Epithelial-mesenchymal transition (EMT) is a process
whereby epithelial cells gradually acquire a mesenchymal cell
phenotype. EMT is associated with cancer progression, and is
involved in its metastasis and treatment resistance as well as
embryonic development and inflammatory process (1,2).
During EMT, cells lose their epithelial proteins and acquire
mesenchymal proteins (3). This
transition allows cancer cells to gain the ability to pass through
the basement membrane and achieve increased invasive ability
(4–6).
Through the process of EMT, cancer cells not only
lose their cell-cell adhesion of epithelial phenotype and exhibit
elevated motility and invasion, but also gain increased resistance
to chemotherapeutic drugs (1). In
addition, activation of EMT leads to the generation of cancer cells
with stem cell-like characteristics (7–10).
Therefore, cancer cells undergoing EMT may become drug-resistant
cancer cell progenitors, or cancer stem cells (CSCs) (11). Pulmonary stem cells were first
identified at the bronchioalveolar duct junction and these cells
were termed bronchioalveolar stem cells (12). In recent years, lung CSC research
has gained considerable momentum for both basic and clinical
applications (11).
The present review provides an overview of the EMT
of lung cancer in regards to the tumor microenvironment, CSCs,
related cytokines or chemical mediators, related transcription
factors, EMT epigenetics, invasion/metastasis and putative
therapeutic applications.
Inflammation in the lung microenvironment
contributes to tumor initiation and invasion, promoting cancer cell
EMT through its ability to induce the downregulation of epithelial
cell proteins and upregulation of mesenchymal cell proteins
(13,14). The continuous and bilateral
crosstalk, which occurs in the tumor microenvironment, is mediated
by molecules secreted by either tumor or microenvironment stromal
cells (15). This crosstalk of
inflammatory mediators between tumors and their stroma results in
tumor cell invasion, angiogenesis and metastasis (16).
Carcinoma-associated fibroblasts (CAFs) are known as
major components of the tumor microenvironment and are involved in
cancer cell growth and survival, including angiogenesis and
invasion (17). CAFs in the tumor
microenvironment are present at the invasive front of the tumor
(17). The conversion of
fibroblasts into CAFs is driven by cancer cell-derived cytokines,
such as transforming growth factor-β1 (TGF-β1) (15). CAFs produce extracellular matrix
molecules and growth factors, including TGF-β, FGF2 and VEGF,
leading to a conversion from a normal to a cancer-supporting
microenvironment, a process known as tumor stromatogenesis
(22). Bonde et al (23) reported that analysis of non-small
cell lung cancer revealed a positive correlation between
intratumoral CAF densities, EMT markers, intraepithelial TGF-β
levels and tumor grade.
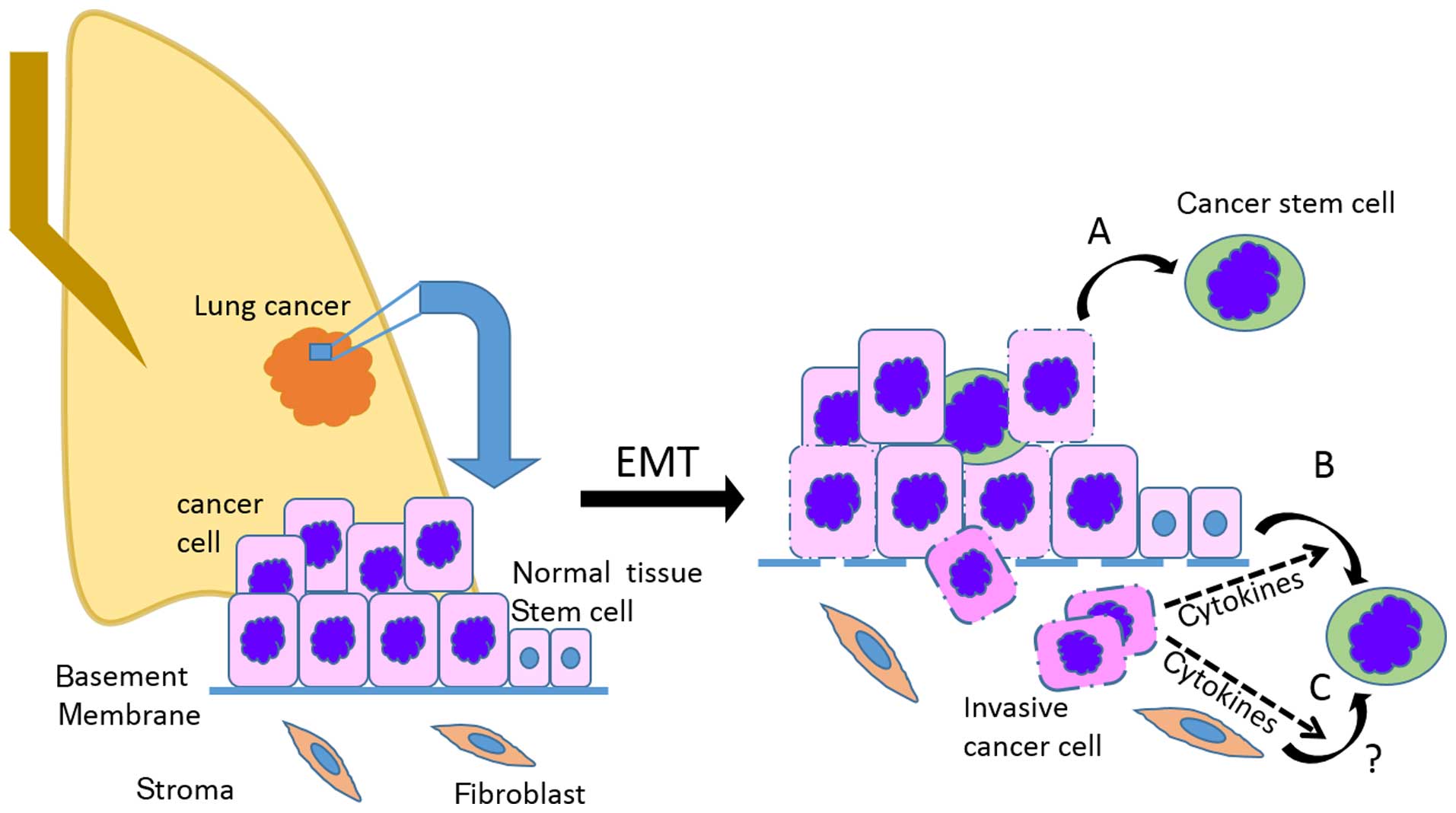
Numerous components of the tumor microenvironment
have been associated with CSCs and carcinogenesis, suggesting that
the developing tumor microenvironment drives expansion and possible
malignant conversion of normal tissue stem cells into CSCs via
induction of EMT (Fig. 1). The
precise origin of lung CSCs still remains to be resolved, but the
term CSCs refers to tumorigenic cells with the ability to
self-renew and to generate the diverse cell types present within
the tumor (24,25). Since most tumors are clonal in
origin, tumorigenic CSCs must have the ability to give rise to a
phenotypically diverse progeny (24) resulting in different degrees of
proliferation, differentiation and invasiveness (26,27).
