-34a and
miR-15a/16 are co-regulated in non-small cell lung cancer and
control cell cycle progression in a synergistic and Rb-dependent
manner. Mol Cancer. 10:552011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dotto GP and Karine L: miR-34a/SIRT6 in
squamous differentiation and cancer. Cell Cycle. 13:1055–1056.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cole KA, Attiyeh EF, Mosse YP, Laquaglia
MJ, Diskin SJ, Brodeur GM and Maris JM: A functional screen
identifies miR-34a as a candidate neuroblastoma tumor suppressor
gene. Mol Cancer Res. 6:735–742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li L, Yuan L, Luo J, Gao J, Guo J and Xie
X: MiR-34a inhibits proliferation and migration of breast cancer
through downregulation of Bcl-2 and SIRT1. Clin Exp Med.
13:109–117. 2013. View Article : Google Scholar
|
|
21
|
Li L, Xie X, Luo J, Liu M, Xi S, Guo J,
Kong Y, Wu M, Gao J, Xie Z, et al: Targeted expression of miR-34a
using the T-VISA system suppresses breast cancer cell growth and
invasion. Mol Ther. 20:2326–2334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu X, Lv XB, Wang XP, Sang Y, XU S, Hu K,
Wu M, Liang Y, Liu P, Tang J, et al: MiR-138 suppressed
nasopharyngeal carcinoma growth and tumorigenesis by targeting the
CCND1 oncogene. Cell Cycle. 11:2495–2506. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen HZ, Tsai SY and Leone G: Emerging
roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pignataro L, Pruneri G, Carboni N,
Capaccio P, Cesana BM, Neri A and Buffa R: Clinical relevance of
cyclin D1 protein overexpression in laryngeal squamous cell
carcinoma. J Clin Oncol. 16:3069–3077. 1998.PubMed/NCBI
|
|
27
|
Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R,
Sun Z and Zheng X: Downregulation of CCND1 and CDK6 by miR-34a
induces cell cycle arrest. FEBS Lett. 582:1564–1568. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahn YH, Gibbons DL, Chakravarti D,
Creighton CJ, Rizvi ZH, Adams HP, Pertsemlidis A, Gregory PA,
Wright JA, Goodall GJ, et al: ZEB1 drives prometastatic actin
cytoskeletal remodeling by downregulating miR-34a expression. J
Clin Invest. 122:3170–3183. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jirawatnotai S, Hu Y, Michowski W, Elias
JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB,
et al: A function for cyclin D1 in DNA repair uncovered by protein
interactome analyses in human cancers. Nature. 474:230–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leemans CR, Braakhuis BJ and Brakenhoff
RH: The molecular biology of head and neck cancer. Nat Rev Cancer.
11:9–22. 2011. View
Article : Google Scholar
|
|
31
|
Gao J, Li L, Wu M, Liu M and Xie X, Guo J,
Tang H and Xie X: MiR-26a inhibits proliferation and migration of
breast cancer through repression of MCL-1. PLoS One. 8:e651382013.
View Article : Google Scholar : PubMed/NCBI
|