It has been revealed that normal tissue stem cells and CSCs share
several stem cell-associated characteristics, such as the
expression of primitive stem cell markers (28).
The employment of stem cell markers remains the most
widely used approach for identifying lung CSCs (11). The markers currently used to isolate
lung CSCs, are CD44, CD24, CD133 and ALDH (28–30).
CD44 plays an important role in tumor cells undergoing an EMT-like
process and is associated with cancer progression (31,32).
CD44-positive NSCLC cells were shown to be enriched in CSCs and
resistant to cisplatin treatment (33). Sullivan et al (30) discovered that ALDH selects for a
subpopulation of self-renewing NSCLC stem-like cells with increased
tumorigenic potential, and NSCLC patients with ALDH1 tumor cells
were found to have a worse prognosis (17). Since a single definite lung CSC
marker has not yet been identified, a combination of markers are
required for a reliable quantification of CSCs (11).
Several studies have investigated the potential
clinical impact of lung CSC markers (11). The identification of new CSC
biomarkers for diagnosis and treatment would be crucial for a
better understanding of lung CSCs.
The molecular markers involved in EMT help to
distinguish an epithelial cell from a mesenchymal cell. Cells
undergoing EMT typically show an increase in the protein abundance
of vimentin, N-cadherin, fibronectin, and a decrease in E-cadherin,
cytokeratins and occludin (36).
Early molecular signs of EMT involve the
dissociation of cell-cell junctions induced by downregulation or
disassembly of junctional components, such as occludins, ZO-1,
claudins and E-cadherin (2,4). Loss of E-cadherin expression is
considered to be a hallmark of EMT (11). This loss is followed by upregulation
of several mesenchymal markers, which is associated with an
increased invasive potential (37).
The regulators involving mesenchymal differentiation, such as Snail
and Twist, are considered to regulate the EMT-induced decrease in
E-cadherin expression (38,39).
Vimentin is a widely expressed protein that is
constitutively expressed in mesenchymal cells, including
endothelial cells, macrophages, neutrophils, fibroblasts and
leukocytes (40,41). Vimentin is a typical marker of EMT
by which epithelial cells acquire a mesenchymal phenotype that
causes them to alter their shape and motility (42). In normal lung tissue, vimentin
expression is restricted to the basal and columnar cells of the
bronchial epithelium (43).
However, in lung cancer, increased vimentin expression is
associated with epithelial-derived tumor cells (44), and is used as a diagnostic marker
for invasive and metastatic tumor cells (45,46).
Other cytoskeletal changes involve the expression of
fibroblast-specific protein (FSP) in the bronchial epithelium,
which is associated with a more invasive phenotype and worse
prognosis in lung cancer (47).
The Notch signaling pathway participates in the
establishment of motile and invasive mesenchymal phenotypes from
polarized epithelial properties, involving downregulation of
epithelial markers and upregulation of mesenchymal markers
(48,49). The involvement of Notch signaling in
lung cancer was experimentally confirmed in a transgenic mouse
model (50). Clinical studies have
observed that Notch signaling impacts survival in lung cancer
patients (51). A recent study by
Donnem et al assessed the prognostic impact of Notch ligands
and receptors in NSCLC and found that high Notch expression was
statistically associated with poor outcomes in lung adenocarcinoma
patients (52).
TGF-β1, a ubiquitous cytokine with profound growth
inhibitory effects on epithelial and other tissues, orchestrates an
intricate signaling network that is crucial to determine cell
differentiation and proliferation, leading to not only tumor
suppression but also EMT related with tumor invasion (53). TGF-β1 mediates the interaction of
Smad to the promoters of Snail, contributing to the development of
EMT in NSCLC (54,55). This could be blocked by
pharmacological inactivation of Notch, indicating the key role of
Notch signaling in TGF-β-induced EMT (56). Meanwhile, inhibition of Notch
signaling significantly inhibited TGF-β1-induced expression of SMA,
suggesting that Notch induces EMT through a TGF-β1-Smad pathway
that activates SMA gene transcription (57).
It is well established that during the physiological
repair process after injury, loosening of epithelial-epithelial
cell contacts, alteration of cell-extracellular matrix contacts,
and morphological changes occur during wound repair (58). These phenotypic changes are thought
to be mediated by EMT transcription factors (59). Key transcription factors driving EMT
in lung cancer include Snail, Slug, Twist and ZEB family (Table I), which are repressors of
E-cadherin transcription.
The ZEB family (ZEB1 and ZEB2), another group of
transcription factors, contributes to EMT in lung cancer (61,62).
Immunohistochemical study revealed that ZEB1 and Twist are commonly
expressed in lung tumors (63).
Downregulation of claudin 1 by Snail and Slug has been observed of
lung cancer in EMT (64,65).
The transcriptional repressors of E-cadherin can be
regulated by hypoxia in lung cancer (17). Hypoxia-inducible factor-1 (HIF-1), a
major regulator of the cellular response to hypoxia, has been shown
to regulate vimentin gene expression (66). Therefore, vimentin transcriptional
regulation by HIF-1 may be a potential driver of EMT in lung cancer
(42).
The expression of CSC-associated transcription
factors could provide prognostic information of lung cancers
(11). Among them, Bmi-1 has been
shown to sustain stem cell properties in normal and cancerous lung
tissues (67,68). An increased expression of Oct4 and
Nanog was found to be associated with a worse prognosis in lung
adenocarcinoma patients (69).
Thyroid transcription factor-1 (TTF-1) is a transcription factor
that is expressed in ~75% of lung adenocarcinomas (3). Loss of expression of TTF-1 was
observed to facilitate the generation of EMT by tumor cells
resulting in a worse prognosis of patients.
Recently, researchers have focused their attention
on the roles of epigenetic regulation in the EMT process due to the
hypothesis that epigenetic alterations have important influences on
tumorigenesis (71). Histone
acetylation, microRNA (miRNA), and DNA methylation refer to three
major epigenetic modifications contributing to tumorigenesis
including EMT and cancer metastasis (72,73).
Aberrations of any of the three major histone
proteins, HATs, HDACs and histone readers, have been demonstrated
to be closely correlated with lung cancer (74). The microenvironment, such as
hypoxia, is demonstrated to be correlated with histone acetylation
which consequently affects the EMT process (74). Increasing evidence suggests that
histone acetylation/deacetylation may regulate E-cadherin during
EMT in several types of cancers, including lung cancer (75). The understanding of the mechanisms
of histone acetylation and its interaction with other epigenetic
modifications in EMT may provide the means by which to develop
potential therapeutic strategies with higher efficiency and fewer
side-effects (74).
miRNAs are a class of small non-coding RNAs that
negatively regulate gene expression by binding to homologous
regions in target mRNAs inducing mRNA degradation (76,77).
miRNAs play important roles in essential cellular processes
including cell growth, differentiation, apoptosis and immune
response (17). miRNAs serve as key
administrators of post-transcriptional regulation and participate
in EMT (78). Aberrant expression
of various miRNAs is related to tumor growth and metastasis
(79,80).
One of the first characterized miRNA families
relevant in carcinogenesis was the miR-17-92 cluster (81). It has been demonstrated that
miR-17-92 cluster expression is upregulated by the protooncogene,
c-myc, which itself is commonly dysregulated in human malignancies
(82). Among other c-myc-induced
miRNAs are miR-221 and miR-222, which target proteins involved in
cell cycle arrest (82). miR-9
suppresses E-cadherin expression and promotes metastasis (17). Overall, the c-myc-induced miRNA
network is reported to be directly related to tumor aggressiveness
in various types of cancers (83).
The role of EMT mediators in the regulation of
miRNAs is just beginning to be revealed (17). Transcriptional repressors, such as
Snail, Slug and Twist that are induced by inflammation are involved
in this regulation (17). For
example, Snail is able to upregulate miR-661 increasing the
metastatic potential of breast cancer cells (93). Twist upregulates a positive
regulator of cancer cell migration and invasion.
In recent years, miRNA studies have demonstrated
that miRNAs implicated in EMT may serve as diagnostic and
prognostic markers for various types of cancer (94). Further studies are needed to define
the detailed mechanisms to verify the role of individual miRNAs in
lung cancer. This may allow the development of novel therapeutic
strategies targeting oncogenic miRNAs in lung cancer.
EMT is largely thought to play an important role in
invasion and metastasis. EMT may enable cancer cells to lose their
cell polarity and cell-cell adhesive interactions, allowing the
cells to escape from the primary tumor (95). Cancer cells, that have acquired
mesenchymal characteristics, can more effectively invade
surrounding tissues and migrate to distant sites. Since tumor
metastasis is the main obstacle for long-term survival,
identification of molecular markers related to metastasis may
predict the prognosis of patients with lung cancer (3). Increased E-cadherin expression was
found to markedly decreased the invasion/migration of tumor cells
(96). In contrast, the
upregulation of N-cadherin expression is linked to the metastasis
of NSCLC (97).
The association between EMT and metastasis comes
from clinical observations that distant metastasis derived from a
variety of primary carcinomas resemble an epithelial phenotype
(98). For example, metastases in
distal organs derived from a variety of primary types of tumors
exhibit overt epithelial phenotypes (99,100).
These observations raise the possibility that tumor cells may
disseminate without switching to a mesenchymal phenotype, thereby
providing controversy to the requirement of EMT for metastasis
formation (98). In contrast, if
cancer cells must undergo EMT to disseminate, an important question
is why the resulting metastases closely resemble, at the
histopathologic level, the primary carcinomas from which they have
originated (98). This question has
led to the possibility that the disseminated mesenchymal tumor
cells recruited to target organs may undergo a reverse phenotypic
transition from a mesenchymal back to an epithelial phenotype by a
process called mesenchymal-to-epithelial transition (MET) (98). Xue et al (101) reported that disseminated breast
tumor cells expressed mesenchymal marker Fsp-1, suggesting that EMT
had occurred, which could shift back to an Fsp-1-negative
phenotype, suggesting MET. This finding suggests that cancer cells
may undergo MET in the secondary metastatic organ (98). E-cadherin was expressed at a higher
level in metastatic lesions in the brain from the primary lung
cancer (100). These results
indicate that EMT occurs during lung carcinogenesis as well as the
inverse process MET in metastatic sites (74).
Numerous strategies are being explored to develop
noninvasive methods by which to detect cancer (17). These include investigations into the
detection of circulating cancer cells, as well as identifying
biomarkers in bronchial, oral or nasal samples (17). Profiling serum-based miRNAs is also
under investigation and shows promise in identifying cancer
patients vs. non-cancer individuals (102,103). miRNAs, particularly the miR-200
family, have been implicated in EMT/MET transitions in cancer
(104). miR-200c was found to
inhibit EMT and induce an epithelial phenotype (105). In lung cancer, the number of
circulating tumor cells (CTCs) expressing epithelial marker EpCAM
was found to be lower compared with the number in other solid
tumors (106). However, when CTC
isolation is not based on epithelial markers, the CTC numbers are
similar to those of other solid tumors and have strong prognostic
value (107). These data suggest
that lung cancer CTCs lose their epithelial characteristics having
undergone EMT (108).
EMT has also been related to therapy resistance in
cancer, with both preclinical and clinical evidence (95). Therapeutic refractory lung cancer
cells frequently reveal an EMT phenotype, and signaling to block
EMT has been shown to enhance chemotherapy sensitivity (109,110). Taken together, EMT, metastasis and
drug resistance are intertwined in lung cancer, and lead to
aggressiveness and poor prognosis (51).
Although the underlying mechanisms have been studied
for many years, the overall survival rates have not been
significantly improved for patients receiving targeted therapy as
compared with chemotherapy (111,112). Currently, the major therapeutic
obstacles are tumor recurrence and metastasis even after surgical
resection, which are the main causes of mortality (74) in lung cancer patients. Studies
concerning the molecular biology of EMT have elucidated the
processes of invasion and metastasis, and it is expected that these
basic biological findings can be eventually translated into new
therapeutic approaches (95).
Receptor-specific approaches such as monoclonal
antibodies or siRNAs directed against Notch may be useful in
reducing the tumorigenicity and invasiveness of lung cancer
(113). For instance, nanoparticle
(NP) technology has been applied to deliver specific siRNAs to
knock down Notch1 to arrest tumor growth and reverse EMT by the
upregulation of miR-200 and downregulation of the transcription
factors ZEB1, ZEB2, Snail and Slug (114).
Intermediate filaments (IFs) are an attractive
potential therapeutic target for lung cancer, due to their
involvement in cellular motility, transcriptional regulation, and
association with EMT and tumor metastasis (42). Vimentin may be a key regulator of
several tumorigenic pathways, as it forms a complex that may
prevent the dephosphorylation of proteins in the complex,
inhibiting antitumor activity within tumor cells (42). Furthermore, inhibition of vimentin
expression by RNA interference has been shown to reduce metastatic
cell invasiveness and decrease tumor volume (115).
As Wnt signaling pathways have been shown to be
important in the pathogenesis of lung cancer (116), they could serve as another
promising therapeutic target. At present, there are multiple
therapeutic approaches targeting Wnt signaling in lung cancer which
may be applied in the near future (117,118).
Since CSCs share several stem-like characteristics
with normal stem cells, targeted CSC therapies should be designed
to preserve normal stem cells and to 'hit' only CSC-specific
signaling pathways (119). The
final aim is to develop a CSC-targeted therapy that will result in
the complete elimination of CSCs. Presumably, this could be
achieved through the disruption of signaling pathways that control
the self-renewal, proliferation and differentiation of CSCs
(11). Since most studies on the
therapeutic efficacy of CSC targeting drugs are still in their
early phases, more information must be gathered to verify the
clinical importance of these drugs (11).
In conclusion, much research has highlighted the
involvement of EMT in lung cancer. The precise clinical importance
of the association of EMT and tumor invasion/metastasis,
chemoresistance of tumor cells, de novo generation of CSCs,
and tumor microenvironment remains to be determined. However, the
development of drugs that target chemical mediators known to
promote EMT suggest that regulation of EMT processes in the
clinical setting may be possible. We anticipate the identification
of novel molecular targets to facilitate the development of
therapeutic agents for lung cancer as the relationships between
EMT, tumor microenvironment, and CSCs are further explored in the
near future.
The present study was supported by a grant from
Xavier, Catholic University of Daegu (2015).
|
1
|
Xiao D and He J: Epithelial mesenchymal
transition and lung cancer. J Thorac Dis. 2:154–159.
2010.PubMed/NCBI
|
|
2
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Y, Wu H, Zhang M, Ding L, Meng F and
Fan X: Expression of the epithelial-mesenchymal transition-related
proteins and their clinical significance in lung adenocarcinoma.
Diagn Pathol. 8:892013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gupta GP and Massagué J: Cancer
metastasis: Building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klymkowsky MW and Savagner P:
Epithelial-mesenchymal transition: A cancer researcher's conceptual
friend and foe. Am J Pathol. 174:1588–1593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morel AP, Lièvre M, Thomas C, Hinkal G,
Ansieau S and Puisieux A: Generation of breast cancer stem cells
through epithelial-mesenchymal transition. PLoS One. 3:e28882008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Battula VL, Evans KW, Hollier BG, et al:
Epithelial-mesenchymal transition-derived cells exhibit
multilineage differentiation potential similar to mesenchymal stem
cells. Stem Cells. 28:1435–1445. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koren A, Motaln H and Cufer T: Lung cancer
stem cells: A biological and clinical perspective. Cell Oncol.
36:265–275. 2013. View Article : Google Scholar
|
|
12
|
Kim CF, Jackson EL, Woolfenden AE,
Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT and Jacks T:
Identification of bronchioalveolar stem cells in normal lung and
lung cancer. Cell. 121:823–835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Willis BC and Borok Z: TGF-beta-induced
EMT: Mechanisms and implications for fibrotic lung disease. Am J
Physiol Lung Cell Mol Physiol. 293:L525–L534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Wever O and Mareel M: Role of tissue
stroma in cancer cell invasion. J Pathol. 200:429–447. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heinrich EL, Walser TC, Krysan K, Liclican
EL, Grant JL, Rodriguez NL and Dubinett SM: The inflammatory tumor
microenvironment, epithelial mesenchymal transition and lung
carcinogenesis. Cancer Microenviron. 5:5–18. 2012. View Article : Google Scholar :
|
|
18
|
Nizet V and Johnson RS: Interdependence of
hypoxic and innate immune responses. Nat Rev Immunol. 9:609–617.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fitzpatrick SF, Tambuwala MM, Bruning U,
Schaible B, Scholz CC, Byrne A, O'Connor A, Gallagher WM, Lenihan
CR, Garvey JF, et al: An intact canonical NF-κB pathway is required
for inflammatory gene expression in response to hypoxia. J Immunol.
186:1091–1096. 2011. View Article : Google Scholar
|
|
20
|
Semenza GL: Defining the role of
hypoxia-inducible factor 1 in cancer biology and therapeutics.
Oncogene. 29:625–634. 2010. View Article : Google Scholar :
|
|
21
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giatromanolaki A, Sivridis E and
Koukourakis MI: The pathology of tumor stromatogenesis. Cancer Biol
Ther. 6:639–645. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bonde AK, Tischler V, Kumar S, Soltermann
A and Schwendener RA: Intratumoral macrophages contribute to
epithelial-mesenchymal transition in solid tumors. BMC Cancer.
12:352012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu WH, You N, Zhang N, Yan HT, Wang T,
Huang Z, Liu HB and Tang LJ: Interpretation of interlocking key
issues of cancer stem cells in malignant solid tumors. Cell Oncol.
35:397–409. 2012. View Article : Google Scholar
|
|
26
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: Accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sullivan JP, Minna JD and Shay JW:
Evidence for self-renewing lung cancer stem cells and their
implications in tumor initiation, progression, and targeted
therapy. Cancer Metastasis Rev. 29:61–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kitamura H, Okudela K, Yazawa T, Sato H
and Shimoyamada H: Cancer stem cell: Implications in cancer biology
and therapy with special reference to lung cancer. Lung Cancer.
66:275–281. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sullivan JP, Spinola M, Dodge M, Raso MG,
Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, et
al: Aldehyde dehydrogenase activity selects for lung adenocarcinoma
stem cells dependent on notch signaling. Cancer Res. 70:9937–9948.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leung EL, Fiscus RR, Tung JW, Tin VP,
Cheng LC, Sihoe AD, Fink LM, Ma Y and Wong MP: Non-small cell lung
cancer cells expressing CD44 are enriched for stem cell-like
properties. PLoS One. 5:e140622010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eramo A, Lotti F, Sette G, Pilozzi E,
Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C and De
Maria R: Identification and expansion of the tumorigenic lung
cancer stem cell population. Cell Death Differ. 15:504–514. 2008.
View Article : Google Scholar
|
|
35
|
Hilbe W, Dirnhofer S, Oberwasserlechner F,
Schmid T, Gunsilius E, Hilbe G, Wöll E and Kähler CM: CD133
positive endothelial progenitor cells contribute to the tumour
vasculature in non-small cell lung cancer. J Clin Pathol.
57:965–969. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Weber CE, Li NY, Wai PY and Kuo PC:
Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound
healing and tissue remodeling after injury. J Burn Care Res.
33:311–318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Castro Alves C, Rosivatz E, Schott C,
Hollweck R, Becker I, Sarbia M, Carneiro F and Becker KF: Slug is
overexpressed in gastric carcinomas and may act synergistically
with SIP1 and Snail in the down-regulation of E-cadherin. J Pathol.
211:507–515. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aamodt R, Bondi J, Andersen SN, Bakka A,
Bukholm G and Bukholm IR: The prognostic impact of protein
expression of E-cadherin-catenin complexes differs between rectal
and colon carcinoma. Gastroenterol Res Pract. 2010:616–623. 2010.
View Article : Google Scholar
|
|
40
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Crystal RG, Randell SH, Engelhardt JF,
Voynow J and Sunday ME: Airway epithelial cells: Current concepts
and challenges. Proc Am Thorac Soc. 5:772–777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kidd ME, Shumaker DK and Ridge KM: The
role of vimentin intermediate filaments in the progression of lung
cancer. Am J Respir Cell Mol Biol. 50:1–6. 2014.
|
|
43
|
Schoumacher M, Goldman RD, Louvard D and
Vignjevic DM: Actin, microtubules, and vimentin intermediate
filaments cooperate for elongation of invadopodia. J Cell Biol.
189:541–556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Helfand BT, Mendez MG, Murthy SN, Shumaker
DK, Grin B, Mahammad S, Aebi U, Wedig T, Wu YI, Hahn KM, et al:
Vimentin organization modulates the formation of lamellipodia. Mol
Biol Cell. 22:1274–1289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Geiger TR and Peeper DS: Metastasis
mechanisms. Biochim Biophys Acta. 1796:293–308. 2009.PubMed/NCBI
|
|
46
|
Gilles C, Polette M, Zahm JM, Tournier JM,
Volders L, Foidart JM and Birembaut P: Vimentin contributes to
human mammary epithelial cell migration. J Cell Sci. 112:4615–4625.
1999.PubMed/NCBI
|
|
47
|
Kimura K, Endo Y, Yonemura Y, Heizmann CW,
Schafer BW, Watanabe Y and Sasaki T: Clinical significance of
S100A4 and E-cadherin-related adhesion molecules in non-small cell
lung cancer. Int J Oncol. 16:1125–1131. 2000.PubMed/NCBI
|
|
48
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chang AC, Garside VC, Fournier M, Smrz J,
Vrljicak P, Umlandt P, Fuller M, Robertson G, Zhao Y, Tam A, et al:
A Notch-dependent transcriptional hierarchy promotes mesenchymal
transdifferentiation in the cardiac cushion. Dev Dyn. 243:894–905.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Allen TD, Rodriguez EM, Jones KD and
Bishop JM: Activated Notch1 induces lung adenomas in mice and
cooperates with Myc in the generation of lung adenocarcinoma.
Cancer Res. 71:6010–6018. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S,
Chen Y and Wu K: Notch signaling and EMT in non-small cell lung
cancer: Biological significance and therapeutic application. J
Hematol Oncol. 7:87–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Donnem T, Andersen S, Al-Shibli K, Al-Saad
S, Busund LT and Bremnes RM: Prognostic impact of Notch ligands and
receptors in nonsmall cell lung cancer: Coexpression of Notch-1 and
vascular endothelial growth factor-A predicts poor survival.
Cancer. 116:5676–5685. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zavadil J and Böttinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin LC, Hsu SL, Wu CL and Hsueh CM: TGFβ
can stimulate the p38/β-catenin/PPARγ signaling pathway to promote
the EMT, invasion and migration of non-small cell lung cancer (H460
cells). Clin Exp Metastasis. 31:881–895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang HJ, Wang HY, Zhang HT, Su JM, Zhu J,
Wang HB, Zhou WY, Zhang H, Zhao MC, Zhang L, et al: Transforming
growth factor-β1 promotes lung adenocarcinoma invasion and
metastasis by epithelial-to-mesenchymal transition. Mol Cell
Biochem. 355:309–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zavadil J, Cermak L, Soto-Nieves N and
Böttinger EP: Integration of TGF-beta/Smad and Jagged1/Notch
signalling in epithelial-to-mesenchymal transition. EMBO J.
23:1155–1165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Matsuno Y, Coelho AL, Jarai G, Westwick J
and Hogaboam CM: Notch signaling mediates TGF-β1-induced
epithelial-mesenchymal transition through the induction of Snai1.
Int J Biochem Cell Biol. 44:776–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vaughan AE and Chapman HA: Regenerative
activity of the lung after epithelial injury. Biochim Biophys Acta.
1832:922–930. 2013. View Article : Google Scholar
|
|
59
|
Savagner P, Kusewitt DF, Carver EA,
Magnino F, Choi C, Gridley T and Hudson LG: Developmental
transcription factor slug is required for effective
re-epithelialization by adult keratinocytes. J Cell Physiol.
202:858–866. 2005. View Article : Google Scholar
|
|
60
|
Yanagawa J, Walser TC, Zhu LX, Hong L,
Fishbein MC, Mah V, Chia D, Goodglick L, Elashoff DA, Luo J, et al:
Snail promotes CXCR2 ligand-dependent tumor progression in
non-small cell lung carcinoma. Clin Cancer Res. 15:6820–6829. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yang Y, Ahn YH, Chen Y, Tan X, Guo L,
Gibbons DL, Ungewiss C, Peng DH, Liu X, Lin SH, et al: ZEB1
sensitizes lung adenocarcinoma to metastasis suppression by PI3K
antagonism. J Clin Invest. 124:2696–2708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Argast GM, Krueger JS, Thomson S,
Sujka-Kwok I, Carey K, Silva S, O'Connor M, Mercado P, Mulford IJ,
Young GD, et al: Inducible expression of TGFβ, snail and Zeb1
recapitulates EMT in vitro and in vivo in a NSCLC model. Clin Exp
Metastasis. 28:593–614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Merikallio H, Kaarteenaho R, Pääkkö P,
Lehtonen S, Hirvikoski P, Mäkitaro R, Harju T and Soini Y: Zeb1 and
twist are more commonly expressed in metastatic than primary lung
tumours and show inverse associations with claudins. J Clin Pathol.
64:136–140. 2011. View Article : Google Scholar
|
|
64
|
Kojima T, Takano K, Yamamoto T, Murata M,
Son S, Imamura M, Yamaguchi H, Osanai M, Chiba H, Himi T, et al:
Transforming growth factor-beta induces epithelial to mesenchymal
transition by down-regulation of claudin-1 expression and the fence
function in adult rat hepatocytes. Liver Int. 28:534–545. 2008.
View Article : Google Scholar
|
|
65
|
Martínez-Estrada OM, Cullerés A, Soriano
FX, Peinado H, Bolós V, Martínez FO, Reina M, Cano A, Fabre M and
Vilaró S: The transcription factors Slug and Snail act as
repressors of Claudin-1 expression in epithelial cells. Biochem J.
394:449–457. 2006. View Article : Google Scholar :
|
|
66
|
Krishnamachary B, Berg-Dixon S, Kelly B,
Agani F, Feldser D, Ferreira G, Iyer N, LaRusch J, Pak B, Taghavi
P, et al: Regulation of colon carcinoma cell invasion by
hypoxia-inducible factor 1. Cancer Res. 63:1138–1143.
2003.PubMed/NCBI
|
|
67
|
Dovey JS, Zacharek SJ, Kim CF and Lees JA:
Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem
cell expansion. Proc Natl Acad Sci USA. 105:11857–11862. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wu KJ: Direct activation of Bmi1 by
Twist1: Implications in cancer stemness, epithelial-mesenchymal
transition, and clinical significance. Chang Gung Med J.
34:229–238. 2011.PubMed/NCBI
|
|
69
|
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong
CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS, et al:
Coexpression of Oct4 and Nanog enhances malignancy in lung
adenocarcinoma by inducing cancer stem cell-like properties and
epithelial-mesenchymal transdifferentiation. Cancer Res.
70:10433–10444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yu M, Smolen GA, Zhang J, Wittner B,
Schott BJ, Brachtel E, Ramaswamy S, Maheswaran S and Haber DA: A
developmentally regulated inducer of EMT, LBX1, contributes to
breast cancer progression. Genes Dev. 23:1737–1742. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sandoval J and Esteller M: Cancer
epigenomics: Beyond genomics. Curr Opin Genet Dev. 22:50–55. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial-mesenchymal transition in cancer: Parallels between
normal development and tumor progression. J Mammary Gland Biol
Neoplasia. 15:117–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Giudice FS, Pinto DS Jr, Nör JE, Squarize
CH and Castilho RM: Inhibition of histone deacetylase impacts
cancer stem cells and induces epithelial-mesenchyme transition of
head and neck cancer. PLoS One. 8:e586722013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang L, Liu Z, Ma W and Wang B: The
landscape of histone acetylation involved in epithelial-mesenchymal
transition in lung cancer. J Cancer Res Ther. 9(Suppl 2): S86–S91.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang Y and Shang Y: Epigenetic control of
epithelial-to-mesenchymal transition and cancer metastasis. Exp
Cell Res. 319:160–169. 2013. View Article : Google Scholar
|
|
76
|
Fire A, Xu S, Montgomery MK, Kostas SA,
Driver SE and Mello CC: Potent and specific genetic interference by
double-stranded RNA in Caenorhabditis elegans. Nature. 391:806–811.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gibbons DL, Lin W, Creighton CJ, Rizvi ZH,
Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y,
Pertsemlidis A, et al: Contextual extracellular cues promote tumor
cell EMT and metastasis by regulating miR-200 family expression.
Genes Dev. 23:2140–2151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ma L, Young J, Prabhala H, Pan E, Mestdagh
P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S,
et al: miR 9, a MYC/MYCN activated microRNA, regulates E cadherin
and cancer metastasis. Nat Cell Biol. 12:247–256. 2010.PubMed/NCBI
|
|
80
|
Valastyan S, Reinhardt F, Benaich N,
Calogrias D, Szász AM, Wang ZC, Brock JE, Richardson AL and
Weinberg RA: A pleiotropically acting microRNA, miR 31, inhibits
breast cancer metastasis. Cell. 137:1032–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tanzer A and Stadler PF: Molecular
evolution of a microRNA cluster. J Mol Biol. 339:327–335. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kim JW, Mori S and Nevins JR: Myc-induced
microRNAs integrate Myc-mediated cell proliferation and cell fate.
Cancer Res. 70:4820–4828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Mestdagh P, Fredlund E, Pattyn F, Schulte
JH, Muth D, Vermeulen J, Kumps C, Schlierf S, De Preter K, Van Roy
N, et al: MYCN/c-MYC-induced microRNAs repress coding gene networks
associated with poor outcome in MYCN/c-MYC-activated tumors.
Oncogene. 29:1394–1404. 2010. View Article : Google Scholar
|
|
84
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Schramedei K, Mörbt N, Pfeifer G, Läuter
J, Rosolowski M, Tomm JM, von Bergen M, Horn F and Brocke-Heidrich
K: MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4.
Oncogene. 30:2975–2985. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mongroo PS and Rustgi AK: The role of the
miR-200 family in epithelial-mesenchymal transition. Cancer Biol
Ther. 10:219–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Manavalan TT, Teng Y, Litchfield LM,
Muluhngwi P, Al-Rayyan N and Klinge CM: Reduced expression of
miR-200 family members contributes to antiestrogen resistance in
LY2 human breast cancer cells. PLoS One. 8:e623342013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hill L, Browne G and Tulchinsky E:
ZEB/miR-200 feedback loop: At the crossroads of signal transduction
in cancer. Int J Cancer. 132:745–754. 2013. View Article : Google Scholar
|
|
89
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Burk U, Schubert J, Wellner U, Schmalhofer
O, Vincan E, Spaderna S and Brabletz T: A reciprocal repression
between ZEB1 and members of the miR-200 family promotes EMT and
invasion in cancer cells. EMBO Rep. 9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang B, Herman-Edelstein M, Koh P, Burns
W, Jandeleit-Dahm K, Watson A, Saleem M, Goodall GJ, Twigg SM,
Cooper ME, et al: E-cadherin expression is regulated by miR-192/215
by a mechanism that is independent of the profibrotic effects of
transforming growth factor-beta. Diabetes. 59:1794–1802. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lamouille S, Subramanyam D, Blelloch R and
Derynck R: Regulation of epithelial-mesenchymal and
mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell
Biol. 25:200–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Vetter G, Saumet A, Moes M, Vallar L, Le
Béchec A, Laurini C, Sabbah M, Arar K, Theillet C, Lecellier CH, et
al: miR-661 expression in SNAI1-induced epithelial to mesenchymal
transition contributes to breast cancer cell invasion by targeting
Nectin-1 and StarD10 messengers. Oncogene. 29:4436–4448. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Brase JC, Wuttig D, Kuner R and Sültmann
H: Serum microRNAs as non-invasive biomarkers for cancer. Mol
Cancer. 9:3062010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Creighton CJ, Gibbons DL and Kurie JM: The
role of epithelial-mesenchymal transition programming in invasion
and metastasis: A clinical perspective. Cancer Manag Res.
5:187–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mateen S, Raina K, Agarwal C, Chan D and
Agarwal R: Silibinin synergizes with histone deacetylase and DNA
methyltransferase inhibitors in upregulating E-cadherin expression
together with inhibition of migration and invasion of human
non-small cell lung cancer cells. J Pharmacol Exp Ther.
345:206–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhang X, Liu G, Kang Y, Dong Z, Qian Q and
Ma X: N-cadherin expression is associated with acquisition of EMT
phenotype and with enhanced invasion in erlotinib-resistant lung
cancer cell lines. PLoS One. 8:e576922013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gao D, Vahdat LT, Wong S, Chang JC and
Mittal V: Microenvironmental regulation of epithelial-mesenchymal
transitions in cancer. Cancer Res. 72:4883–4889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yates C: Prostate tumor cell plasticity: A
consequence of the microenvironment. Adv Exp Med Biol. 720:81–90.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Prudkin L, Liu DD, Ozburn NC, Sun M,
Behrens C, Tang X, Brown KC, Bekele BN, Moran C and Wistuba II:
Epithelial-to-mesenchymal transition in the development and
progression of adenocarcinoma and squamous cell carcinoma of the
lung. Mod Pathol. 22:668–678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xue C, Plieth D, Venkov C, Xu C and
Neilson EG: The gatekeeper effect of epithelial-mesenchymal
transition regulates the frequency of breast cancer metastasis.
Cancer Res. 63:3386–3394. 2003.PubMed/NCBI
|
|
102
|
Houbaviy HB, Murray MF and Sharp PA:
Embryonic stem cell-specific MicroRNAs. Dev Cell. 5:351–358. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lu Y, Thomson JM, Wong HY, Hammond SM and
Hogan BL: Transgenic over-expression of the microRNA miR-17–92
cluster promotes proliferation and inhibits differentiation of lung
epithelial progenitor cells. Dev Biol. 310:442–453. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bullock MD, Sayan AE, Packham GK and
Mirnezami AH: MicroRNAs: Critical regulators of epithelial to
mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in
cancer progression. Biol Cell. 104:3–12. 2012. View Article : Google Scholar
|
|
105
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Krebs MG, Sloane R, Priest L, Lancashire
L, Hou JM, Greystoke A, Ward TH, Ferraldeschi R, Hughes A, Clack G,
et al: Evaluation and prognostic significance of circulating tumor
cells in patients with non-small-cell lung cancer. J Clin Oncol.
29:1556–1563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Hofman V, Bonnetaud C, Ilie MI, Vielh P,
Vignaud JM, Fléjou JF, Lantuejoul S, Piaton E, Mourad N, Butori C,
et al: Preoperative circulating tumor cell detection using the
isolation by size of epithelial tumor cell method for patients with
lung cancer is a new prognostic biomarker. Clin Cancer Res.
17:827–835. 2011. View Article : Google Scholar
|
|
108
|
Bartis D, Mise N, Mahida RY, Eickelberg O
and Thickett DR: Epithelial-mesenchymal transition in lung
development and disease: Does it exist and is it important? Thorax.
69:760–765. 2014. View Article : Google Scholar
|
|
109
|
Buonato JM and Lazzara MJ: ERK1/2 blockade
prevents epithelial-mesenchymal transition in lung cancer cells and
promotes their sensitivity to EGFR inhibition. Cancer Res.
74:309–319. 2014. View Article : Google Scholar :
|
|
110
|
Wilson C, Nicholes K, Bustos D, Lin E,
Song Q, Stephan JP, Kirkpatrick DS and Settleman J: Overcoming
EMT-associated resistance to anti-cancer drugs via Src/FAK pathway
inhibition. Oncotarget. 5:7328–7341. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fukuoka M, Wu YL, Thongprasert S,
Sunpaweravong P, Leong SS, Sriuranpong V, Chao TY, Nakagawa K, Chu
DT, Saijo N, et al: Biomarker analyses and final overall survival
results from a phase III, randomized, open-label, first-line study
of gefitinib versus carboplatin/paclitaxel in clinically selected
patients with advanced non-small-cell lung cancer in Asia (IPASS).
J Clin Oncol. 29:2866–2874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó
L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al:
Crizotinib versus chemotherapy in advanced ALK-positive lung
cancer. N Engl J Med. 368:2385–2394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Li Y, Burns JA, Cheney CA, Zhang N,
Vitelli S, Wang F, Bett A, Chastain M, Audoly LP and Zhang ZQ:
Distinct expression profiles of Notch-1 protein in human solid
tumors: Implications for development of targeted therapeutic
monoclonal antibodies. Biologics. 4:163–171. 2010.PubMed/NCBI
|
|
114
|
Sureban SM, May R, Mondalek FG, Qu D,
Ponnurangam S, Pantazis P, Anant S, Ramanujam RP and Houchen CW:
Nanoparticle-based delivery of siDCAMKL-1 increases microRNA-144
and inhibits colorectal cancer tumor growth via a Notch-1 dependent
mechanism. J Nanobiotechnology. 9:402011. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Paccione RJ, Miyazaki H, Patel V, Waseem
A, Gutkind JS, Zehner ZE and Yeudall WA: Keratin down-regulation in
vimentin-positive cancer cells is reversible by vimentin RNA
interference, which inhibits growth and motility. Mol Cancer Ther.
7:2894–2903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Bartis D, Csongei V, Weich A, Kiss E,
Barko S, Kovacs T, Avdicevic M, D'Souza VK, Rapp J, Kvell K, et al:
Down-regulation of canonical and up-regulation of non-canonical Wnt
signalling in the carcinogenic process of squamous cell lung
carcinoma. PLoS One. 8:e573932013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Henderson WR Jr, Chi EY, Ye X, Nguyen C,
Tien YT, Zhou B, Borok Z, Knight DA and Kahn M: Inhibition of
Wnt/β-catenin/CREB binding protein (CBP) signaling reverses
pulmonary fibrosis. Proc Natl Acad Sci USA. 107:14309–14314. 2010.
View Article : Google Scholar
|
|
118
|
Tennis MA, Van Scoyk M, Heasley LE,
Vandervest K, Weiser-Evans M, Freeman S, Keith RL, Simpson P,
Nemenoff RA and Winn RA: Prostacyclin inhibits non-small cell lung
cancer growth by a frizzled 9-dependent pathway that is blocked by
secreted frizzled-related protein 1. Neoplasia. 12:244–253. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Eramo A, Haas TL and De Maria R: Lung
cancer stem cells: Tools and targets to fight lung cancer.
Oncogene. 29:4625–4635. 2010. View Article : Google Scholar : PubMed/NCBI
|